
Systémové amyloidózy v biopsiích ledvin
Systemic Amyloidoses in Renal Biopsy Samples
The kidneys are one of the most frequent sites of amyloid deposition during systemic AL, AA, and several hereditary amyloidoses. Distinguishing between different forms of amyloids is clinically important because of their different treatment.
Material and methods:
We present a 5-year retrospective study of amyloidoses diagnosed in renal biopsy samples. The classification of amyloidosis was made by immunofluorescence and immunohistochemical staining with antibodies to kappa and lambda immunoglobulin light chains, and for serum amyloid A protein.
Results:
From January 2003 to December 2007, 996 renal biopsy samples from one centre were evaluated. Amyloidosis was diagnosed in 62 samples (6.2%); 33 (53.2%) were classified as AL and 25 (40.3%) as AA amyloidosis. Four cases have remained unclassified. We did not identify any difference in the distribution of deposits among cases with AL and AA amyloidosis, respectively. The majority of patients underwent the renal biopsy due to severe proteinuria or nephrotic syndrome. Three patients had very low proteinuria, less than 0.5g/day. Diagnosis of amyloidosis was suspected by nephrologists in 48 patients (77.4%).
Conclusion:
Diagnosis of amyloidosis involves detection of amyloid deposits and classification of the amyloid form, which represent the basic step for appropriate therapy. Alltogether it is not an easy task for pathologists, and with the emergence of markedly different treatments depending on the specific type of amyloid, the precise typing is of increasing clinical importance and should be performed with great care. Immunofluorescence can be very useful in daily practice for classification of the type of amyloidosis.
Key words:
systemic amyloidosis - diagnosis of amyloidosis - renal amyloidosis
:
L. Bauerová 1; E. Honsová 1,2; R. Ryšavá 3; C. Povýšil 1
:
Ústav patologie, 1. LF UK a VFN, Praha
1; Pracoviště klinické a transplantační patologie, IKEM, Praha
2; Klinika nefrologie, 1. LF UK a VFN, Praha
3
:
Čes.-slov. Patol., 45, 2009, No. 3, p. 64-68
:
Original Article
Ledviny bývají jedním z pravidelně postižených orgánů v průběhu systémové amyloidózy (AA, AL a některých typů hereditárních amyloidóz). Rozpoznání přesného typu amyloidózy je velmi důležité, protože každý typ vyžaduje odlišnou léčbu.
Soubor a metodika:
Prezentujeme retrospektivní soubor renálních biopsií s diagnózou amyloidózy z období 2003–2007. Klasifikace typu amyloidu byla provedena na základě imunofluorescenčního a imunohistochemického vyšetření, s použitím protilátek proti kappa a lambda lehkým řetězcům a sérovému proteinu A.
Výsledky:
Z celkového počtu 996 renálních biopsií dospělých pacientů byla amyloidóza diagnostikována v 62 vzorcích (6,2 %), z toho 33 (53,2 %) případů bylo zařazeno jako AL a 25 (40,3 %) jako AA amyloidóza; 4 případy zůstaly nezařazeny. Závislost rozložení depozit ve tkáni na typu amyloidózy nebyla v našem souboru prokázána. V době diagnózy mělo v klinickém obraze nefrotický syndrom nebo významnou proteinurii 57 (91,9 %) pacientů. Ve 3 případech byla proteinurie pouze do 0,5 g/den. Klinické údaje vedly k podezření na amyloidózu v 77,4 % případů. U čtvrtiny pacientů byla diagnóza amyloidózy neočekávaná a u 2 pacientů klinickým datům neodpovídal typ amyloidu.
Závěr:
Nezbytným předpokladem pro kauzální léčbu pacientů s amyloidózou je stanovení typu amyloidu. Určení typu amyloidu není jednoduchou záležitostí a vyžaduje použití různých technik, mezi kterými má v praxi důležité postavení imunofluorescenční vyšetření.
Klíčová slova:
systémová amyloidóza – amyloidóza ledvin – diagnóza amyloidózy
Amyloidózy představují skupinu nesourodých onemocnění (získaných i vrozených), která spojuje extracelulární ukládání původně rozpustných proteinů nebo jejich fragmentů ve formě amyloidu, tj. abnormálních nerozpustných fibril s charakteristickou ß-strukturou skládaného listu (zodpovědnou za afinitu ke Kongo červeni). Fibrily amyloidu jsou výsledkem změny konfigurace různých prekurzorových proteinů, které po nejasném impulsu vytvářejí protofilamenta a následně fibrily amyloidu. U všech typů amyloidóz dochází k interakci fibril s nefibrilárními složkami (glykosaminoglykany a apolipoproteiny E a J), které jednak podporují ukládání fibril a jednak stabilizují fibrilární formace ve tkáni (9,10).Další nefibrilární složkou je P komponenta, jejímž prekurzorem je sérový amyloidní protein P (SAP) a jejíž radioaktivní značení umožňuje sledovat lokalizaci depozit amyloidu během života pacienta. Amyloidózy se v současnosti klasifikují podle typu prekurzorových proteinů, kterých je dnes známo více než 20. Z klinického pohledu jsou v popředí zájmu systémové amyloidózy, které představují vždy závažné, život ohrožující onemocnění a kde časná diagnóza je klíčová pro kauzální léčbu. Ve skupině systémových amyloidóz jsou v praxi nejčastější a tím klinicky nejvýznamnější AL (podle dřívější terminologie primární) a AA (dříve sekundární) amyloidóza.
AL amyloidóza
Vzniká při klonálním onemocnění plazmatických buněk event. lymfoplazmocytů schopných vytvářet imunoglobuliny. Fibrily se skládají z monoklonálních lehkých řetězců nebo častěji z fragmentů různých částí lehkých řetězců. U většiny nemocných nedochází k rozvoji maligního nemocnění (mnohotný myelom), ale pouze k monoklonální gamapatii označované jako MGUS (monoclonal gammopathy ofundetermined significance). Lehké řetězce λ jsou více amyloidogenní než řetězce κ (9). Vzácně je popisována amyloidóza vznikající z těžkých řetězců imunoglobulinů (6). AL amyloidóza je nejčastějším typem amyloidózy v USA a v Evropě.
AA amyloidóza
Je komplikací chronických zánětlivých procesů, v Evropě a USA nejčastěji revmatoidní artritidy, zánětlivého onemocnění střev a tzv. familiární středozemní horečky (familial Mediterranean fever). Prekurzorovým proteinem je N-terminální fragment sérového amyloidu A (SAA), apolipoprotein syntetizovaný játry jako reaktant akutní fáze zánětu (3). AA amyloidózaje častá v rozvojových zemích v souvislosti s chronickými infekcemi, např. TBC. Představuje 45% všech systémových amyloidóz celosvětově a její zvýšený výskyt byl zaznamenán u uživatelů drog a nemocných AIDS (8).
Diagnóza amyloidózy s určením typu amyloidu je možná pouze na základě bioptického vyšetření a patří tedy plně do rukou patologa. Klasickým průkazem, společným pro všechny typy amyloidů, je pozitivita v barvení Kongo červení, s příslušným zeleným dvojlomem v polarizovaném světle (obr. 1, 2). Také ultrastrukturální průkaz nevětvených, náhodně uspořádaných fibril amyloidu je pro všechny typy stejný. V průběhu systémových amyloidóz bývají ledviny jedním z pravidelně postižených orgánů. Onemocnění ledvin vede nejčastěji k rozvoji významné proteinurie či k plně vyvinutému nefrotickému syndromu s velkými ztrátami bílkovin (i více než 20 g/den), což je v mnoha případech postupně následováno selháním ledvin.
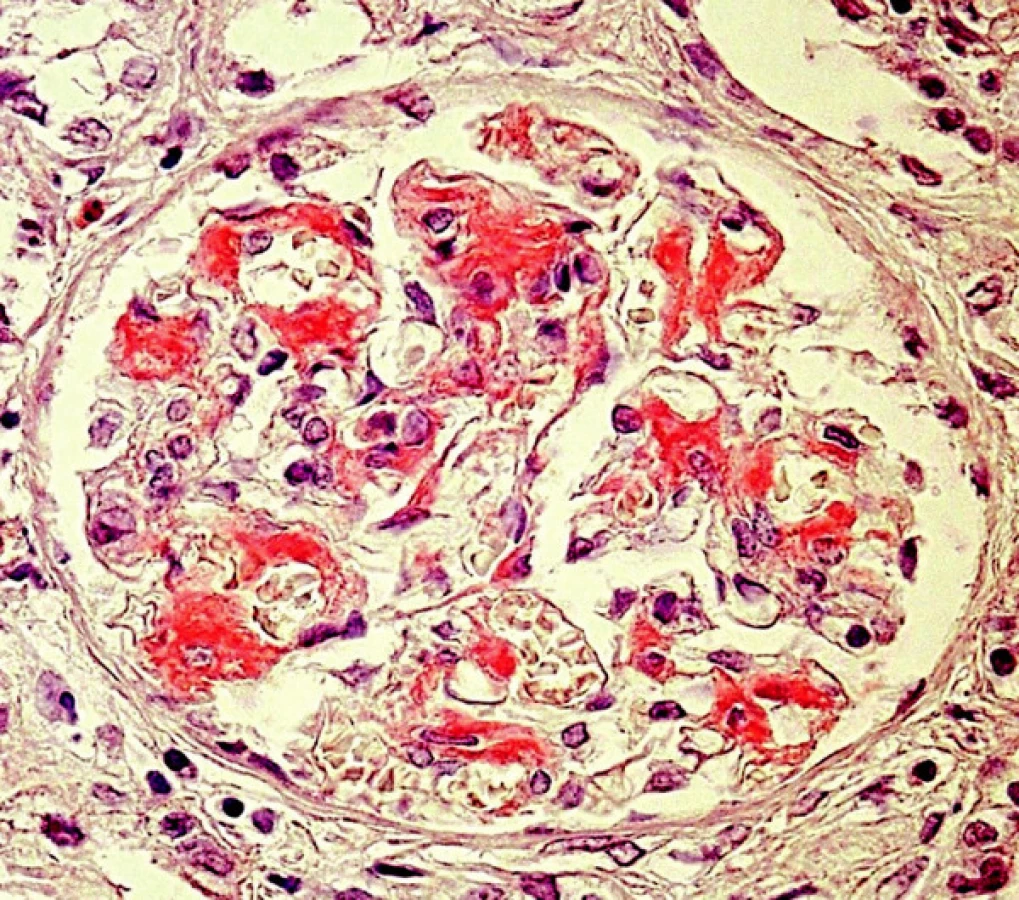
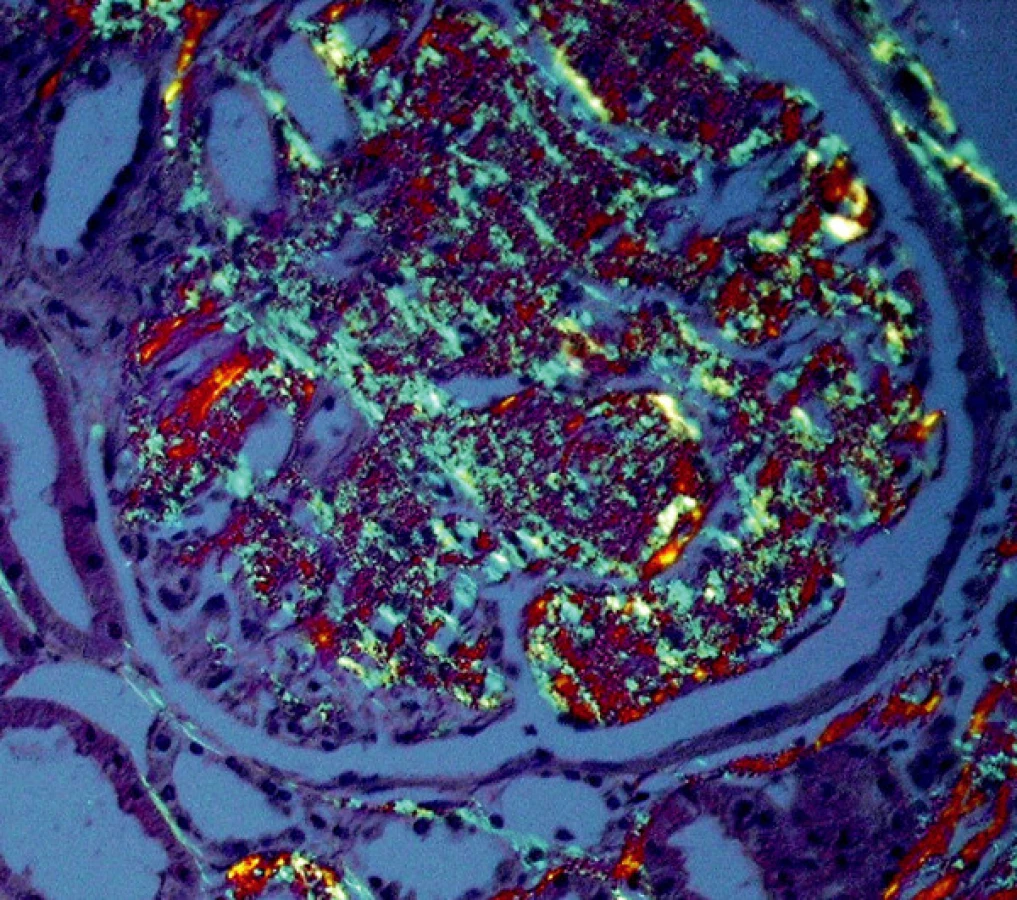

Materiál a metody
V retrospektivní studii jsme hodnotili renální biopsie vyšetřované v 5letém období (od ledna 2003 do prosince 2007). Každý vzorek byl standardně zpracován podle protokolu naší laboratoře. Vzorky byly rozděleny na 3 části; pro imunofluorescenci (IF), světelnou mikroskopii a ultrastrukturální vyšetření (ELMI). IF vyšetření zahrnovalo průkaz imunoglobulinů (IgG, IgA, IgM), frakcí komplementu (C3, C1q), A komponenty amyloidu a průkaz lehkých řetězců kappa a lambda (obr. 3, 4). Pro detekci řetězců κ a λ byla použita přímá fluorescenční technika, s polyklonálními králičími primárními protilátkami značenými fluoresceinem ( F0198 resp. F0199 DAKO; Glostrup, Dánsko).
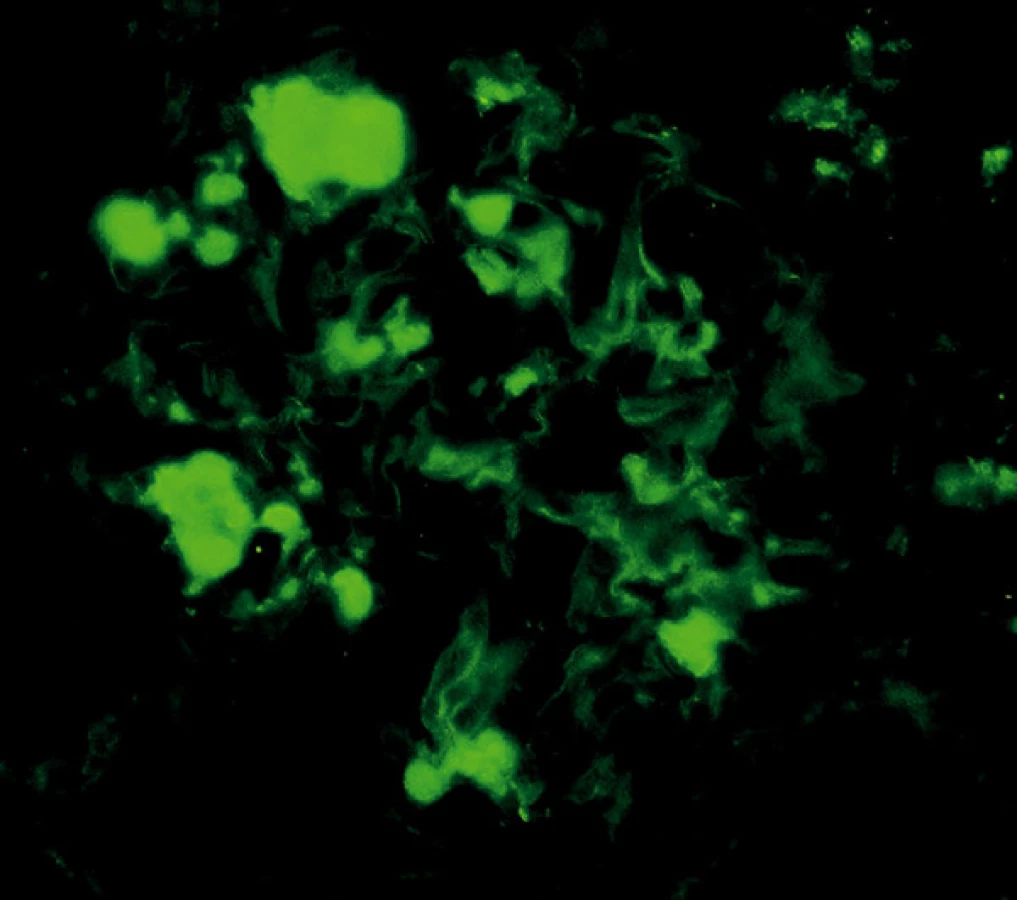
Pro průkaz AA amyloidu byla použita dvoustupňová nepřímá technika; primární protilátka je myší monoklonální (DAKO, Glostrup, Dánsko; M0759), sekundární prasečí je značená fluoresceinem (SwAM/FITC, Sevapharma, Praha).
Protokol pro parafinové zpracování zahrnoval: HE s elastikou, sirius red s elastikou (Srel), PAS/alciánová modř, periodic acid – silver methenamine (PASM), aldehyd fuchsin/oranž G (AFOG), HE, Kongo červeň a Saturnová červeň. Imunohistochemický průkaz (IH) AA amyloidu byl proveden u všech případů s AA pozitivní IF a u všech případů amyloidu nejistého zařazení. Všechny amyloidózy nejistého zařazení byly vyšetřeny IH s protilátkou proti transtyretinu (TTR). Použili jsme dvoustupňovou nepřímou techniku. Primární protilátky jsou polyklonální králičí (A-amyloid M0759; DAKO, Glostrup, Dánsko). Protilátky detekujeme univerzálním imuno-peroxidázovým polymerem (Histofine, Nichirei, Japonsko) a reakci vizualizujeme 3,3-diaminobenzidinem.
Ultrastrukturální vyšetření bylo provedeno z 57 bioptických vzorků.
Výsledky
Celkem bylo vyšetřeno 996 biopsií vlastních ledvin, z toho bylo 62 pacientů s diagnózou systémové amyloidózy (6,2 %). Jeden pacient s diagnózou amyloidózy nejistého zařazení podstoupil opakovanou biopsii ledviny v odstupu jednoho roku.
Z celkového počtu renálních biopsií tvořily biopsie s diagnózou amyloidózy za rok 2003 5,7 %, za rok 2004 5,3 %, za rok 2005 5,2 %, za rok 2006 7,5 % a za rok 2007 7,5%. AL amyloidóza byla diagnostikována v 33 (53,2 %) případech, 25 (40,3 %) pacientů mělo AA amyloidózu a 4 (6,4 %) případy zůstaly nezařazeny.
Klinický diferenciálně diagnostický závěr uváděl amyloidózu jako možnou příčinu renálního postižení ve 48 (77,4 %) případech. V době diagnózy mělo v klinickém obraze nefrotický syndrom nebo významnou proteinurii 57 (91,9 %) pacientů, z toho u 7 pacientů byl nefrotický syndrom kombinován s hematurií. Ve 3 případech byla proteinurie pouze 0,5 g/den.
Biopsie s diagnózou AL amyloidózy
Věkové rozmezí pacientů bylo 37–79 let, medián 67 let (graf 1). V souboru 33 nemocných bylo 20 (60,6 %) mužů a 13 (39,4 %) žen. Ve 28 (84,8 %) případech byla depozita tvořena lehkými řetězci λ, v 5 (15,15 %) případech šlo o lehké řetězce (poměr λ:κ 5,6 : 1). Pouze u 14 (42,4 %) pacientů byl v době biopsie současně diagnostikován paraprotein v moči nebo v séru, z toho u 5 pacientů byl diagnostikován mnohotný myelom.

Biopsie s diagnózou AA amyloidózy
Věkové rozmezí bylo 45–78 let, medián 57 let. V souboru 25 nemocných bylo 13 (52 %) mužů a 12 (48 %) žen. Pouze u 20 pacientů bylo v době diagnózy známo primární chronické zánětlivé onemocnění (v 10 případech revmatoidní artritida). U 2 (8 %) pacientů s AA amyloidózou byl současně zjištěn paraprotein v séru.
Biopsie s diagnózou nezařazené amyloidózy
Ve 4 případech bylo IF vyšetření nejisté, se slabou, matnou nediagnostickou pozitivitou v AA i λ a κ řetězcích. IF i IH průkaz TTR byl negativní. IH průkaz AA byl ve dvou případech slabě pozitivní. U jednoho pacienta byla renální biopsie provedena opakovaně s odstupem jednoho roku se stejně nepříznačným nálezem. V klinickém obraze byl v jednom případě uváděn mnohotný myelom a v jednom případě revmatoidní artritida.
Žádný z našich pacientů neměl v biopsii ledviny prokázanou TTR amyloidózu.
Rozložení depozit amyloidu
Nebyl zaznamenán rozdíl v postižení mezangiálních oblastí a glomerulárních bazálních membrán (GBM) u obou typů amyloidóz. Postižení glomerulů mělo různou kombinaci současného postižení mezangia a kapilár, odlišnost byla pouze v množství depozit u jednotlivých případů, bez ohledu na typ amyloidu. V případech AL amyloidózy byly ve 12 (36,4 %) případech postiženy glomeruly, cévy i intersticium, v 18 (54,5 %) případech byly postiženy cévy a glomeruly, ve 2 případech byly postiženy pouze glomeruly a v jednom případě byla depozita amyloidu pouze v cévách a v intersticiu. Jeden případ AL amyloidózy κ měl neobvyklá difuzní globální depozita pouze podél GBM, bez ložiskově akcentovaného postižení mezangia. Ve vzorcích s AA amyloidózou byly v 7 (28 %) případech postiženy glomeruly, cévy i intersticium. V 18 (72 %) případech byly postiženy cévy a glomeruly.
Diskuse
V poslední době zaznamenala péče o pacienty s amyloidózou zásadní změny. Ještě před několika lety byla možná pouze podpůrná léčba, ale v současnosti léčebný postup zahrnuje imunosupresivní léčbu zánětu, onkologickou terapii s možnou transplantací kmenových buněk kostní dřeně, nebo transplantaci jater. Výběr jednotlivé léčebné metody je závislý na typu amyloidu, a to je hlavním důvodem, proč klinik vyžaduje jasně formulovaný diagnostický závěr s přesným určením typu amyloidózy. Vzhledem k tomu, že terapie amyloidózy bude úspěšná jen u pacientů s nepříliš pokročilým onemocněním, vzrůstá ještě tlak na včasnou diagnózu. V takových případech může být ve tkáni jen velmi malé množství depozit amyloidu a jejich detekce může být v rutinních technikách obtížná. V praxi lze vedle barvení Kongo červení použít také barvení Saturnovou červení, které neinterferuje s kolagenem a umožňuje spolehlivě prokázat i velmi malé množství depozit amyloidu (obr. 5). V současné době je určení typu amyloidu plně v rukou patologa a nutno poznamenat, že vedle vzácných hereditárních typů i relativně běžná AL amyloidóza představuje stále diagnostický problém. Bohužel na trhu neexistují komerčně dostupné protilátky, které by umožnily jednoznačně spolehlivou diagnostiku AL amyloidu z parafinových bloků. Podle doporučení některých zahraničních center (8), která jsou v souhlase s našimi zkušenostmi, je nejvýhodnější pracovat s nativním zmraženým materiálem a provést vždy IF vyšetření. Protože určení typu amyloidu představuje velmi závažné rozhodnutí s jasně definovanými terapeutickými důsledky, ověřujeme IF průkaz dalšími dostupnými technikami (zvl. IH a ELMI). Je třeba, aby vyhodnocení bralo vždy v úvahu možné limitace vyšetření, které mohou být dané:
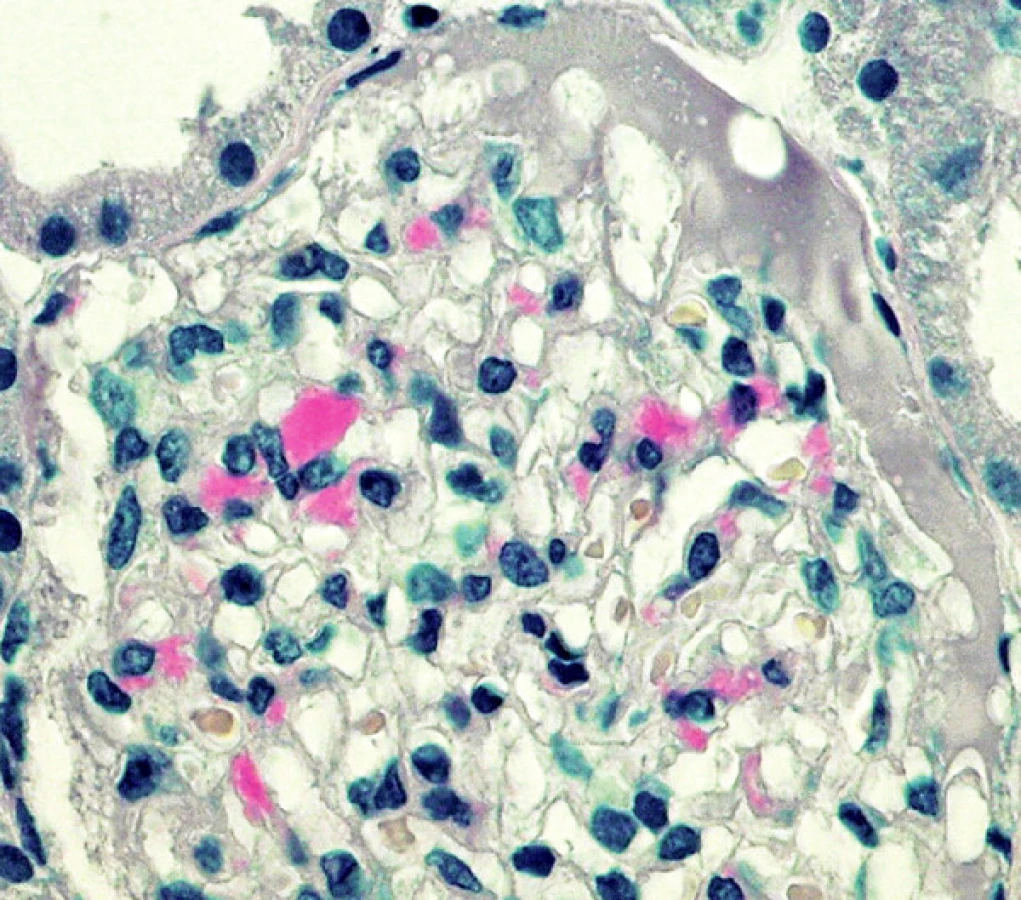
Typem použité protilátky. AL amyloidové fibrily mohou obsahovat intaktní lehké řetězce, stejně tak jako fragmenty s variabilními úseky. Komerčně dostupné protilátky jsou vyvinuty proti konstantní oblasti. Proto některé případy AL amyloidu mohou být v IF a zvl. v IH vyšetření negativní. Komerčně dostupné protilátky proti AA amyloidu jsou spolehlivější.
Kontaminací. Kontaminací se rozumí přibarvování sérovými proteiny, které mohou v různé míře pronikat nebo obklopovat depozita a vytvářet falešnou pozitivitu, která může být příčinou nejasného nebo mylného závěru, zvláště při použití IH průkazů.
Podhodnocením hereditárních typů amyloidóz. Velmi pravděpodobně hereditární amyloidózy a kombinace různých typů amyloidóz představují i u nás podhodnocenou skupinu (2, 5).
Malou zkušeností patologa. Vyhodnocení výsledků zde, stejně jako v jiných oborech, vyžaduje zkušenost.
K problémům s diagnostikou AL amyloidózy se přidává i dobře známý fakt, že část pacientů nemá v době biopsie paraprotein v moči nebo v séru (9); v našem souboru to byla více než polovina pacientů. Na druhé straně, nezanedbatelná část starších pacientů s AA amyloidózou má současně při chronickém zánětlivém onemocnění i paraprotein v séru (v našem souboru 8 % pacientů). Ačkoliv je amyloidóza v renální biopsii považována za diagnózu relativně málo častou, tím že ve vysokém procentu případů umožní rozlišení typu amyloidu, stává se pro klinika jedním z důležitých diagnostických postupů. V našem souboru tvořily renální biopsie s diagnózou amyloidózy průměrně 12,4 (6,2 %) případů ročně. AA a AL typ amyloidózy jsme odlišili v 93,5% případů.
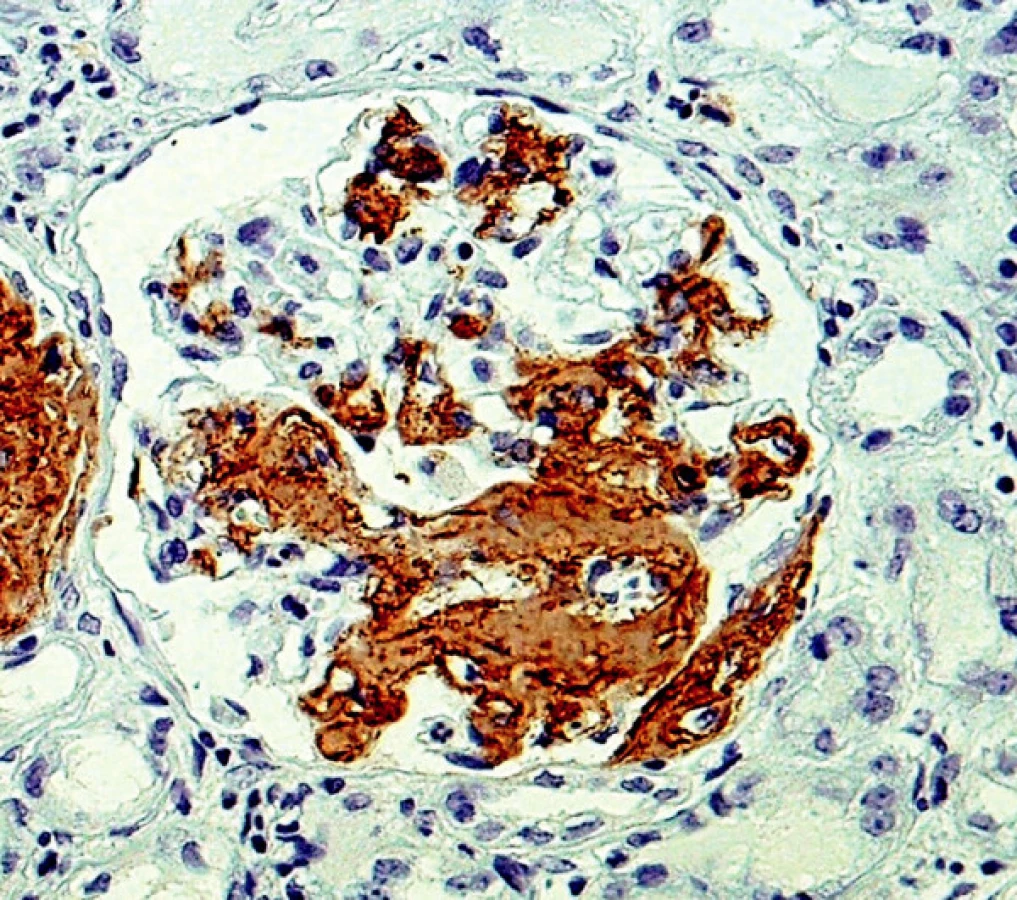
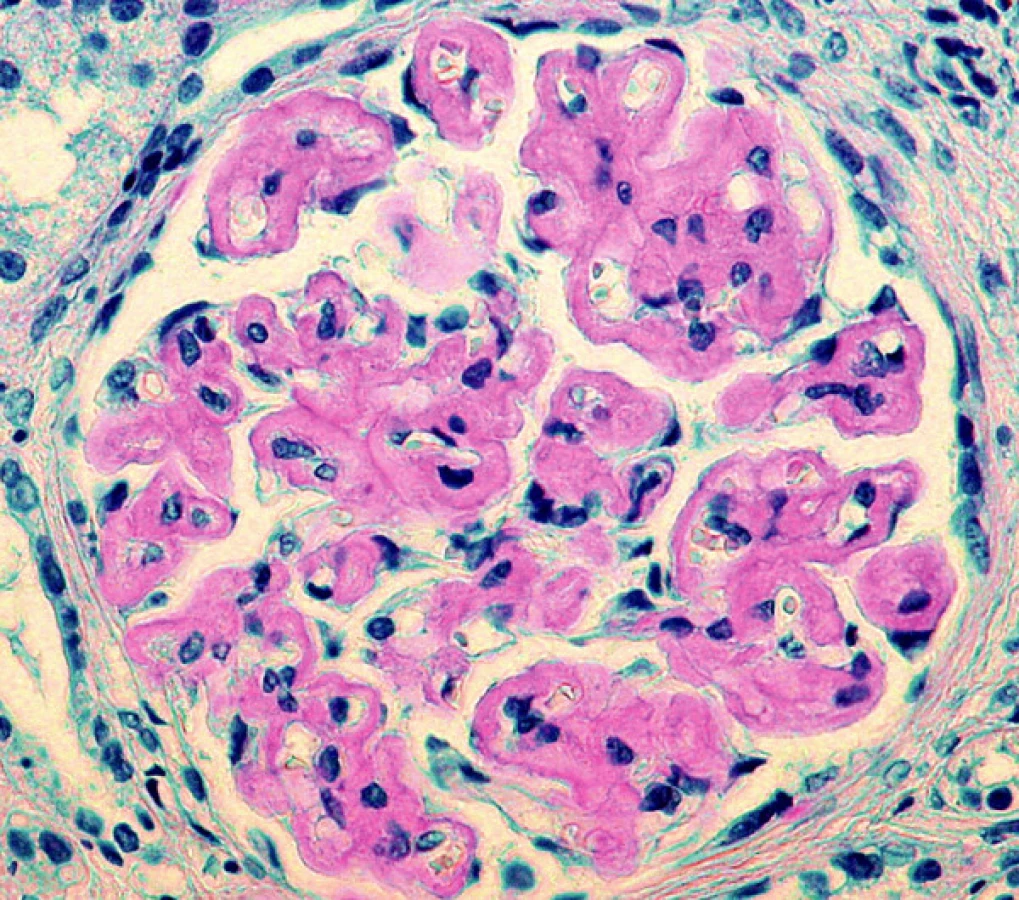
Další nezodpovězenou otázkou zůstává vztah rozložení depozit v různých orgánech u různých typů amyloidóz. Empiricky je známo, že ledviny jsou běžně postiženy v rámci AL a AA amyloidózy, stejně jako v průběhu amyloidóz, kde prekurzorovým proteinem je lysozym nebo fibrinogen. Na druhé straně TTR amyloidóza postihuje ledviny méně často, stejně tak jako v malém rozsahu postihuje játra, kde bývají jen minimální depozita ve stěnách arteriol. Z genetických studií vyplynulo, že určité typy mutací mají opakovaně stejný charakter rozložení depozit ve tkáních (3). Například u familiární TTR amyloidózy záměna methioninu za valin v pozici 30 (Val30Met) má v klinice dominující polyneuropatii, zatímco u mutace Val122Ile je převládající manifestací kardiomyopatie (4). Rozložení depozit v ledvinné tkáni při AA nebo AL amyloidóze není specifické pro jednotlivé typy amyloidu. Lze jen konstatovat, že většinou bývají postiženy arterioly (často v ledvinném hilu), dále mezangium a různou měrou se depozita objevují kolem glomerulárních bazálních membrán. V našem souboru jsme zaznamenali nejrůznější variace rozložení depozit, jak co do lokalizace, tak co do množství deponovaného amyloidu, včetně velmi neobvyklého postižení v jednom případě AL amyloidu κ, kdy depozity amyloidu byly rovnoměrně difuzně povšechně lemovány kapilární kličky glomerulů (obr. 6). Taková morfologie je pro amyloidózu velmi netypická a může vést k mylnému diagnostickému závěru, protože napodobuje jiné afekce (membranoproliferativní glomerulonefritidu, kryoglobulinémii atd.). Stejný typ uložení depozit popsal v jednom případě Veeramachaneni a spol. také u amyloidu AL κ (11).
Vedle AL a AA amyloidózy je klinicky významná TTR amyloidóza, která představuje nejčastější formu familiárních amyloidóz (až 4 % Afroameričanů jsou nositeli abnormálního TTR). Ostatní formy hereditárních amyloidóz jsou vzácné (7). Není jasné, jakou přesně roli hraje v rozvoji TTR amyloidózy časový faktor. Výskyt tohoto typu amyloidu přichází ve středním a vyšším věku a rodinná anamnéza je často němá, protože v dřívějších generacích nebylo onemocnění diagnostikováno. Navíc klinický obraz může být značně variabilní a část těchto nemocných má současně paraprotein v séru (podobně jako pacienti s AA typem amyloidu). To vše přispívá nepochybně k tomu, že jejich amyloid je někdy mylně diagnostikován jako AL (1,2,5). Na druhé straně pacienti s mutací TTR mohou mít monoklonální gamapatii a AL amyloidózu, stejně jako chronické zánětlivé onemocnění a AA amyloid.
Závěrem můžeme shrnout, že s tím, jak se rozšiřují různorodé léčebné možnosti amyloidózy, se současně zvyšují požadavky na časnou diagnózu a přesné stanovení typu amyloidu. Podle klinických údajů ani podle charakteru rozložení depozit amyloidu nelze spolehlivě určit typ systémové amyloidózy. V praxi by měla být používána imunofluorescence a imunohistochemie jako standardní postupy při určování typu amyloidu. Do budoucna lze nepochybně i v rutinní diagnostice očekávat biologicky nebo chemicky zaměřené průkazy proteinů. Tyto metodiky v současnosti ještě nemají validaci a jsou dostupné pouze v několika výzkumných laboratořích. Určení typu amyloidu dosud není jednoduchou záležitostí. Představuje ale klíčový krok, který je naprosto nezbytný pro kauzální léčbu pacienta.
Práce byla podpořena Výzkumným záměrem IKEM MZO 00023001.
Korespondující autor:
MUDr. Eva Honsová, PhD.
Pracoviště klinické a transplantační patologie
IKEM
Vídeňská 1958/9
140 21 Praha 4
E mail: eva.honsova@ikem.cz
Sources
1. Anjali, A., Satoskar, A.A., Burdge, K. et al..: Typing of amyloidosis in renal biopsies. Arch. Pathol. Lab. Med., 131, 2007, s. 917–922.
2. Comenzo, R.L., Zhou, P., Fleisher, M., et al.: Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood, 107, 2006, s. 3489-3491.
3. Dember, L.M.: Amyloidosis-associated kidney disease. J. Am. Soc. Nephrol., 17, 2006, s. 3458–3471.
4. Jacobson, D.R., Pastore, R.D., Yaghoubian, R., et al.: Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med., 336, 1997, s. 466–473.
5. Lachmann, H.J., Booth, D.R., Booth, S.E. et al.: Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis., N. Engl. J. Med., 346, 2002, s. 1786–1791.
6. Mai, H.L., Sheikh-Hamad D., Herrera G.A.: Immunoglobulin heavy chain can be amyloidogenic. Am. J. Surg. Pathol., 27, 2003, s. 541–545.
7. Olafsson, I., Grubb, A.: Hereditary cystatin C amyloid angiopathy. Amyloid, 7, 2000, s. 70–79.
8. Picken, M.M.: New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr. Opin. Nephrol. Hypertens., 16, 2007, s. 196–203.
9. Ryšavá, R.: AL Amyloidosis with renal involvement. Kidney Blood Press. Res., 30, 2007, s. 359–364.
10. Sanchorawala, V.: Light-chain (AL) amyloidosis: diagnosis and treatment. Clin. J. Am. Soc. Nephrol., 1, 2006, s. 1331–1341.
11. Veeramachaneni, R., Gu, X., Herrera, G.A.: Atypical amyloidosis: diagnostic challenges and the role of immunoelectron microscopy in diagnosis. Ultrastructural Pathology, 28, 2004, s. 75–82.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2009 Issue 3
Most read in this issue
- Neuroendocrine Tumours of the Alimentary Tract – History and at Present
- Perforating Folliculitis. Case Report and Differential Diagnosis
- Systemic Amyloidoses in Renal Biopsy Samples
- Retiform Hemangioendotelioma in a 8-Year-Old Girl – Case Report