
Systémová sklerozující choroba spojená s imunoglobuliny IgG4 – současné poznatky
IgG4-related Systemic Sclerosing Disease: a Review
IgG4-related systemic sclerosing disease (SSD) is a multisystemic condition characterized by an increased number of IgG4-producing plasma cells which occurs mainly in older men. SSD involves particularly pancreas, hepatobiliary system, salivary glands and retroperitoneum. Microscopically, the findings include lymphoplasmacytic inflammation, fibrosis and vascular changes in the form of obliterative phlebitis. Using immunohistochemistry, an increased number of IgG4-positive plasma cells might be detected in affected tissues. Since SSD frequently mimicks a malignancy both clinically and radiologically, this inflammatory sclerosing condition should be considered in the differential diagnosis of neoplastic and/or pseudoneoplastic lesions.
Key words:
sclerosing disease – IgG4 – sclerosing pancreatitis – sclerosing sialadenitis – retroperitoneal fibrosis
:
J. Laco
:
Fingerlandův ústav patologie, Lékařská fakulta UK a Fakultní nemocnice, Hradec Králové
:
Čes.-slov. Patol., 46, 2010, No. 4, p. 82-85
:
Reviews Article
Systémová sklerozující choroba spojená s imunoglobuliny IgG4 (SSCh) je multisystémové onemocnění charakterizované zvýšeným množstvím plazmatických buněk produkujících IgG4, které se vyskytuje převážně u mužů staršího věku. K nejčastěji postiženým orgánům a tkáním patří pankreas, žlučové cesty, slinné žlázy a retroperitoneum. Mikroskopický nález u SCCh zahrnuje lymfoplazmacelulární zánětlivý infiltrát, fibrózu a cévní změny ve formě obliterativní flebitidy. Pomocí imunohistochemických metod je možné v zánětlivém infiltrátu prokázat zvýšené množství IgG4-pozitivních plazmatických buněk. Vzhledem k tomu, že SSCh může v řadě orgánů klinicky a radiologicky napodobovat maligní nádor, je nutné na toto zánětlivě sklerozující onemocnění v rámci diferenciální diagnostiky nádorů pomýšlet, zejména s ohledem na skutečnost, že většina pacientů se SSCh reaguje dobře na terapii kortikosteroidy.
Klíčová slova:
sklerozující choroba – IgG4 – sklerozující pankreatitida – sklerozující sialadenitida – retroperitoneální fibróza
Systémová sklerozující choroba spojená s imunoglobuliny IgG4 (angl. IgG4-related systemic sclerosing disease – SSCh) je multisystémové onemocnění, které může postihovat téměř kterýkoli orgán a které je charakterizováno zvýšeným množstvím plazmatických buněk produkujících imunoglobuliny třídy IgG4 (2, 15). Vzhledem k tomu, že SSCh v některých případech může klinicky, radiologicky a mikroskopicky napodobovat maligní nádorový proces, je znalost tohoto onemocnění důležitá. Navíc, většina pacientů se SSCh příznivě reaguje na terapii kortikosteroidy, což často snižuje nutnost případného chirurgického zákroku (2, 15).
Historie
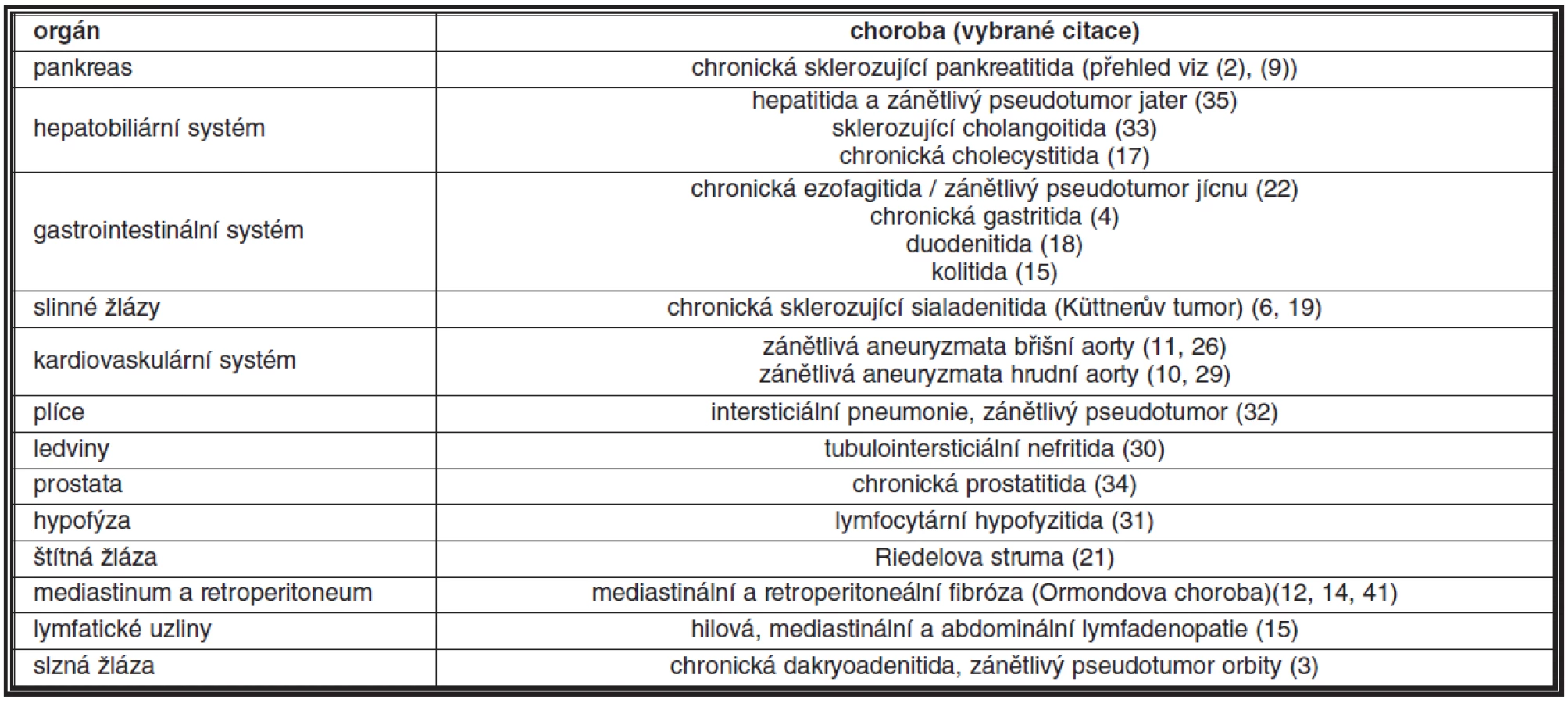
Historie rozpoznání SSCh jako imunopatologického procesu začíná v roce 1995, ve kterém Yoshida et al. (39) poprvé popsali zvýšené množství IgG4 produkujících plazmatických buněk u chronické sklerozující / autoimunitní pankreatitidy (tvořící přibližně 2–6 % chronických zánětů pankreatu), u které byla pravděpodobná etiologická účast imunitních mechanismů uvažována již v roce 1961 (27). Následně se objevilo mnoho studií, které popisovaly přítomnost IgG4 produkujících plazmatických buněk u chronických sklerozujících zánětů v celé řadě orgánů a tkání, jejichž přehled uvádí tabulka 1; přehled viz (2, 15). Mezi nejznámější příklady patří, kromě již zmíněné chronické sklerozující pankreatitidy, např. retroperitoneální fibróza (Ormondova choroba), mediastinální fibróza, chronická sklerozující sialadenitida (Küttnerův tumor), chronická sklerozující tyreoiditida (Riedelova struma) a zánětlivé pseudotumory vyskytující se v některých orgánech, zejména v játrech, plicích a v orbitě. Vzhledem k možnému multiorgánovému postižení je v názvu onemocnění zdůrazňován systémový charakter choroby (13).
Etiopatogeneze
Etiopatogeneze SSCh je pravděpodobně dvou - či vícestupňový proces, který ještě není ve všech detailech objasněn a ve kterém hlavní úlohu hraje porucha regulačních mechanismů buněk imunitního systému, a to zejména u pacientů s genotypem HLA-DRB1 a HLA-ABCF1 (25). Ve fázi indukce dochází, společně s poklesem CD45RA-pozitivních regulačních T-lymfocytů, k autoimunitní reakci namířené proti některým autoantigenům, např. karboanhydráze II a IV, laktoferinu, pankreatickému inhibitoru trypsinu a α-foldrinu, která je následována aktivací Th1 podtřídy T-lymfocytů produkujících prozánětlivé faktory INF-α, TNF-α a IL-2. Ve fázi progrese pak dochází naopak k aktivaci Th2 podtřídy T-lymfocytů, které svými působky stimulují B-lymfocyty k diferenciaci v plazmatické buňky. Ne zcela vysvětlen zůstává zvýšený počet CD25-pozitivních regulačních T-lymfocytů, které jsou schopny produkcí IL-10 imunitní reakci naopak tlumit (2).
Co je však spouštěčem výše popsané imunopatologické reakce zůstává nejasné. V úvahu přichází zejména zkřížená reakce namířená původně proti exogenním antigenům, které sdílejí jistou sekvenční homologii s výše uvedenými autoantigeny. V této souvislosti je nejčastěji zmiňována infekce Helicobacter pylori, jímž produkovaná karboanhydráza II vykazuje částečnou homologii s humánní formou tohoto enzymu (8).
Klinické příznaky
SSCh se vyskytuje zejména u pacientů středního a staršího věku mezi 50-70 lety, a to 2-3krát častěji u mužů než u žen (15). Avšak s ohledem na skutečnost, že většina zejména prvotních publikací pochází z pera japonských či obecně asijských autorů nelze vyloučit, že jsou tyto charakteristiky do jisté míry ovlivněny i genetickými, rasovými a environmentálními faktory.
Ačkoliv jakousi „klasickou“ manifestací SSCh zůstává chronická sklerozující / autoimunitní pankreatitida, u části pacientů zůstává pankreas nepoškozen a SSCh se projeví postižením jiných orgánů či tkání. Proto mohou být klinické příznaky onemocnění u jednotlivého pacienta mimořádně pestré. I pokud klinickému nálezu dominuje postižení jednoho orgánu, lze pomocí pozitronové emisní tomografie prokázat zvýšené vychytávání 18F-fluorodeoxyglukózy i v orgánech, v nichž onemocnění není klinicky patrné, což svědčí o multisystémovém charakteru choroby (23).
U části pacientů mohou být přítomny obecné klinické a laboratorní příznaky zánětu, a to včetně zvýšené hodnoty C-reaktivního proteinu; zvýšené teploty však nebývají. Postižení parenchymatózních orgánů, z nichž nejčastější je již výše zmíněná chronická sklerozující / autoimunitní pankreatitida, se projevuje většinou jejich zduřením, které může být difúzní, ale i ložiskové. Zejména v těchto případech může SSCh klinicky i při vyšetření zobrazovacími metodami napodobovat nádor (13).
Sérologické nálezy
U přibližně 75 % pacientů se SSCh je zvýšena hladina imunoglobulinu IgG4 v séru (13), kterou je možné stanovit metodou radiální imunodifuze a která je však do jisté míry závislá na věku nemocného, a to zejména u dětí do 15 let. U pacientů starších 15 let je za horní hranici normy v imunologické laboratoři FN HK považována hodnota 0,93 g/l. U pacientů s autoimunitní pankreatitidou hodnoty sérové koncentrace IgG4 často převyšují 1,35 g/l (16). IgG4 představuje nejméně početnou podtřídu imunoglobulinů IgG, tvořící pouze 3–6 % celkového IgG v séru (1, 37). Vzhledem k nízké síle kovalentní vazby mezi těžkými řetězci vedoucí k dynamickým výměnným interakcím mezi Fab fragmenty je IgG4 funkčně v podstatě bispecifickou monovalentní protilátkou, která – na rozdíl od IgG1, IgG2 a IgG3 – nemá schopnost aktivovat komplement a která vykazuje nízkou afinitu vazby k antigenům (36).
V současné době není jasné, zda se při SSCh plazmatické buňky produkující IgG4 přímo účastní patogenetického procesu či zda představují pouze diagnosticky cenný marker. Zvýšené hladiny této podtřídy IgG je možné navíc prokázat i u jiných, patogeneticky zcela odlišných onemocnění, např. u parazitárních infekcí či u některých kožních chorob, zejména u atopické dermatitidy, pemphigus vulgaris a pemphigoid bullosus (28).
Mikroskopický obraz
Mikroskopický obraz SSCh zahrnuje obecně zánětlivý infiltrát, fibrózu a změny cév, jejichž intenzita je však dosti variabilní a závislá na době trvání onemocnění a případně rovněž na protizánětlivé léčbě (pro přehled viz (2)). Zatímco v časném stadiu dominuje přítomnost intenzivního zánětlivého infiltrátu, v pozdějších fázích tento ustupuje a do popředí nálezu se dostává fibróza s již nečetnými zánětlivými buňkami. V této situaci může být stanovení diagnózy SSCh obtížné. Níže uvedený popis mikroskopických změn odpovídá nálezům popisovaným při SSCh v exokrinních žlázách, zejména v pankreatu (24, 40) a ve slinných žlázách (6, 19).
Zánětlivý infiltrát sestává z lymfocytů a plazmatických buněk a například v pankreatu či ve slinných žlázách bývá často lokalizován periduktálně. Z lymfocytů převažují disperzně rozptýlené T-lymfocyty, zatímco B-lymfocyty mohou tvořit lymfatické folikuly se zárodečnými centry. Vzácněji mohou být přítomny lymfocyty přímo v epitelu malých vývodů (tzv. lymfocytární exocytóza), což může ve slinných žlázách vést k mylné diagnóze lymfoepiteliální sialoadenitidy (benigní lymfoepiteliální léze). Na rozdíl od tohoto onemocnění však u SSCh nedochází k výraznější přeměně takto změněných vývodů v lymfoepiteliální ostrůvky. Z plazmatických buněk převažují buňky produkující IgG, z nichž podstatnou část tvoří buňky produkující IgG4, které je možné prokázat pomocí imunohistochemie. Sami máme dobré zkušenosti s monoklonální protilátkou HP6025 od fy Invitrogen. Původně stanovený limit pro diagnózu chronické sklerozující pankreatitidy byl > 10 IgG4-pozitivních plazmatických buněk na 1 zorné pole velkého zvětšení (12). V současné době se uvádí, že – pro diagnózu SSCh – limit > 50 IgG4-pozitivních plazmatických buněk na 3 zorná pole velkého zvětšení zvyšuje senzitivitu na 70 % a specificitu na 100 % (20). Dle našich zkušeností bývá u chronické sklerozující sialadenitidy počet IgG4-pozitivních plazmatických buněk běžně vyšší než 50 buněk na 1 zorné pole velkého zvětšení. Elektronmikroskopicky byla v postižených orgánech prokázána přítomnost elektrondenzních imunokomplexů / depozit, v nichž byl následně imunohistochemicky prokázán IgG4 (5). V některých případech může být přítomno různé množství eozinofilních granulocytů, což by mohlo souviset i s jistou úlohou IgE v patogenezi SSCh. Méně často mohou být přítomny i neutrofilní granulocyty, a to převážně v luminu malých vývodů (tzv. granulocytární epiteliální léze). Tento nález však dle našeho názoru v podstatě odpovídá akutní katarální dochitidě. Ojediněle byly pozorovány i periduktální granulomy sestávající z histiocytů.
Fibróza bývá tvořena většinou více buněčnou vazivovou tkání s četnými fibroblasty tvořícími kolagen. V pozdějších stadiích dochází k náhradě funkčního parenchymu postiženého orgánu právě tímto vazivem, což může vést k poruše jeho funkce, např. ve formě snížení sekrece pankreatických šťáv či slin. Vzácněji jsou v rámci SSCh v některých orgánech nalézány drobné konkrementy.
Cévní změny je možné pozorovat převážně v žilách malé a střední velikosti. Dosti charakteristickou změnou pro SSCh je tzv. obliterativní flebitida, vznikající jako důsledek perivenulárního zánětu spojeného s destrukcí stěny cévy. Vzhledem k tomu, že tento diagnosticky cenný nález může být překryt okolním zánětem a fibrózou, je s výhodou použít v těchto případech barvení na elastiku. Daleko méně častá obliterativní arteritida bývá přítomna u závažně probíhajícího onemocnění.
Prognóza
Pacienty se SSCh je nutné celkově vyšetřit a dlouhodobě dispenzarizovat, protože u nich může dojít ke vzniku víceložiskového postižení, a to jak synchronně, tak i metachronně, s variabilním časovým odstupem (15). Z klinického a léčebného hlediska je velmi důležitý poznatek, že většina pacientů se SSCh reaguje příznivě na léčbu kortikosteroidy, jejichž podání může vést ke zmenšení obtíží a z toho důvodu již nemusí být nutná chirurgická léčba (15).
Případný onkologický potenciál SSCh zůstává nejasný. V literatuře jsou publikovány jednotlivé případy popisující vznik duktálního adenokarcinomu v terénu chronické sklerozující pankreatitidy (38), lymfomu v terénu chronické sklerozující dakryoadenitidy (3) a salivárního duktálního karcinomu v terénu chronické sklerozující sialadenitidy (7).
Závěr
SSCh je relativně nově zavedená nozologická jednotka, jejíž správné rozpoznání má důležité důsledky. Patolog vstupuje do diagnostického procesu při vyšetřování bioptického materiálu z ložiska, které může klinicky a radiologicky připomínat maligní nádor. Správně stanovená diagnóza zánětlivě fibrotizujícího procesu se zvýšeným množstvím IgG4-pozitivních plazmatických buněk ve tkáni nejenže vyvrátí podezření z nádorového procesu, ale současně umožní ošetřujícímu lékaři indikovat odpovídající léčbu.
Práce byla podpořena Výzkumným záměrem Ministerstva zdravotnictví ČR 00179906.
MUDr. Jan Laco, Ph.D.
Fingerlandův ústav patologie
Fakultní nemocnice Hradec Králové
Sokolská 581
500 05 Hradec Králové
Tel.: 495 832 548
Fax: 495 832 004
e-mail: lacoj@lfhk.cuni.cz
Sources
1. Aalberse, R.C., Schuurman, J.: IgG4 breaking the rules. Immunology, 105, 2002, s. 9–19.
2. Bateman, A.C., Deheragoda, M.G.: IgG4-related systemic sclerosing disease – an emerging and under-diagnosed condition. Histopathology, 55, 2009, s. 373–383.
3. Cheuk, W., Yuen, H.K., Chan, A.C. et al.: Ocular adnexal lymphoma associated with IgG+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am. J. Surg. Pathol., 32, 2008, s. 1159–1167.
4. Deheragoda, M.G., Church, N.I., Rodriguez-Justo, M. et al.: The use of immunoglobulin G4 immunostaining in diagnosing pancreatic and extrapancreatic involvement in autoimmune pancreatitis. Hepatol., 5, 2007, s. 1229–1234.
5. Deshpande, V., Chicano, S., Finkelberg, D. et al.: Autoimmune pancreatitis: a systemic immune complex disease. Am. J. Surg. Pathol., 30, 2006, s. 1537–1545.
6. Geyer, J.T., Ferry, J.A., Harris, N.L. et al.: Chronic sclerosing sialadenitis (Küttner tumor) is an IgG4-associated disease. Am. J. Surg. Pathol., 34, 2010, s. 202–210.
7. Gill, J., Angelo, N., Yeong, M.L., McIvor, N.: Salivary duct carcinoma arising in IgG4-related autoimmune disease of the parotid gland. Hum. Pathol., 40, 2009, s. 881–886.
8. Guarneri, F., Guarneri, C., Benvenga, S.: Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimickry? J. Cell. Mol. Med., 9, 2005, s. 741–744.
9. Honsová, E., Lodererová, A., Kostolná, E., Oliverius, M: Autoimunní pankreatitida s postižením žlučovodů a jater jako součást IgG4 pozitivního autoimunního onemocnění (IgG4-related autoimmune sclerosing disease). Čes.-slov. Patol., 46, 2010, s. 65–67.
10. Ishida, M., Hotta, M., Kushima, R., Asai, T., Okabe, H.: IgG4-related inflammatory aneurysm of the aortic arch. Pathol. Int., 59, 2009, s. 269–273.
11. Ito, H., Kaizaki, Y., Noda, Y., Fujii, S., Yamamoto, S.: IgG4-related inflammatory abdominal aortic aneurysm associated with autoimmune pancreatitis. Pathol. Int., 58, 2008, s. 421–426.
12. Kamisawa, T., Funata, N., Hayashi, Y. et al.: Close relationship between autoimmune pancreatitis and multifocal fibrosclerosis. Gut, 52, 2003, s. 683–687.
13. Kamisawa, T., Funata, N., Hayashi, Y. et al.: A new clinicopathological entity of IgG4-related autoimmune disease. J. Gastroenterol., 38, 2003, s. 982–984.
14. Kamisawa, T., Matsukawa, M., Ohkawa, M.: Autoimmune pancreatitis associated with retroperitoneal fibrosis. JOP, 6, 2005, s. 260–263.
15. Kamisawa, T., Okamoto, A.: IgG4-related sclerosing disease. World J. Gastroenterol., 14, 2008, s. 3948–3955.
16. Kamisawa, T., Okamoto, A., Funata, N.: Clinicopathological features of autoimmune pancreatitis in relation to elevation of serum IgG4. Pancreas, 31, 2005, s. 28–31.
17. Kamisawa, T., Tu, Y., Nakajima, H. et al.: Sclerosing cholecystitis associated with autoimmune pancreatitis. World J. Gastroenterol., 12, 2006, s. 3736–3739.
18. Kamisawa, T., Tu, Y., Nakajima, H., Egawa, N., Tsuruta, K., Okamoto, A.: Usefulness of biopsying the major duodenal papilla to diagnose autoimmune pancreatitis: a prospective study using IgG4 immunostaining. World J. Gastroenterol., 12, 2006, s. 2031–2033.
19. Kitagawa, S., Zen, Y., Harada, K. et al.: Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Küttner tumor). Am. J. Surg. Pathol., 29, 2005, s. 783–791.
20. Kloppel, G., Sipos, B., Zamboni, G., Kojima, M., Morohoshi, T.: Autoimmune pancreatitis: histo - and immunopathological features. J. Gastroenterol., 42 (Suppl. XVIII), 2007, s. 28–31.
21. Komatsu, K., Hamano, H., Ochi, Y. et al. High prevalence of hypothyroidism in patients with autoimmune pancreatitis. Dig. Dis. Sci., 50, 2005, s. 1052–1057.
22. Lopez, J., Hochwald, S.N., Lancia, N., Dixon, L.R., Ben-David, K.: Autoimmune esophagitis: IgG4-related tumors of the esophagus. J. Gastrointest. Surg., 2010, doi 10.1007/s11605-010-1172-4.
23. Nakajo, M., Jinnouchi, S., Fukukura, Y., Tanabe, H., Tateno, R., Nakajo, M.: The efficacy of whole-body FDG-PET or PET/CT for autoimmune pancreatitis and associated extrapancreatic autoimmune lesions. Eur. J. Nucl. Med. Mol. Imaging, 34, 2007, s. 2088–2095.
24. Notohara, K., Burgart, L.J., Yadav, D., Chari, S., Smryk, T.C.: Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinico-pathologic features of 35 cases. Am. J. Surg. Pathol., 27, 2003, s. 1119–1127.
25. Ota, M., Katsuyama, Y., Hamano, H. et al.: Two critical genes (HLA-DRB1 and ABCF1) in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics, 59, 2007, s. 45–52.
26. Sakata, N., Tashiro, T., Uesugi, N. et al.: IgG4-positive plasma cells in inflammatory abdominal aortic aneurysm: the possibility of an aortic manifestation of IgG4-related sclerosing disease. Am. J. Surg. Pathol., 32, 2008, s. 553–559.
27. Sarles, H., Sarles, J.C., Muratore, R., Guien, C.: Chronic inflammatory sclerosis of the pancreas: an autoimmune pancreatic disease? Am. J. Dig. Dis., 6, 1961, s. 688–698.
28. Shirakata, Y., Shiraishi, S., Sayama, K., Miki, Y.: Subclass characteristics of IgG autoantibodies in bullous pemphigoid and pemphigus. J. Dermatol., 17, 1990, s. 661–666.
29. Stone, J.H., Khosroshahi, A., Hilgenberg, A., Spooner, A., Isselbacher, E.M., Stone, J.R.: IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum., 60, 2009, s. 3139–3145.
30. Takeda, S., Haratake, J., Kasai, T., Takaeda, C., Takazakura, E.: IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol. Dial. Transplant., 19, 2004, s. 474–476.
31. Taniguchi, T., Hamasaki, A., Okamoto, M.: A case of suspected lymphocytic hypophysitis and organizing pneumonia during maintenance therapy for autoimmune pancreatitis associated with autoimmune thrombocytopenia. Endocr. J., 53, 2006, s. 563-566.
32. Taniguchi, T., Ko, M.: Interstitial pneumonia associated with autoimmune pancreatitis. Gut, 53, 2004, s. 770–774.
33. Uehara, T., Hamano, H., Kawa, S., Sano, K., Honda, T., Ota, H.: Distinct clinico-pathological entity ”autoimmune pancreatitis-associated sclerosing cholangitis”. Pathol. Int., 55, 2005, s. 405–411.
34. Uehara, T., Hamano, H., Kawakami, M. et al.: Autoimmune pancreatitis associated prostatis: distinct clinico-pathological entity. Pathol. Int., 58, 2008, s. 118–125.
35. Umemura, T., Zen, Y., Hamano, H., Kawa, S., Nakanuma, Y., Klyosawa, K.: Immunoglobulin G4-hepatopathy: association of immunoglobulin G4-bearing plasma cells in liver with autoimmune pancreatitis. Hepatology, 46, 2007, s. 463–471.
36. Van der Neut Kolfschoten, M., Schuurman, J., Losen, M. et al.: Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science, 317, 2007, s. 1554–1557.
37. Van der Zee, J.S., Aalberse, R.C.: Immunochemical characteristics of IgG4 antibodies. N. Engl. Reg. Allergy Proc., 9, 1988, s. 31–33.
38. Witkiewicz, A.K., Kennedy, E.P., Kennyon, L., Yeo, C.J., Hruban, R.H.: Synchronous autoimmune pancreatitis and infiltrating ductal adenocarcinoma: a case report and review of the literature. Hum. Pathol., 39, 2008, s. 1548–1551.
39. Yoshida, K., Toki, F., Takeuchi, T., Watanabe, S., Shiratori, K., Hayashi, N.: Chronic pancreatitis caused by an autoimmune abnormality: proposal of the concept of autoimmune pancreatitis. Dig. Dis. Sci., 40, 1995, s. 1561–1568.
40. Zamboni, G., Luttges, J., Capelli, P. et al.: Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch., 445, 2004, s. 552–563.
41. Zen, Y., Sawazaki, A., Miyayama, S., Notsumata, K., Tanaka, N., Nakanuma, Y.: A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis). Hum. Pathol., 37, 2006, s. 239–243.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2010 Issue 4
Most read in this issue
- Muir-Torre Syndrome – a Phenotypic Variant of Lynch Syndrome
- Lymphatic System: Morphology and Pathology Update
- IgG4-related Systemic Sclerosing Disease: a Review
- Prolonged Treatment of Chronic Renal Insufficiency, Acquired Cystic Kidney Disease, Simultaneous Precancerous Lesions and Multiple Tumors of Left Kidney