
Lynchův syndrom v rukách patologa
Lynch syndrome in the hands of pathologists
Lynch syndrome (formerly hereditary non-polyposis colorectal cancer) is the most common familial colorectal cancer syndrome with a known molecular genetic background. The syndrome is caused by a germline mutation of one of the genes encoding mismatch repair (MMR) proteins that are responsible for DNA replication errors repair. Impaired function of these proteins leads to microsatellite instability (MSI) and forms a suitable background for the development and progression of tumors, mainly colorectal cancer. Traditionally, Lynch syndrome was regarded to be responsible for 2 % of all cases of colorectal cancer, however recent estimates reach even 5 %. Due to this relatively high frequency, familial occurence, the absence of the premorbid phenotype and the development of malignant tumors during the productive years of life, the correct diagnosis becomes not only a medical, but also a socioeconomical problem. Unfortunately, clinical means of diagnostics of Lynch syndrome (like the Amsterdam criteria and Bethesda guidelines) lack sensitivity. It was shown that predictive models based on histological signs of MSI are more sensitive than the clinical criteria used to detect patients suspicious of Lynch syndrome. Of all MSI-H colorectal cancers, 1/5 is caused by Lynch syndrome, the rest being only sporadic cancers caused by epigenetic inactivation of a MMR protein. To rule out the sporadic cases, molecular genetic investigation of the BRAF gene and methylation analysis of MLH1 is used in the diagnostic workup of Lynch syndrome. The suspicion of Lynch syndrome, based on the results of the assortment of diagnostic methods mentioned above, should be proven by detection of a germline mutation of an MMR gene in peripheral blood, and followed by screening of family members, which is a necessary condition for efficient prevention.
Keywords:
colorectal cancer – Lynch syndrome – HNPCC – MSI – microsatellite instability
:
Ondřej Daum 1
![]() ; Zdeněk Beneš 2; Ladislav Hadravský 1; Jan Stehlík 3; Kateřina Černá 3; Martin Dušek 1,3; Bohuslava Kokošková 1; Michal Michal 1,3
; Zdeněk Beneš 2; Ladislav Hadravský 1; Jan Stehlík 3; Kateřina Černá 3; Martin Dušek 1,3; Bohuslava Kokošková 1; Michal Michal 1,3
![]()
:
Šiklův ústav patologie LF UK a FN Plzeň
1; Interní oddělení Fakultní Thomayerovy nemocnice, Praha
2; Bioptická laboratoř, s. r. o., Plzeň
3
:
Čes.-slov. Patol., 50, 2014, No. 1, p. 18-24
:
Reviews Article
Lynchův syndrom (dříve hereditární nepolypózní kolorektální karcinom) je nejčastější příčinou familiárního výskytu kolorektálního karcinomu se známým molekulárně genetickým podkladem. Příčinou je germinální mutace některého z genů kódujících takzvané MMR proteiny, které opravují chyby ve struktuře DNA vznikající při její replikaci. Tyto mutace způsobují dysfunkci opravného komplexu, která vede k rozvoji nestability mikrosatelitů (MSI) a ke vzniku a progresi nádorů, zejména kolorektálního karcinomu. Tradičně se jeho frekvence odhaduje na 2 % všech kolorektálních karcinomů, v současné době však odhady dosahují až 5 %. Vzhledem k této poměrně vysoké četnosti, absenci premorbidního fenotypu, familiárnímu výskytu a prezentaci maligních nádorů v produktivním věku se z jeho diagnostiky stává problém nejen medicínský, ale i socioekonomický. Bohužel, Bethesda guidelines, natož Amsterdamská kritéria, která byla sestavena především ke klinickému záchytu pacientů se suspekcí na Lynchův syndrom, nejsou dostatečně senzitivní. Ukázalo se, že vyšší senzitivitu vykazuje histologická detekce karcinomů s morfologickými znaky asociovanými s MSI. Jedna pětina karcinomů s MSI by měla být podmíněna Lynchovým syndromem, zbytek tvoří sporadické MSI-H karcinomy způsobené epigenetickou inaktivací opravného proteinu. K vyloučení těchto sporadických případů z dalšího testování slouží vyšetření genu BRAF a analýza methylace promotoru genu MLH1. Podezření na Lynchův syndrom vyplývající z výsledků výše uvedeného komplexu vyšetření by mělo být nakonec potvrzeno detekcí germinální mutace některého z MMR genů v periferní krvi s následným vyšetřením rodinných příslušníků pro zajištění účinné prevence.
Klíčová slova:
kolorektální karcinom – Lynchův syndrom – HNPCC – MSI – nestabilita mikrosatelitů
Lynchův syndrom - SOUČASNÁ DEFINICE
Lynchův syndrom (LS) je autozomálně dominantně dědičné onemocnění vytvářející predispozici ke vzniku maligních nádorů, jehož podkladem je (až na výjimky) germinální mutace některého z genů odpovědných za opravy replikačních chyb v DNA, takzvaných mismatch repair (MMR) genů. Nejčastějším karcinomem vznikajícím při Lynchově syndromu je kolorektální karcinom (CRC), je však zvýšené riziko vzniku i extrakolonických malignit, zejména karcinomů endometria, žaludku, tenkého střeva, ovaria, ledvinné pánvičky a močovodu, nádorů mozku a kůže.
Na podkladě LS vzniká pravděpodobně až 5 % CRC. Na rozdíl od familiární adenomatózní polypózy (FAP) tyto nádory nevznikají v terénu polypózy (definované jako >100 polypů), což však neznamená, že nemohou být přítomny žádné polypy. Dalším důležitým znakem LS je absence „premorbidního fenotypu“, tedy přítomnosti benigních změn, které by umožňovaly diagnostikovat tento syndrom ještě před vznikem maligního tumoru (jako je tomu třeba v případě FAP, neurofibromatózy 1. typu a dalších familiárních karcinomových syndromů). Výjimkou z tohoto pravidla je fenotypická varinta LS projevující se vznikem kožních sebaceózních nádorů, označovaná jako Muir-Torreho syndrom (MTS) (1). V ostatních případech může být LS diagnostikován prakticky až při nálezu maligního tumoru, případně při genetickém vyšetření rodinných příslušníků již diagnostikovaného probanda.
HISTORIE A VÝVOJ TERMINOLOGIE
První známou dokumentovanou rodinou s (tehdy samozřejmě ještě nepojmenovaným) LS je „karcinomová rodina G“, kterou v roce 1913 popsal prof. Warthin (2). Pozorování podobně významného rodinného výskytu kolorektálního karcinomu při absenci polypózy (3) přivedlo Lynche ke studiu a doplnění materiálů o „karcinomové rodině G“ a zaznamenání základních znaků tohoto syndromu:
- zvýšená incidence adenokarcinomů, zejména kolorektálních a endometriálních,
- zvýšené riziko multiplicity nádorů,
- autosomálně dominantní dědičnost, a
- vznik karcinomů v mladším věku (4).
Ve svých prvních publikacích Lynch používal k jeho označení termín „syndrom karcinomové rodiny“, v pozdějších „hereditární nepolypózní kolorektální karcinom“ (HNPCC) (5). Zároveň se však začalo synonymicky používat označení „Lynchův syndrom“ (6). V současné době se upouští od termínu HNPCC, přičemž označení „Lynchův syndrom“ se používá pro případy splňující Amsterdamská kritéria (viz níže) a zároveň nesoucí germinální mutaci některého z MMR genů, zatímco pro případy splňující Amsterdamská kritéria, ale bez prokazatelné germinální mutace některého z MMR genů, se doporučuje termín „familiární kolorektální karcinom typu X“ (7).
MOLEKULÁRNĚ GENETICKÝ PODKLAD
Příčinou LS je ve většině případů germinální mutace v některém z genů zodpovědných za opravy replikačních chyb v DNA, takzvaných mismatch repair (MMR) genů (8,9). Fyziologicky se MMR proteiny kódované těmito geny spojují do funkčních komplexů, a to zejména v heterodimery MSH2-MSH6 (příp. MSH2-MSH3) a MLH1-PMS2. První z těchto dimerů rozpoznává chyby v DNA (nespárované nebo špatně spárované nukleotidy) a signalizuje poškození, druhý chyby opravuje. Zároveň se podílí na zastavení buněčného cyklu a indukci apoptózy v reakci na poškození DNA. Pokud některý z proteinů není funkční následkem inaktivace obou alel jeho genu, dochází ke vzniku nádorů charakteristických vysokým stupněm nestability v takzvaných mikrosatelitech, tedy krátkých tandemových repeticích (short tandem repeats, STR) (10,11).
Mikrosatelity jsou úseky DNA tvořené několikanásobným opakováním jednoho, dvou, tří, čtyř a vzácněji více nukleotidů, které se vyskytují v celém genomu poměrně hojně. Délky jednotlivých mikrosatelitů (tedy počty opakování těchto sekvencí) jsou v rámci jedince stejné, ale mohou se lišit mezi jednotlivci. Mikrosatelitní sekvence jsou velmi snadno zranitelné při replikaci DNA, protože DNA polymeráza na takovýchto místech snadněji sklouzává a dochází tak ke vzniku delších (inzerce sekvence) či kratších (delece sekvence) úseků. Kolísá-li délka mikrosatelitů v rámci jednoho jedince, hovoříme o nestabilitě mikrosatelitů (microsatellite instability, MSI). Stanovení stupně MSI spočívá ve stanovení nestability mezinárodně kodifikovaných markerů, přičemž na základě počtů postižených markerů se rozlišují stavy (resp. nádory) se stabilními mikrosatelity (microsatellite stable, MSS), s nízkým stupněm nestability (microsatellite instability – low, MSI-L) a s vysokým stupněm nestability mikrosatelitů (microsatellite instability – high, MSI-H) (12). Přehled používaných markerů a algoritmus stanovení stupně MSI shrnuje tabulka 1.

MSI-H tumory mohou vznikat dvěma různými základními způsoby. Buď jde o sporadické nádory vyvolané genetickými a/nebo epigenetickými změnami v somatické buňce, nebo o familiárně se vyskytující nádory v rámci LS způsobené nejčastěji germinální mutací některého z MMR genů. Mutovaná alela je děděna autozomálně dominantně. Jelikož ale MMR geny patří mezi typické tumor supresorové geny, k vlastnímu vzniku nádoru dochází až somatickou inaktivací druhé alely téhož genu (13). LS je z více než 80 % asociován se zárodečnými mutacemi genů MLH1 a MSH2 (14), přibližně 10 % objevených mutací je lokalizováno v genu MSH6 a ostatní mutace připadají na další MMR geny (PMS2, PMS1, MSH3, MLH3). Většinou se jedná o mutace malého rozsahu jako jsou substituce, inzerce nebo delece, rozsáhlé delece a duplikace celých exonů bývají nacházeny hlavně v MSH2 (až 1/3 mutací) a poměrně často také v MLH1 (15). U posledně jmenovaného genu byly už popsány i germinální hypermetylace promotoru vedoucí k jeho epigenetické inaktivaci (16,17). V nedávné době byly popsány i germinální delece 3´ konce genu EPCAM (TACSTD1), které vedou k epigenetické inaktivaci MSH2 (18,19). U některých mutací a polymorfizmů MMR genů zatím nebyl prokázán jejich klinický význam, nicméně poznatky o variantách genů jsou neustále aktualizovány na stránkách International Society for Gastrointestinal Hereditary Tumours (http://www.insight-group.org/).
Je-li u osob nesoucích jednu germinálně mutovanou alelu MMR genu během jejich života inaktivována i alela druhá, dochází ke vzniku různých malignit, jako například CRC, karcinomů dalších částí GIT, urotelu a dalších. Důvodem může být somatická mutace v druhé alele genu, ztráta heterozygozity (LOH), nebo také metylace promotoru, jak tomu často bývá u genu MLH1 (20). Velmi vzácně se vyskytuje germinální bialelická mutace genů MMR, a to zejména jako následek incestu. Tento stav je charakteristický vznikem CRC již v mladém věku, hematologickými malignitami a fenotypickým obrazem připomínajícím neurofibromatózu 1. typu (21,22).
Klinické projevy mohou být u mutací jednotlivých genů různé. Například nosiči germinální mutace MLH1 mají riziko vzniku kolorektálního karcinomu v nízkém věku nižší, než je tomu u nosičů mutací v ostatních MMR genech, a také u nich často bývá kolorektální karcinom jedinou diagnostikovanou malignitou. Ostatní nádorová onemocnění vyskytující se v souvislosti s Lynchovým syndromem jsou detekována častěji u pacientů s germinální mutací v MSH2 (23). Mutace v MSH6, které mají poměrně nízkou penetranci, jsou 6x častěji asociovány s karcinomy endometria než s nádory kolorekta, navíc jsou u těchto pacientů kolorektální karcinomy (v porovnání s Lynchovými syndromy způsobenými mutacemi jiných MMR genů) častěji levostranné. Důležité také je, že mutace v MSH6 nevedou vždy ke vzniku nestability mikrosatelitů, pravděpodobně díky tvorbě alternativního heterodimeru MSH2-MSH3 (24,25). Naopak mutace v PMS2 vedou většinou ke vzniku MSI-H tumorů a jsou asociovány jak s časným kolorektálním, tak endometriálním karcinomem, avšak ani zde penetrance není příliš vysoká (26-28). Na vzniku MTS se podílí především germinální mutace v genech MLH1 a MSH2, přičemž mutace v genu MSH2 je převažující (29,30).
Turcotův syndrom, který je charakterizován společným výskytem nádorů mozku (většinou gliomů) a CRC, může být také způsoben zárodečnou mutací některého z MMR genů, mutace byly detekovány zejména v MLH1 a PMS2 (31).
Nádory pacientů s LS a MTS jsou v 89 %, resp. 70 % MSI-H, na rozdíl od sporadických kolorektálních karcinomů, kde se MSI-H status vyskytuje pouze u 15 % pacientů a v naprosté většině případů bývá asociován s hypermetylací promotoru genu MLH1, nikoli se somatickými mutacemi MMR genů (10,32,33).
Mutační analýza je prováděna v ideálním případě z periferní krve pacienta, lze k ní využít ale i nenádorovou tkáň pacienta. K detekci rozsáhlých delecí a duplikací, které zahrnují celé exony nebo několik exonů, je v poslední době stále častěji využívána metoda MLPA (multiple ligation probe amplification). Drobné mutace mohou být detekovány pomocí SSCP (single strand conformation polymorphism) nebo přímou sekvenací celých exonů a exon-intronových oblastí.
ZÁKLADY KLINICKÉ DIAGNOSTIKY LYNCHOVA SYNDROMU
V roce 1990 byla formulována klinická kritéria k diagnostice HNPCC, která se označují jako Amsterdamská kritéria (34). Pro zvýšení senzitivity, zejména s přihlédnutím k možnosti prezentace syndromu extrakolonickou malignitou, byla tato kritéria v roce 1998 revidována na Amsterdamská kritéria II (Tabulka 2) (35).

Bethesda guidelines
K identifikaci pacientů s CRC, u kterých by měla být vyšetřena nestabilita mikrosatelitů (MSI), případně provedeno molekulárně genetické vyšetření, byla v roce 1996 vypracována a v roce 2002 revidována tzv. Bethesda guidelines (BG, resp. RBG), která berou v potaz nejen klinická kritéria, ale i morfologické znaky tumoru (Tabulka 3) (36). Bohužel ani tato širší kritéria nezachytí všechny případy LS (37), zejména v případě postižení MSH6 a PMS2 (27, 38-41). Odhaduje se, že až čtvrtina pacientů s LS propadne sítem kriterií RBG. Vzhledem k tomu, že falešná negativita v případě LS nemá za následek nerozpoznání tohoto syndromu pouze u vyšetřovaného pacienta, ale i u jeho případných příbuzných, nelze tuto senzitivitu pokládat za dostatečnou.

ROLE MORFOLOGA VE ZVÝŠENÍ ZÁCHYTU LYNCHOVA SYNDROMU
Současný nárůst role morfologických vyšetření v diagnostice Lynchova syndromu je způsoben potřebou zvýšení senzitivity, byť i za cenu snížení specificity, které je v tomto případě možné familiární tendence ke vzniku maligních nádorů ospravedlnitelné jak z hlediska etického, tak ekonomického.
Mezi hlavní argumenty pro zvýšení senzitivity systému depistáže patří fakt, že 1 ze 660 lidí je nositelem geminální mutace některého z MMR genů (42), riziko vzniku CRC u LS je 60 - 80 % (43,44), dále že k progresi z adenomu do karcinomu pravděpodobně dochází během 2-3 let, narozdíl od 8 - 10 let u sporadických případů (45,46), a zejména že průměrný věk v době diagnózy je 45 let, tedy asi o 20 let méně než u sporadického CRC, navíc se zvýšeným rizikem synchronního a metachronního CRC (45).
1. Konvenční histologie
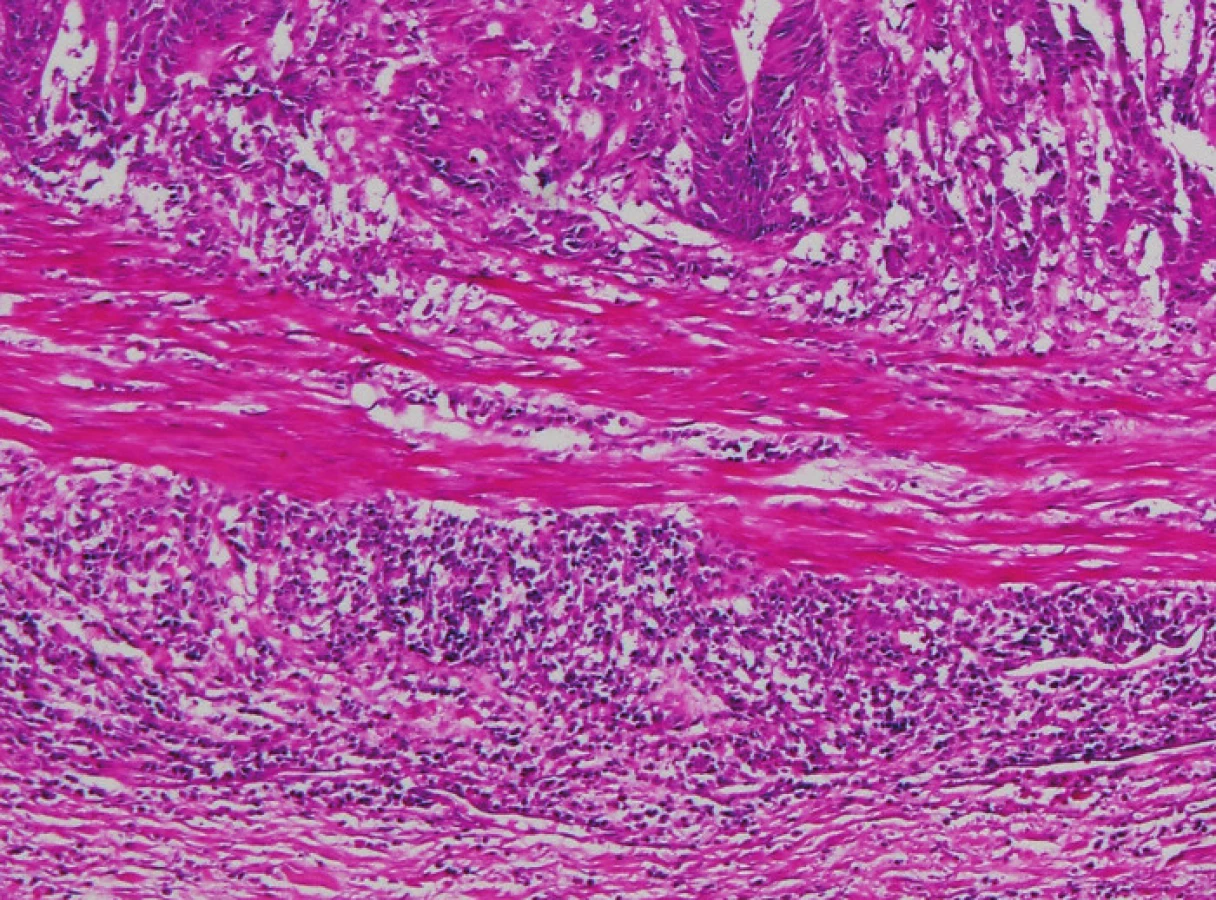
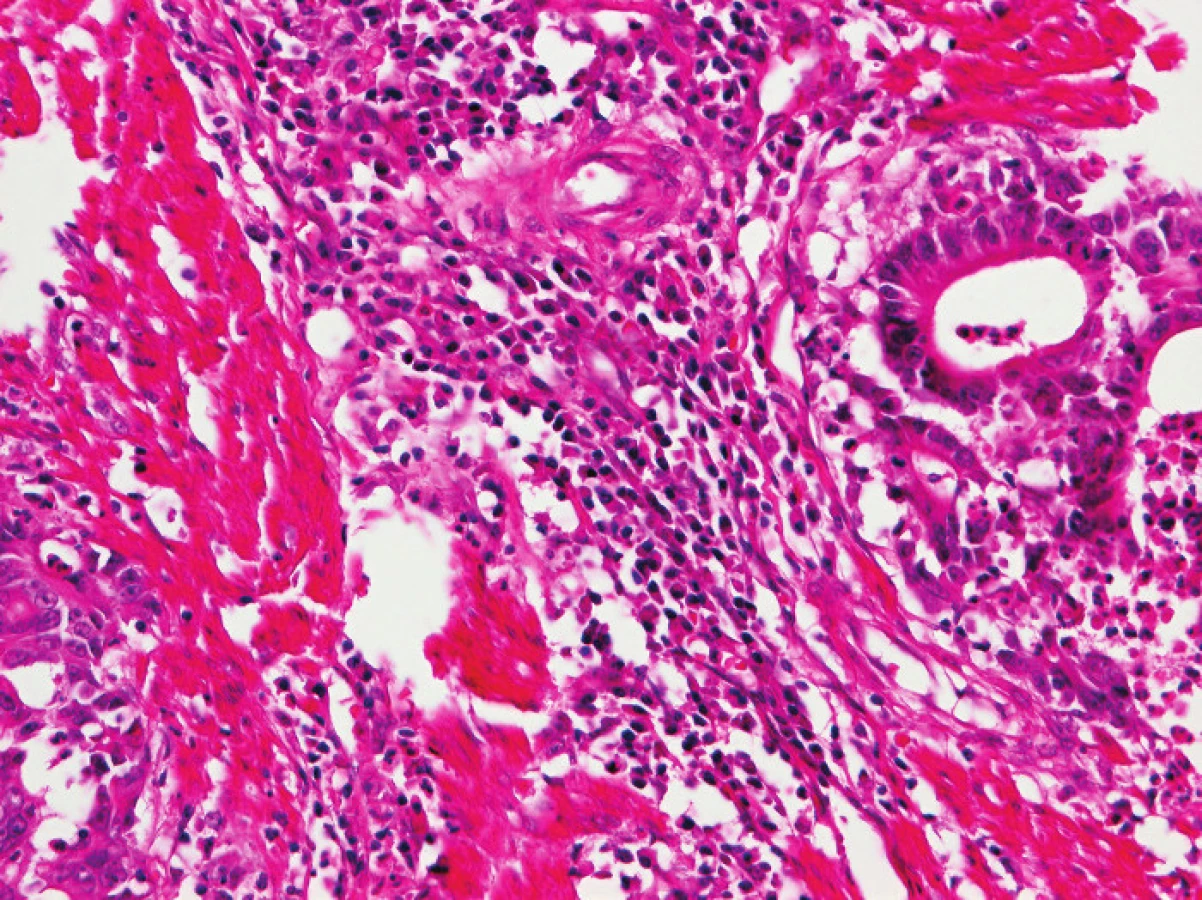
Zapojení histopatologa do vyhledávání pacientů s LS je založeno na detekci histologických znaků charakteristických pro CRC s vysokým stupněm nestability mikrosatelitů, tedy takzvané „MSI-H histologii“. Tyto morfologické rysy sice nejsou charakteristické pouze pro LS, protože se vyskytují i u sporadických MSI-H karcinomů způsobených epigenetickou inaktivací MLH1, ale odhaduje se, že 1 z 5 MSI-H CRC je CRC v rámci LS. Tento jistě ospravedlnitelný pokles specificity je daní za fakt, že samotná MSI-H morfologie má vyšší senzitivitu než souhrn zbývajících 4 kritérií RBG (47). Ačkoli byly morfologické znaky vídané v MSI-H CRC popsány již v RBG (konkrétně byly uváděny karcinomy se špatnou diferenciací, karcinomy mucinózní, medulární nebo „z buněk pečetního prstenu“, přítomnost Crohn-like lymfoidních nodulů a tumor-infiltrujících lymfocytů (TIL)), nebyla dostatečně známa ani prediktivní hodnota jednotlivých znaků, ani jejich reprodukovatelná kvantitativní definice, která by byla použitelná v rutinní praxi (36,48). Proto v posledních letech proběhlo několik studií (49), jejichž cílem bylo nalézt racionální algoritmus detekce MSI-H karcinomů, z nichž se v současné době jako nejužitečnější jeví model Model PREDICT (Pathological RolE in the Determination of Instability in Colorectal Tumors) (47), zejména ve své zjednodušené formě jako Semi PREDICT skóre (Tabulka 4). Histologickými znaky, na nichž je tento model založen jsou: přítomnost mucinu dissekujícího stroma v jakémkoli množství (obr. 1), přítomnost tumor-infiltrujících lymfocytů (TIL) (obr. 1, 2), peritumorální lymfoidní lem (obr. 3) a zastoupení plazmatických buněk mezi buňkami zánětlivého infiltrátu ve stromatu převyšující 25 % (obr. 4).
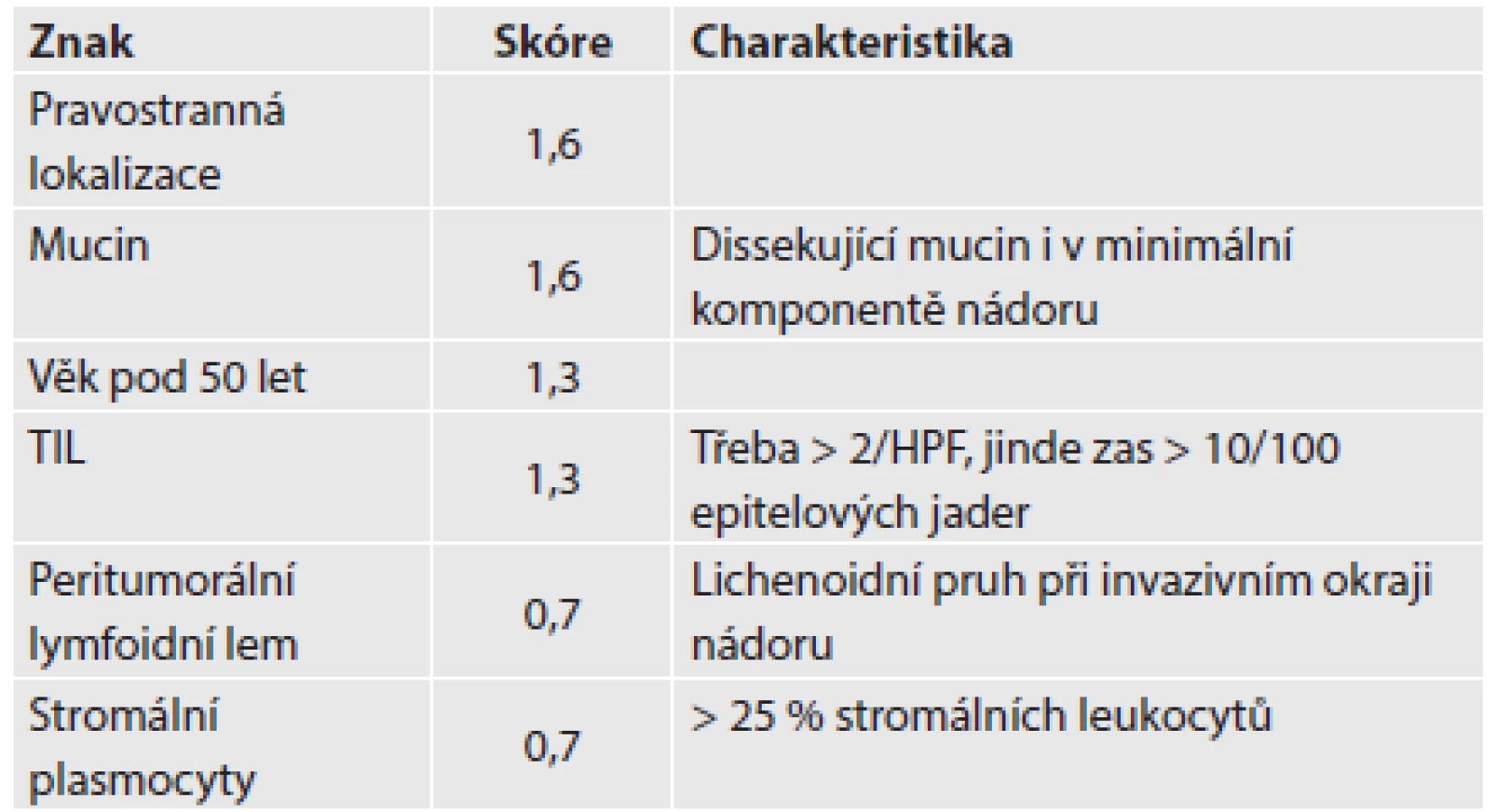
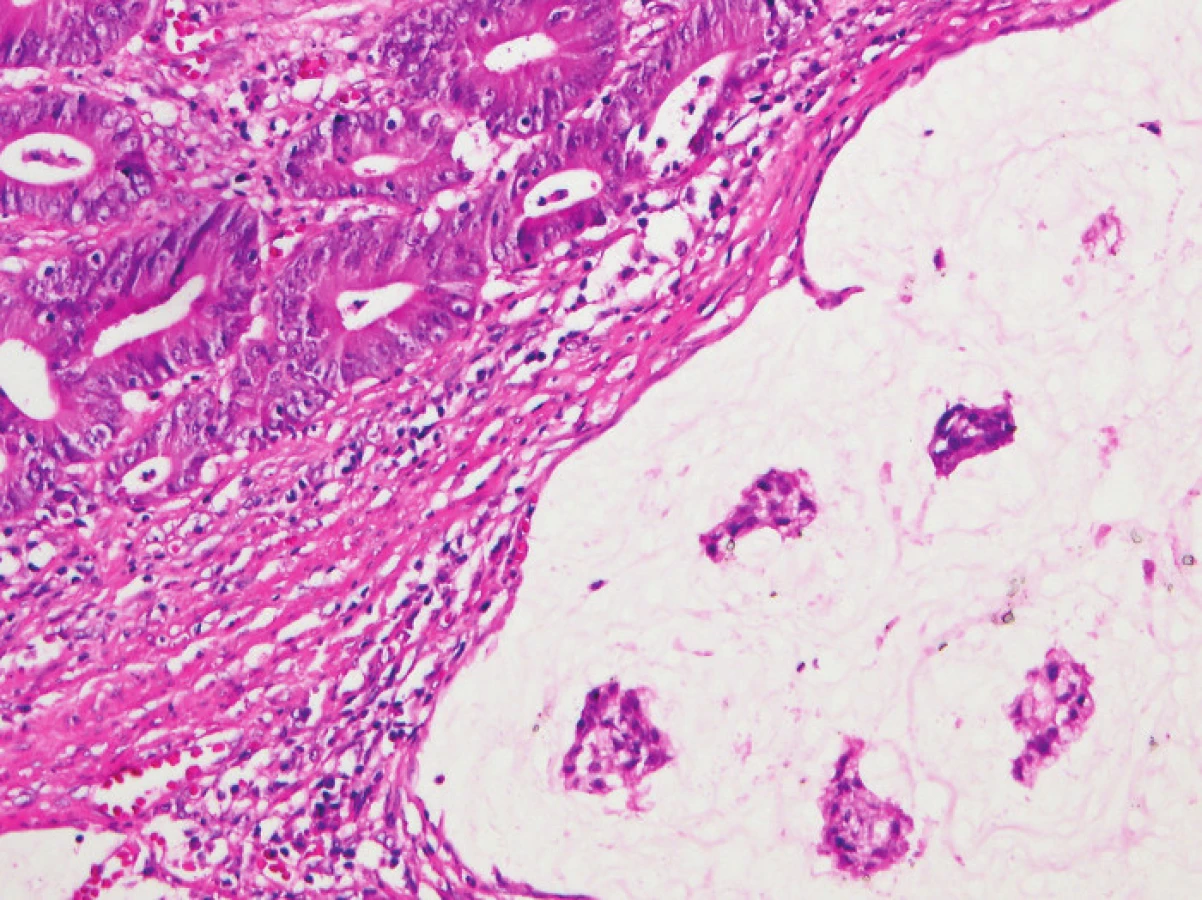
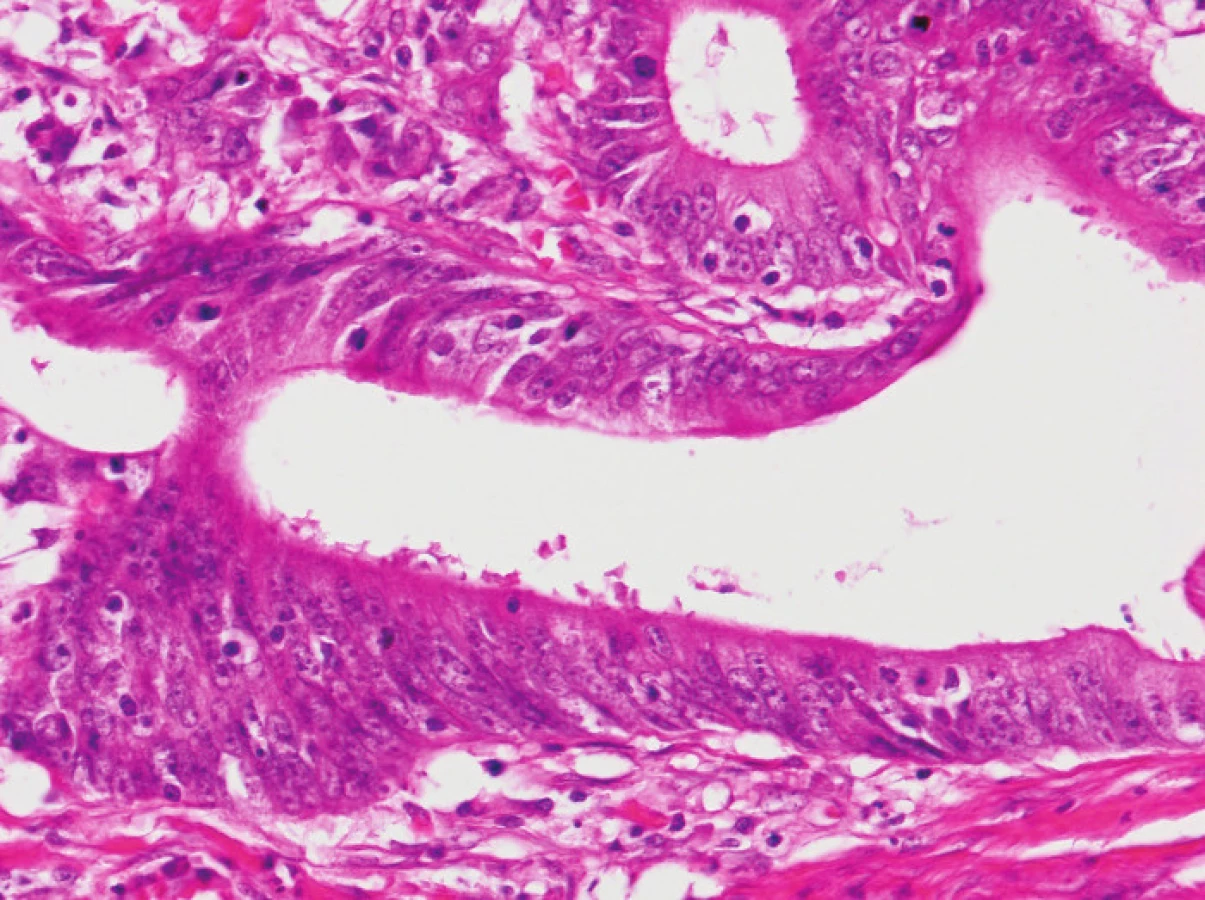
2. Imunohistochemie a stanovení MSI
Méně zprostředkovanou informaci o funkci MMR komplexu lze získat pomocí imunohistochemického vyšetření exprese hlavních MMR proteinů (tedy MLH1, PMS2, MSH2 a MSH6) nebo prostřednictvím vyšetření MSI Bethesda panelem mikrosatelitních markerů (viz výše a Tabulka 1). Zpočátku, když byla imunohistochemicky stanovována pouze exprese MLH1 a MSH2, zdála se senzitivita imunohistochemického vyšetření ve srovnání s MSI nedostatečná. Avšak po zavedení dalších dvou protilátek, které detekují partnery MLH1 a MSH2 pro tvorbu heterodimerů (tedy proteiny PMS2 a MSH6), je senzitivita imunohistochemického vyšetření a stanovení MSI srovnatelná (50). Význam detekce těchto funkčně minoritních partnerů pro tvorbu heterodimerů spočívá nejen v průkazu jejich vlastních strukturálních abnormalit, ale i v nepřímém průkazu ztráty aktivity jejich dominantních partnerů. Některé mutace totiž vedou ke ztrátě funkce při zachování antigenicity, zejména v případě MLH1, což se ale projeví ztrátou exprese PMS2 (50). Tento efekt závislosti exprese funkčně minoritních proteinů (PMS2 a MSH6) na intaktní funkci jejich dominantních partnerů (MLH1 a MSH2) vedl i k vytvoření imunohistochemického detekčního modelu založeného pouze na dvou protilátkách detekujících PMS2 a MSH6. Podle něj by ztráta exprese PMS2 měla detekovat mutace MLH1 i PMS2, ztráta exprese MSH6 detekuje i poškození MSH2 (51). Další studie však tento model zpochybnila, navíc dokonce navrhla semikvantitativní model místo dosavadního binárního (pozitivní/negativní) (52).
V současné době jsou obě metody (stanovení MSI a imunohistochemické vyšetření) vnímány jako komplementární, protože v kombinaci mají vyšší senzitivitu než při samostatném použití (53,54). Typickým příkladem přínosu imunohistochemie jsou případy s mutací MSH6, které mohou uniknout při detekci MSI pomocí PCR, protože MSH2 může také tvořit komplex s MSH3 (50,55). Imunohistochemická detekce MMR proteinů navíc (narozdíl od stanovení MSI) umožňuje predikovat postižený gen pro molekulárně genetické vyšetření.
3. Rozlišení mezi karcinomem při Lynchově syndromu a sporadickým MSI-H karcinomem
Běžná světelná mikroskopie může být nápomocna i v diferenciální diagnostice mezi sporadickým MSI-H karcinomem a MSI-H karcinomem při LS. Zatímco sporadické MSI-H karcinomy s methylací promotoru MLH1 vznikají z tzv. „sesilních serrated adenomů“ (do češtiny někdy nešťastně překládaných jako „přisedlé pilovité adenomy“), prekurzorovou lézí karcinomů v terénu LS je „konvenční“ (tubulární, tubulovilózní nebo vilózní) adenom. Z toho lze odvodit, že najdeme-li v periferii tumoru zbytky prekurzorového sesilního serrated adenomu, jde s největší pravděpodobností o sporadický MSI-H karcinom (56). Nicméně tento jednoduchý diagnostický znak vylučující LS nemusí být vždy dostupný, ať již z důvodu nedostatečného samplingu nebo destrukce adenomu pokročilým adenokarcinomem, což vyžaduje použití dalších metod k diferenciální diagnostice.
V současné době umožňuje rozlišení sporadických a LS-asociovaných MSI-H karcinomů zapojení dvou metod molekulární patologie do managementu CRC. První z nich je analýza genu BRAF, konkrétně průkaz mutace V600E, která je přítomna až u poloviny sporadických MSI-H CRC, ale (téměř) nikdy u LS. Druhou metodu představuje průkaz hypermethylace promotoru MLH1, která je markerem sporadických MSI-H CRC a naopak až na výjimky nebývá přítomna u LS (57). S vědomím určitého statistického zjednodušení lze tedy tyto dvě metody využít k vyřazení pacientů z dalšího (nákladného) diagnostického managementu, pokud tomu nebrání jiné okolnosti (např. nízký věk, výrazné familiární postižení, multiplicita nádorů).
Spíše teoretickou hodnotu má fakt, že MSI je přítomna již asi v polovině případů adenomů u LS, zatímco ve sporadických adenomech není prakticky nikdy, takže průkaz MSI v adenomu je celkem specifický pro LS.
4. Komplexní diagnostický management CRC
Ke zvýšení záchytu LS využíváme v současné době diagnostický algoritmus, na jehož počátku stojí mikroskopické vyšetření histologických řezů obarvených hematoxylinem a eozinem, na něž v případě přítomnosti znaků „MSI-H histologie“ navazuje sada metod imunohistochemických a molekulárně patologických, jejichž cílem je potvrdit přítomnost MSI-H, vyloučit možnost sporadického MSI-H karcinomu a určit pravděpodobně postižený gen k detekci germinální mutace.
Nádory, jejichž Semi PREDICT skóre vzbuzuje podezření na MSI-H, vykazují ztrátu exprese některého (nebo některých) z MMR proteinů, nemají hypermethylovaný promotor genu MLH1 a nebyla u nich prokázána substituce V600E genu BRAF, jsou určeny k detekci germinální mutace MMR genu určeného na základě imunohistochemického vyšetření. Celý algoritmus je znázorněn na obr. 5. Zatímco imunohistochemické vyšetření a analýza genu BRAF se běžně provádí v materiálu z parafínových bločků tumoru (ačkoli pro molekulárně genetickou analýzu by byla vhodnějším materiálem čerstvá nádorová tkáň, jejíž získání je však z logistických důvodů kromě specifických situací poměrně obtížné), analýza hypermethylace promotoru MLH1 a stanovení MSI vyžaduje srovnání s nenádorovou tkání. Tato sice bývá běžně dostupná v případě chirurgických resekátů (většinou jsou k tomuto účelu používány vzorky z chirurgických okrajů resekátu), většinou však není k dispozici při vyšetřování endoskopických biopsií. V těchto případech je nutná domluva se zainteresovaným gastroenterologem, aby zajistil kontrolní tkáň, ať už v podobě endoskopického vzorku z nenádorové sliznice nebo nesrážlivé periferní krve. Pokud jde o finální analýzu MMR genů, zde je optimálním materiálem nesrážlivá periferní krev, protože průkaz mutace mimo nádorovou tkáň potvrzuje její germinální povahu, přičemž z důvodu technické náročnosti analýzy těchto genů je výtěžnost z krve výrazně vyšší než z nenádorové tkáně fixované ve formolu.
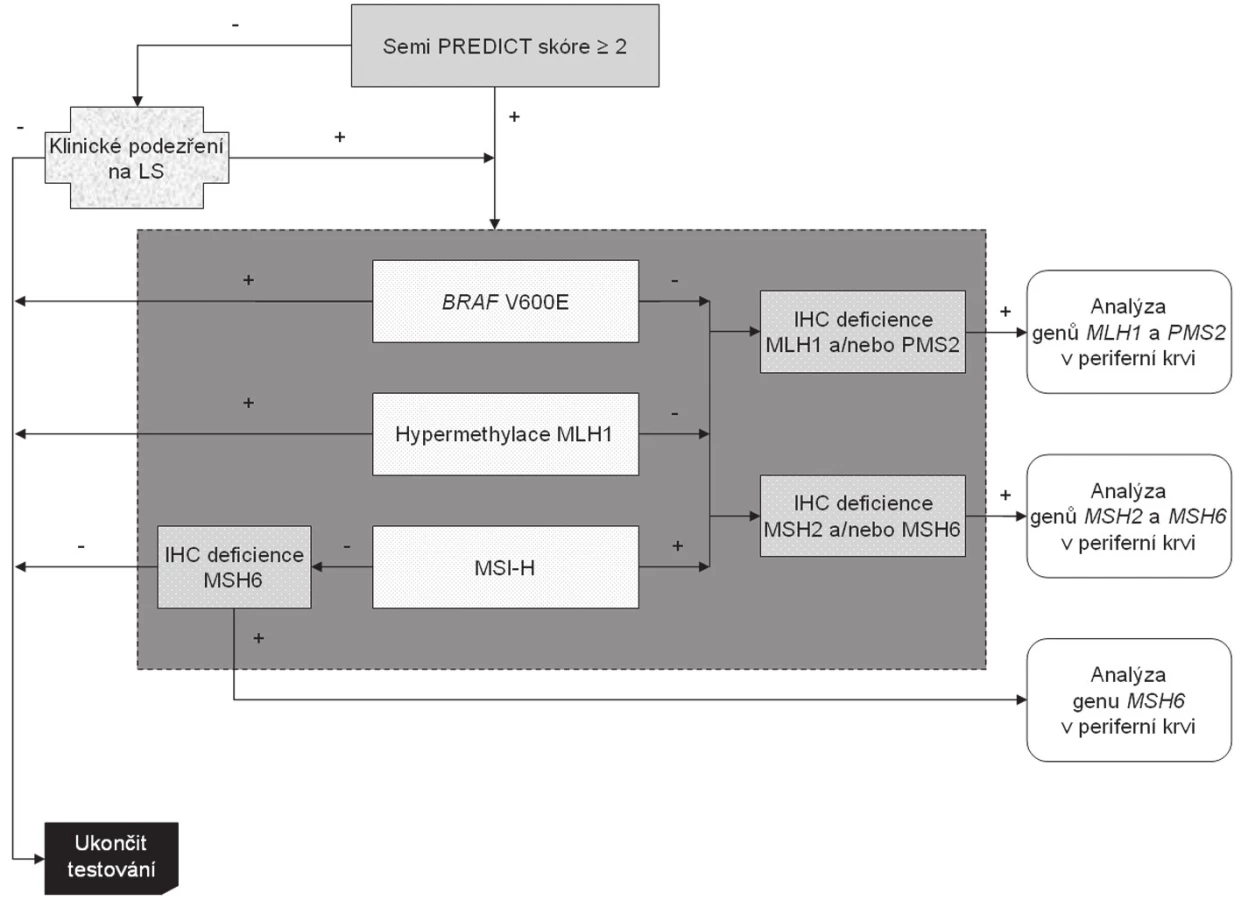
Ačkoli jsou možné i odlišné přístupy (např. primární plošné vyšetřování MSI u všech CRC), plnohodnotný postup by měl zahrnovat všechny uvedené diagnostické modality. Důvodem je jejich vzájemná komplementárnost. Kupříkladu samotné vyšetření MSI bez analýzy genu BRAF a stavu methylace promotoru MLH1 nedokáže odlišit sporadické CRC od syndromových, dále značná část karcinomů s mutací MSH6 nemusí vykazovat MSI-H, a konečně bez imunohistochemického vyšetření nelze zúžit spektrum MMR genů, jejichž stav má být analyzován.
PREZENTACE LYNCHOVA SYNDROMU MIMO KOLOREKTÁLNÍ KARCINOM
Ačkoli germinální mutace způsobující LS může být teoreticky zodpovědná za vznik maligního tumoru ve kterékoli lokalizaci (viz výše), zvláštní zřetel zasluhuje endometriální karcinom a syndromologické varianty LS.
Endometriální karcinom je totiž u žen druhou nejčastější malignitou asociovanou s LS, přičemž riziko vzniku je podle literárních údajů 40 - 60 % (44,58,59). Odhaduje se, že 1,8% všech nově zjištěných karcinomů endometria je součástí LS, přičemž častěji než u typických případů LS je u nich zastoupena mutace MSH6 (24). S tím jde ruku v ruce nižší penetrance LS, která může zakrýt familiární vazbu endometriálního karcinomu, prezentace ve vyšším věku než je pro LS typické, absence MSI-H a morfologických znaků svědčících pro LS. Z toho důvodu se jeví jako nutnost provádění imunohistochemického vyšetření exprese MMR proteinů u všech diagnostikovaných endometriálních karcinomů (60).
Ze syndromologických variant byl samostatně popsán Turcotův syndrom 1. typu, který (ačkoli použití internetového vyhledávače odhaluje v této oblasti značný terminologický chaos) je v našich podmínkách definován jako asociace CRC a nádoru mozku, zejména astrocytárního. Ačkoli je tento syndrom závažný z hlediska prognózy pacienta a může hrát i určitou diagnostickou roli, nelze jej významem srovnávat s MTS, který se vyznačuje přítomností sebaceózních kožních nádorů, které jsou snadno přístupné vyšetření, a představuje jedinou variantu LS s premorbidním fenotypem (1).
ZÁVĚR
Lynchův syndrom (LS) je familiární karcinomový syndrom způsobený germinální mutací některého z genů, jehož proteinový produkt se účastní opravy chyb v DNA vzniklých při replikaci. Vzhledem k tomu, že výskyt LS v populaci se nyní odhaduje až na 5% a vede ke vzniku maligních nádorů již v produktivním věku, sílí v současné době tlak na zvýšení senzitivity jeho detekce. Protože klinický přístup k jeho záchytu se ukázal jako nedostatečně efektivní, klade se dnes stále větší důraz na morfologickou a molekulárně patologickou diagnostiku. Ta by měla představovat komplex vyšetření histologického, imunohistochemického a molekulárně biologického, byť vzájemné uspořádání jednotlivých komponent tohoto komplexu může být různé v závislosti na lokálních podmínkách. Podle našich zkušeností je nezbytné zavést jasný algoritmus, aby se diagnostika LS stala rutinní záležitostí a její management se co nejvíce zautomatizoval. Bohužel se dosud nepodařilo v našich podmínkách vytvořit spolehlivě fungující systém zpětné vazby s klinickými lékaři, kteří by měli organizovat další průběh vyšetření rodiny nemocného s LS. V nejbližších letech je tedy nezbytné zainteresovat do diagnostiky LS i klinické lékaře, bez jejichž aktivní účasti není možná ani kompletní diagnostika pacienta ani další vyšetření jeho rodinných příslušníků.
SEZNAM ZKRATEK
BG - Bethesda guidelines
CRC - colorectal carcinoma, kolorektální karcinom
EPCAM - epithelial cell adhesion molecule
FAP - familiární adenomatózní polypóza
HNPCC - hereditary non-polyposis colorectal cancer, hereditární nepolypózní kolorektální karcinom
LS - Lynchův syndrom
MLH1 - mut L homolog 1
MMR - mismatch repair
MSH2 - mut S homolog 2
MSH6 - mut S homolog 6
MSI - microsatellite instability, nestabilita mikrosatelitů
MSI-H - microsatellite instability – high, vysoký stupeň nestability mikrosatelitů
MSS - microsatellite stable, stabilní mikrosatelity
MTS - Muir – Torreho syndrom
PMS2 - postmeiotic segregation increased 2
RBG - revidovaná Bethesda guidelines
Adresa pro korespondenci:
Doc. MUDr. Ondřej Daum, Ph.D.
Šiklův ústav patologie LF UK a FN Plzeň
Edvarda Beneše 13, 305 99 Plzeň
tel.: +420377402523
e-mail: DAUM@fnplzen.cz
Sources
1. Kacerovská D, Kazakov DV, Černá K, et al. Muir-Torre syndrom - fenotypická varianta Lynchova syndromu. Cesk Patol 2010; 46(4): 86-94.
2. Warthin AS. Heredity with reference to carcinoma: as shown by the study of the cases examined in the pathological laboratory of the University of Michigan, 1895-1913. Arch Intern Med 1913; 12(5): 546-555.
3. Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med 1966; 117(2): 206-212.
4. Lynch HT, Krush AJ. Cancer family “G” revisited: 1895-1970. Cancer 1971; 27(6): 1505-1511.
5. Lynch HT, Drouhard TJ, Schuelke GS, et al. Hereditary nonpolyposis colorectal cancer in a Navajo Indian family. Cancer Genet Cytogenet 1985; 15(3-4): 209-213.
6. Boland CR, Troncale FJ. Familial colonic cancer without antecedent polyposis. Ann Intern Med 1984; 100(5): 700-701.
7. Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293(16): 1979-1985.
8. Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75(5): 1027-1038.
9. Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75(6): 1215-1225.
10. Peltomaki P, Lothe RA, Aaltonen LA, et al. Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res 1993; 53(24): 5853-5855.
11. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science 1993; 260(5109): 816-819.
12. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58(22): 5248-5257.
13. Yuen ST, Chan TL, Ho JW, et al. Germline, somatic and epigenetic events underlying mismatch repair deficiency in colorectal and HNPCC-related cancers. Oncogene 2002; 21(49): 7585-7592.
14. Nystrom-Lahti M, Wu Y, Moisio AL, et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet 1996; 5(6): 763-769.
15. Wijnen J, van der Klift H, Vasen H, et al. MSH2 genomic deletions are a frequent cause of HNPCC. Nat Genet 1998; 20(4): 326-328.
16. Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002; 62(14): 3925-3928.
17. Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 2007; 356(7): 697-705.
18. Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet 2009; 41(1): 112-117.
19. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 2009; 30(2): 197-203.
20. Kruse R, Rutten A, Hosseiny-Malayeri HR, et al. “Second hit” in sebaceous tumors from Muir-Torre patients with germline mutations in MSH2: allele loss is not the preferred mode of inactivation. J Invest Dermatol 2001; 116(3): 463-465.
21. Gallinger S, Aronson M, Shayan K, et al. Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology 2004; 126(2): 576-585.
22. Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam Cancer 2005; 4(4): 323-333.
23. Kastrinos F, Stoffel EM, Balmana J, et al. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol Biomarkers Prev 2008; 17(8): 2044-2051.
24. Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006; 66(15): 7810-7817.
25. Berends MJ, Wu Y, Sijmons RH, et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 2002; 70(1): 26-37.
26. Nakagawa H, Lockman JC, Frankel WL, et al. Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res 2004; 64(14): 4721-4727.
27. Hendriks YM, Jagmohan-Changur S, van der Klift HM, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130(2): 312-322.
28. Truninger K, Menigatti M, Luz J, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 2005; 128(5): 1160-1171.
29. Mathiak M, Rutten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol 2002; 26(3): 338-343.
30. Mangold E, Pagenstecher C, Leister M, et al. A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet 2004; 41(7): 567-572.
31. De Rosa M, Fasano C, Panariello L, et al. Evidence for a recessive inheritance of Turcot’s syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene 2000; 19(13): 1719-1723.
32. Kamory E, Kolacsek O, Otto S, Csuka O. hMLH1 and hMSH2 somatic inactivation mechanisms in sporadic colorectal cancer patients. Pathol Oncol Res 2003; 9(4): 236-241.
33. Entius MM, Keller JJ, Drillenburg P, et al. Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res 2000; 6(5): 1784-1789.
34. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34(5): 424-425.
35. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116(6): 1453-1456.
36. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96(4): 261-268.
37. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352(18): 1851-1860.
38. Liu T, Yan H, Kuismanen S, et al. The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res 2001; 61(21): 7798-7802.
39. van der Klift H, Wijnen J, Wagner A, et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 2005; 44(2): 123-138.
40. Dovrat S, Figer A, Fidder HH, et al. Mutational analysis of hMsh6 in Israeli HNPCC and HNPCC-like families. Fam Cancer 2005; 4(4): 291-294.
41. Hegde MR, Chong B, Blazo ME, et al. A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res 2005; 11(13): 4689-4693.
42. de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer 2005; 4(3): 233-237.
43. Jenkins MA, Baglietto L, Dowty JG, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006; 4(4): 489-498.
44. Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005; 42(6): 491-496.
45. Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36(11): 801-818.
46. Jass JR, Stewart SM. Evolution of hereditary non-polyposis colorectal cancer. Gut 1992; 33(6): 783-786.
47. Hyde A, Fontaine D, Stuckless S, et al. A histology-based model for predicting microsatellite instability in colorectal cancers. Am J Surg Pathol 2010; 34(12): 1820-1829.
48. Jenkins MA, Hayashi S, O’Shea AM, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology 2007; 133(1): 48-56.
49. Roman R, Verdu M, Calvo M, et al. Microsatellite instability of the colorectal carcinoma can be predicted in the conventional pathologic examination. A prospective multicentric study and the statistical analysis of 615 cases consolidate our previously proposed logistic regression model. Virchows Arch 2010; 456(5): 533-541.
50. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 2008; 10(4): 293-300.
51. Shia J, Tang LH, Vakiani E, et al. Immunohistochemistry as first-line screening for detecting colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: a 2-antibody panel may be as predictive as a 4-antibody panel. Am J Surg Pathol 2009; 33(11): 1639-1645.
52. Barrow E, Jagger E, Brierley J, et al. Semiquantitative assessment of immunohistochemistry for mismatch repair proteins in Lynch syndrome. Histopathology 2010; 56(3): 331-344.
53. Funkhouser WK, Jr., Lubin IM, Monzon FA, et al. Relevance, pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn 2012; 14(2): 91-103.
54. Halvarsson B, Lindblom A, Rambech E, Lagerstedt K, Nilbert M. Microsatellite instability analysis and/or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer? Virchows Arch 2004; 444(2): 135-141.
55. Boland CR, Koi M, Chang DK, Carethers JM. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer 2008; 7(1): 41-52.
56. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007; 50(1): 113-130.
57. Domingo E, Laiho P, Ollikainen M, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004; 41(9): 664-668.
58. Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997; 6(1): 105-110.
59. Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81(2): 214-218.
60. Clarke BA, Cooper K. Identifying Lynch syndrome in patients with endometrial carcinoma: shortcomings of morphologic and clinical schemas. Adv Anat Pathol 2012; 19(4): 231-238.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2014 Issue 1
Most read in this issue
- Lynch syndrome in the hands of pathologists
- Cell cultures
- Detection of chromosome changes by CGH, array-CGH and SNP array techniques in tumours
- Uterine tumors resembling ovarian sex cord tumors (UTROSCT). Report of a case with lymph node metastasis