
Neurofibromatosis von Recklinghausen typ 1 (NF1) – klinický obraz a molekulárně-genetická diagnostika
Neurofibromatosis von Recklinghausen type 1 (NF1) – clinical picture and molecular-genetics diagnostic
Neurofibromatosis von Recklinghausen type 1 (NF1) is a multisystem, autosomal dominant hereditary neurocutaneous disease characterized by skin, central and peripheral nervous system , eyes , bone, endocrine, gastrointestinal and blood vessel wall involvement. It has an estimated frequency of 1 in 3000. Neurofibromatosis type 1 is caused by mutations in the large NF1 gene located on chromosome 17q11.2, encoding the cytoplasmic protein neurofibromin. It is expressed in multiple cell types but is highly expressed in Schwann cells, oligodendrocytes, neurons, astrocytes and leukocytes. Neurofibromin is known to act as a tumor suppressor via Ras-GTPase activation, which causes down-regulation of cellular signaling via the Ras/mitogen-activated protein kinase (MAPK) pathway. Failure of this function is associated with a tendency to form tumors which are histologically hamartomas as well as benign tumors. Tumors of the central nervous system include low-grade gliomas (pilocytic astrocytomas grade I), especially optic pathway gliomas. They are often clinically asymptomatic. Other intracranial tumors are in the brain stem and also elsewhere in the brain and spinal cord. Hydrocephalus may be a complication of NF1 gliomas or due to stenosis of the distal part of the aqueduct Silvii. Cutaneous and subcutaneous neurofibromas or plexiform neurofibromas are localized in the peripheral nervous system. Plexiform neurofibromas have a significant lifetime risk of malignancy.
The clinical diagnosis of NF1 is defined by diagnostic criteria. The NF1 diagnosis is satisfied when at least two of the seven conditions are met. The method of direct DNA analysis of large NF1 gene (61 exons) is available. The results of studies of genotype - phenotype established few correlations. But predicting the disease by finding mutations is not currently possible. NF1 exhibits a wide range of variability of expression and complete penetrance, even within the same family. About half of cases are new mutations. The treatment of patients with neurofibromatosis is symptomatic. Central nervous system symptomatic low-grade gliomas are most often treated with chemotherapy. For plexiform neurofibromas surgical removal is currently the only treatment option.
Keywords:
NF1 – neurofibromin – glioma – neurofibroma – hydrocephalus - genetics
Authors:
Bořivoj Petrák 1; Šárka Bendová 2; Jiří Lisý 3; Josef Kraus 1; Tomáš Zatrapa 4; Marie Glombová 1; Josef Zámečník 5
Authors‘ workplace:
Klinika dětské neurologie, 2. LF UK a FN Motol, Praha
1; Ústav biologie a lékařské genetiky, 2. LF UK a FN Motol, Praha
2; Klinika zobrazovacích metod, 2. LF UK a FN Motol, Praha
3; I. ortopedická klinika, 1. LF UK a FN Motol, Praha
4; Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
5
Published in:
Čes.-slov. Patol., 51, 2015, No. 1, p. 34-40
Category:
Reviews Article
Overview
Neurofibromatosis von Recklinghausen typ 1 (NF1) je multisystémové, autozomálně dominantně dědičné neurokutánní onemocnění charakterizované postižením kůže, centrálního a periferního nervového systému, oka, kostí a cévní stěny. Incidence je 1 : 3000 živě narozených dětí. Příčinou rozvoje onemocnění je mutace tumor supresorového genu NF1 (17q11.2) a z toho vyplývající porucha tvorby neurofibrominu - cytoplazmatického proteinu, přednostně exprimovaného v neuronech, Schwannových buňkách, oligodendrocytech, astrocytech a leukocytech. Hlavní úkolem neurofibrominu je jeho funkce negativního regulátoru komplexu Ras. Porucha této funkce je u NF1 dávána do souvislosti s výskytem mnohočetných nádorových procesů, které histologicky odpovídají hamartomům nebo benigním nádorům. Nádory centrálního nervového systému jsou především gliomy nízkého gradu (pilocytární astrocytom, grade I), zvláště v průběhu zrakové dráhy. Tyto nádory jsou často klinicky asymptomatické. Gliomy bývají časté také v oblasti mozkového kmene a mohou být nalezeny také jinde v mozku a v míše. Hydrocefalus může být u NF1 komplikací gliomu nebo vzniká při stenóze distální části mokovodu. V periferním nervovém systému se objevují neurofibromy a plexiformní neurofibromy. Plexiformní neurofibromy mají významné celoživotní riziko malignizace.
Klinická diagnóza NF1 je definována sedmi diagnostickými kriterii a ke stanovení diagnózy je nutný nález alespoň dvou z nich. Metodika přímé DNA analýzy rozsáhlého NF1 genu (61 exonů) je vypracována. Výsledky studií genotyp-fenotyp jsou ale málo průkazné a predikce průběhu onemocnění podle nalezené mutace není v současné době možná. Frekvence sporadického výskytu NF1 je vysoká (30 - 50 %). Variabilita fenotypu onemocnění je výrazná i v rámci jedné rodiny. Kauzální terapie není v současné době známa - léčba je symptomatická. Při progredujícím low-grade gliomu je metodou volby chemoterapie, pro plexiformní neurofibromy chirurgické řešení.
Klíčová slova:
NF1 – neurofibromin – gliom – neurofibrom – hydrocefalus – genetika
Diagnózu neurofibromatózy popsal Friedrich Daniel von Recklinghausen v roce 1882 v práci „Über die multiplen Fibrome der Haut und ihre Beziehnung zu den multiplen Neuronomen“ a dedikoval ji svému učiteli Rudolfu Virchowovi (1). V 70. letech 20. století byla vyčleněna skupina pacientů s oboustrannými vestibulárními schwannomy (neurinomy) a diagnóza NF se tím rozdělila na typ 1 a typ 2. Následně v 90. letech 20. století byly nalezeny geny pro NF1 na chromozomu 17 (17q11.2) a pro NF2 na chromozomu 22 (22q12.2). Potvrdilo se tak, že se nejedná o varianty jednoho onemocnění, ale o dvě samostatné jednotky, které mají odlišný klinický obraz a rozdílný věk manifestace (2-4). Obě diagnózy lze tedy určit na základě klinických i molekulárně genetických charakteristik (2). Dříve používané názvy periferní NF (typ 1) a centrální NF (typ 2) nevystihují charakter jednotlivých diagnóz a proto se již nepoužívají (4,5). NF1 může mít charakter mozaiky. Sem lze zařadit již dříve klinicky vyčleněnou segmentální formu NF1 (dříve nazývaná NF typ V), která představuje mozaiku somatické mutace genu NF1 (2,5,6).
Neurokutánní syndromy
Neurofibromatózy jsou řazeny do skupiny neurokutánních syndromů, kde NF1 představuje nejčastější onemocnění. Neurokutánní syndromy tvoří skupina chorob, které postihují především kůži a nervový systém, ale současně se mohou objevit také změny v dalších systémech organismu a jedná se tedy o multisystémová onemocnění. Manifestují se od porodu do dospělosti, mají většinou pomalý progresivní charakter a variabilní klinický projev s rizikem častějšího výskytu nádorů kůže, nervového systému a ostatních orgánů. Původní termín fakomatózy se již nepoužívá. Nejčastější neurokutánní syndromy mají převážně autozomálně dominantní charakter dědičnosti, ale sporadický výskyt je u těchto jednotek také velmi častý (2,4,5). Za jednotící prvek potřebný pro zařazení do této skupiny je v současnosti považována porucha vývoje v oblasti neurální lišty (utváření neurální lišty, migrace nebo závěrečná diferenciace zárodečných buněk) a postižení neuro-ektodermu (2,5,7). Zodpovědné geny mají funkci tumor-suppressor genů (7).
NEUROFIBROMATOSIS VON RECKLINGHAUSEN TYP 1
(neurofibromatosis 1, neurofibromatóza 1. typu, NF1)
Neurofibromatosis von Recklinghausen typ 1 (NF1) je multisystémové onemocnění charakterizované postižením kůže, centrálního a periferního nervového systému, oka a také kostí a cévní stěny. Hlavní příčinou rozvoje NF1 je inaktivace tumor supresorového genu NF1 (HGNC:7765; MIM 613113; RefSeq: NM_000267.3) zárodečnými mutacemi. To je spojeno s výskytem mnohočetných nádorových procesů – především periferního a centrálního nervového systému. Tyto nádorové procesy jsou převážně benigního charakteru a histologicky odpovídají hamartomům a benigním nádorům, současně však existuje sklon k jejich maligní transformaci. NF1 má autozomálně dominantní dědičnost (AD), s variabilní expresivitou a prakticky úplnou penetrancí do dospělého věku. Při incidenci 1 : 3000 živě narozených dětí je jedním z nejčastěji se vyskytujících AD onemocnění u člověka (8). Současně jsou ale časté také nové mutace (30 - 50 %) a téměř polovina případů má tedy sporadický výskyt (5,9). Přitom je častější výskyt nových mutací uváděn na paternálních chromozomech a také výskyt germinální mozaiky je výrazně častější u otců (9). Vysoký sporadický výskyt NF1 nebyl dosud uspokojivě vysvětlen. Jedna z hypotéz uvádí například vliv vyššího věku otce v době početí - na základě dlouhodobé metylace DNA některých spermií se vznikem bodových mutací (9). První příznaky NF1 se většinou manifestují v časném kojeneckém věku. Další diagnostické příznaky jsou věkově vázané a s věkem jejich výskyt stoupá.
Gen NF1 a neurofibromin
Gen NF1 se nachází na dlouhém raménku chromozomu 17 v oblasti q11.2, blízko centromery (17q11.2). Zahrnuje 280 kb genomové DNA, skládá se z 61 exonů a kóduje 12 kb dlouhý transkript mRNA s 9 kb dlouhým otevřeným čtecím rámcem. V sekvenci genu NF1 se nacházejí 2 rozsáhlé introny, 1 a 27b. Intron 27b obsahuje 3 malé, sobě nepříbuzné geny, EVI2A, EVI2B a OMG. Individuální či společný vliv na expresi genu NF1 je dosud neznámý (10). Proteinové produkty jsou exprimovány především v mozku (EVI2A a OMG) a v kostní dřeni (EVI2A). Produkt genu OMG, OMGP, je membránový glykoprotein exprimovaný v centrálním nervovém systému během procesu myelinizace a působí jako inhibitor proliferace neuronů a fibroblastů in vitro (11). Neurofibromin (RefSeq: NP_000258.1, UniProtKB P21359-2) je gigantický cytoplazmatický protein (2818 aminokyselin, 320 kDa) kódovaný genem NF1, přednostně exprimovaný v neuronech, Schwannových buňkách, oligodendrocytech, astrocytech a leukocytech. V nízkých koncentracích se exprimuje i v mnoha jiných tkáních (12-15). Aktivita neurofibrominu je u pacientů s NF1 omezena. Protein zastává různé biochemické funkce včetně asociace s mikrotubuly a účastní se několika signálních drah (MAPK, cAMP) (16). Neurofibromin patří mezi tzv. savčí GTPázu aktivující proteiny (GAP). Ras-specifické GAP regulují biologickou aktivitu mnoha ostatních proteinů. Cytoplazmatický protein p21RAS o velikosti 21 kDa je kódován geny H-Ras, K-Ras (Harvey a Kirsten murine sarcoma onkogeny virového původu) a N-Ras (lidský neuroblastomový onkogen) a hraje klíčovou roli v buněčné diferenciaci a růstu či v komunikaci mezi cytoplazmatickou membránou a jádrem. Aktivovaný p21RAS interaguje s Raf-MEK-ERK (MAPK) signální drahou, která je zodpovědná za regulaci exprese genů podílejících se na kontrole buněčného cyklu, diferenciace a migrace buněk. Mutace genů skupiny RAS spolu s haploinsuficiencí nebo úplnou absencí neurofibrominu jsou příčinou udržování p21RAS v aktivním stavu, čímž dochází ke stálé stimulaci signální dráhy MAPK a následné proliferaci buněk. Snížená exprese neurofibrominu koreluje s množstvím aktivovaného p21RAS v některých typech nádorů (16,17).
Neurofibromin také pozitivně reguluje intracelulární hladinu cAMP. V NF1-deficientních buňkách je hladina cAMP výrazně snížena, což vede, např. v astrocytech, k inhibici buněčného růstu. Exprese NF1 je do značné míry daná genotypem. Za možné modifikátorové geny jsou považovány geny, které sousedí s genem NF1 a jsou spolu s ním inaktivovány mikrodelecemi nebo geny mající vliv na mitotickou rekombinaci, a tím také na míru ztráty heterozygozity (LOH) (18). Vysvětlením vysoké inter - i intra-familiární variability fenotypu pacientů s NF1 mohou být kromě modifikátorových genů také alelická heterogenita, mutace druhého zásahu, somatický mozaicismus, vliv prostředí nebo epigenetické změny. Dosud bylo v genu NF1 nalezeno přes 1300 kauzálních mutací (19). Jedná se o mutace bodové (missense, nonsense), posunové (inserce, delece, duplikace), sestřihové a velké genové přestavby. Žádná mutace regulačního charakteru nebyla doposud nalezena. Přibližně 30 - 50 % mutací v genu NF1 vzniká během embryonálního vývoje probanda (de novo) a manifestují se pak jako sporadické případy onemocnění NF1 v rodině. Způsob manifestace onemocnění nekoreluje s pohlavím ani etnikem probandů. Mezi nejčastější příčiny onemocnění NF1 patří mutace vedoucí ke ztrátě funkce genu NF1. Více než 80% zárodečných mutací způsobuje posun čtecího rámce s následným vznikem předčasného terminačního kodonu (20,21). Tento typ mutací je spojený především s absencí nebo sníženou mírou exprese mRNA (22). Missense, tiché mutace a in-frame varianty (delece resp. inserce jedné nebo více aminokyselin), které nejsou zodpovědné za vznik předčasného terminačního kodonu, s velkou pravděpodobností ovlivňují terciární strukturu neurofibrominu, a tím i jeho správnou funkci (23,24). Mutace jsou, nezávisle na typu, rovnoměrně rozptýleny podél celé kódující oblasti genu. Mnoho studií se u pacientů s NF1 snažilo odhalit jasnou korelaci mezi jejich genotypem a fenotypem (8,25,26). První z nich jsou rozsáhlé delece genu NF1 a přilehlých genů (tzv. mikrodeleční syndromy NF1 typ I a typ II) (27). Mikrodelece typu I o velikosti 1,4 Mb se vyskytuje častěji a zahrnuje gen NF1 a 14 dalších genů (27). Mikrodelece typu II má velikost 1,2Mb, zahrnuje 13 genů a její frekvence je nižší než u typu I a byla popsána pouze u žen. Tyto mikrodelece mají u pacientů s NF1 přibližně 5% výskyt a každá z nich se projevuje specifickým fenotypem. Další mutací se známým vlivem na fenotyp pacientů je delece AAT (3 bp) v exonu 17 (c.2970_2972delAAT) (26). Pacienti s touto delecí mají mírnější projevy fenotypu NF1 a nedochází u nich k rozvoji kožních, podkožních a plexiformních neurofibromů. Somatické mutace se v genu NF1 mohou objevit v průběhu embryonálního vývoje jako mutace prvního zásahu a podílet se tak na vytvoření mozaicismu nebo mohou vzniknout jako mutace druhého zásahu a inaktivovat divokou alelu NF1 u heterozygotního pacienta.
Molekulárně diagnostické metody
V rodinách se dvěma a více členy postiženými NF1 je možné použít nepřímou molekulárně genetickou diagnostiku založenou na vazebné analýze polymorfních DNA markerů (28). Nepřímá diagnostika nevypovídá o konkrétních kauzálních mutacích a neumožňuje detekci mutací de novo. Cílem přímé molekulárně genetické diagnostiky je identifikace kauzální zárodečné mutace v genu NF1 a potvrzení tak diagnózy určené na základě klinických kritérií pacienta a následně prediktivní testování příbuzných nebo prenatální diagnostika. Mutační analýza genu NF1 je ztížena některými jeho vlastnostmi. Patří mezi ně především velikost a vysoká mutační míra genu. Mutace všech typů jsou rozmístěny v celé šíři genu a nenacházíme zde místa se zvýšenou pravděpodobností jejich výskytu. Roli selekční metody zaujala hojně používaná metoda DHPLC (denaturační vysokotlaká kapalinová chromatografie) doplněná přímým sekvenováním a metodou určenou k detekci rozsáhlých genových přestaveb MLPA (multiplex ligation probe-dependent amplification). Druhé, často používané vyšetřovací schéma, je založeno na přímé detekci zárodečných mutací v cDNA genu NF1. Metody založené na analýze RNA umožňují odhalit intronové mutace a neobvyklé varianty sestřihu (29). V současné době vyšetřujeme oblast celé cDNA genu NF1, která byla získaná reverzní transkripcí RNA izolované z leukocytů periferní krve. Použitými metodami jsou RT PCR amplifikace a přímá sekvenace 20 exonů cDNA genu NF1 (obr.1). Nalezené mutace jsou ověřeny oboustrannou sekvenací příslušného exonu genomové DNA genu NF1. Přítomnost mutace je verifikována porovnáním s referenční sekvencí DNA. Pokud kauzální mutace není nalezena, genomová DNA pacientů je vyšetřena metodou MLPA. Tato metoda umožňuje odhalit i takové genové přestavby, které není možné detekovat přímou sekvenací cDNA. K vyhledávání delecí v genomu NF1 pacientů se dále používají metody FISH fluorescenční in situ hybridizace) a array CGH (komparativní genomová hybridizace). V případě nálezu kauzální mutace přímá DNA analýza umožní (pokud má rodina zájem) prenatální diagnostiku také u pacientů se sporadickým výskytem NF1 nebo zpřesní diagnostiku u informativních rodin. Rodinám by mělo být nabídnuto genetické poradenství a současně informace o jeho možnostech a možnostech prenatální diagnostiky.
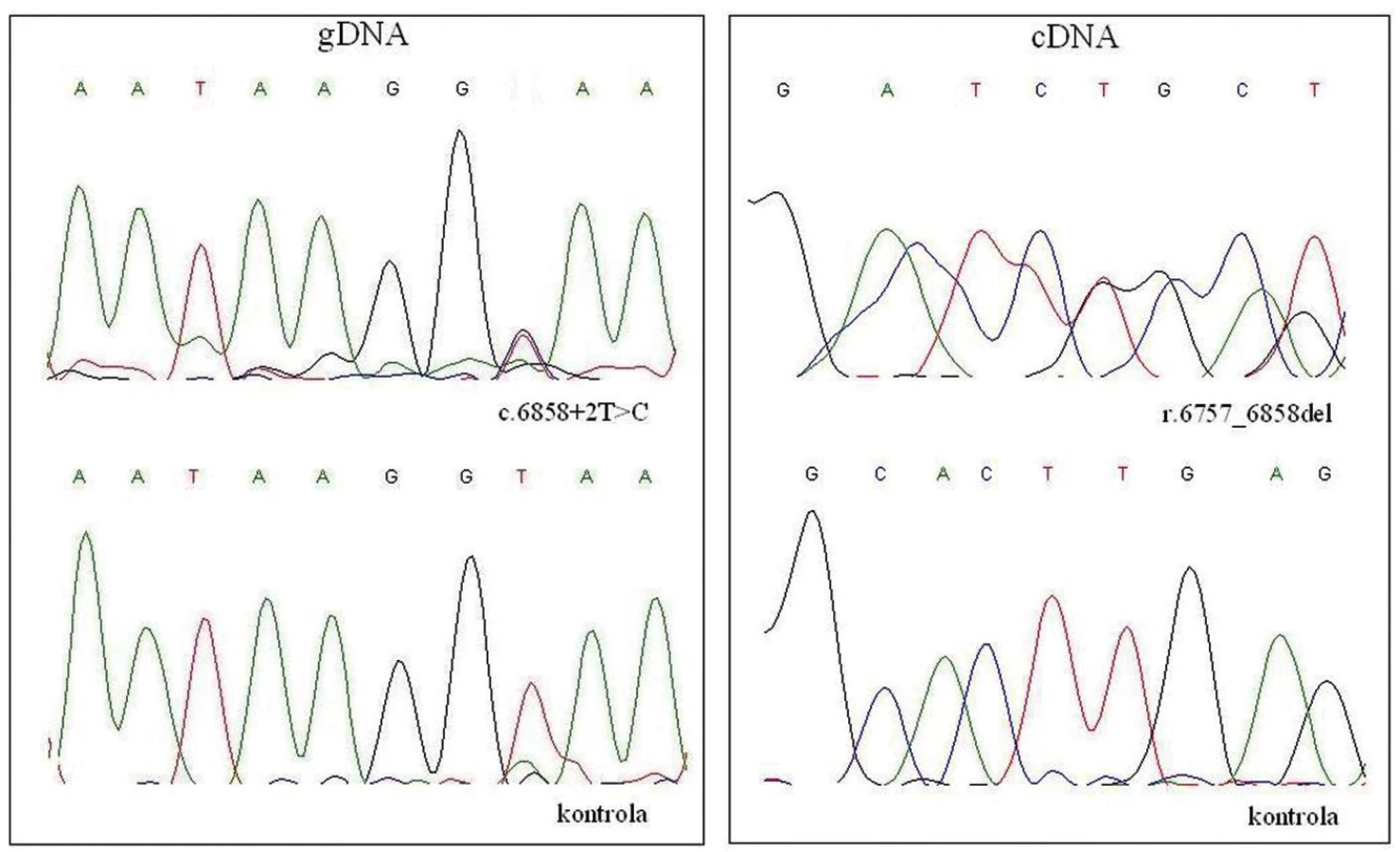
Stanovení diagnózy NF1
Význam DNA analýzy NF1 v diagnostice je zatím limitován. Proto je, navzdory možnostem molekulární biologie, diagnóza NF1 stále zásadně závislá na hodnocení klinického obrazu a diagnostických kriterií. Současná diagnostická kriteria byla stanovena v roce 1988 a platí dosud beze změny (30).
Kritéria National Institute of Health Consensus Development Conference (1988) – pro stanovení diagnózy NF1 je nutná přítomnost 2 nebo více z následujících příznaků:
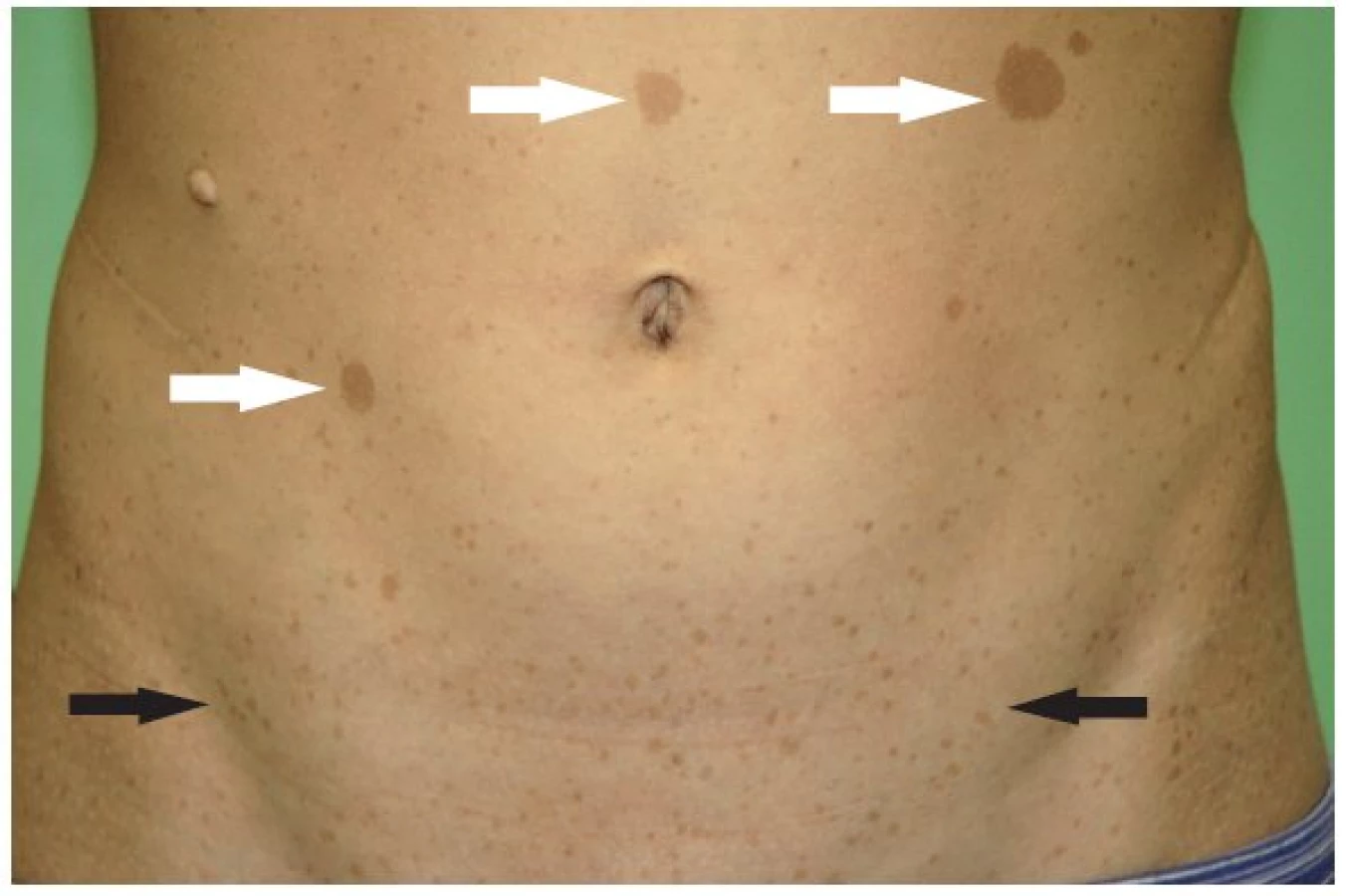
- skvrny barvy bílé kávy na kůži (skvrny café au lait) v počtu 6 a více - do puberty o průměru rovném nebo větším než 5 mm; po pubertě o průměru rovném nebo větším než 15 mm (obr. 2)
- mnohočetný axilární a/nebo inquinální freckling (obr. 2)
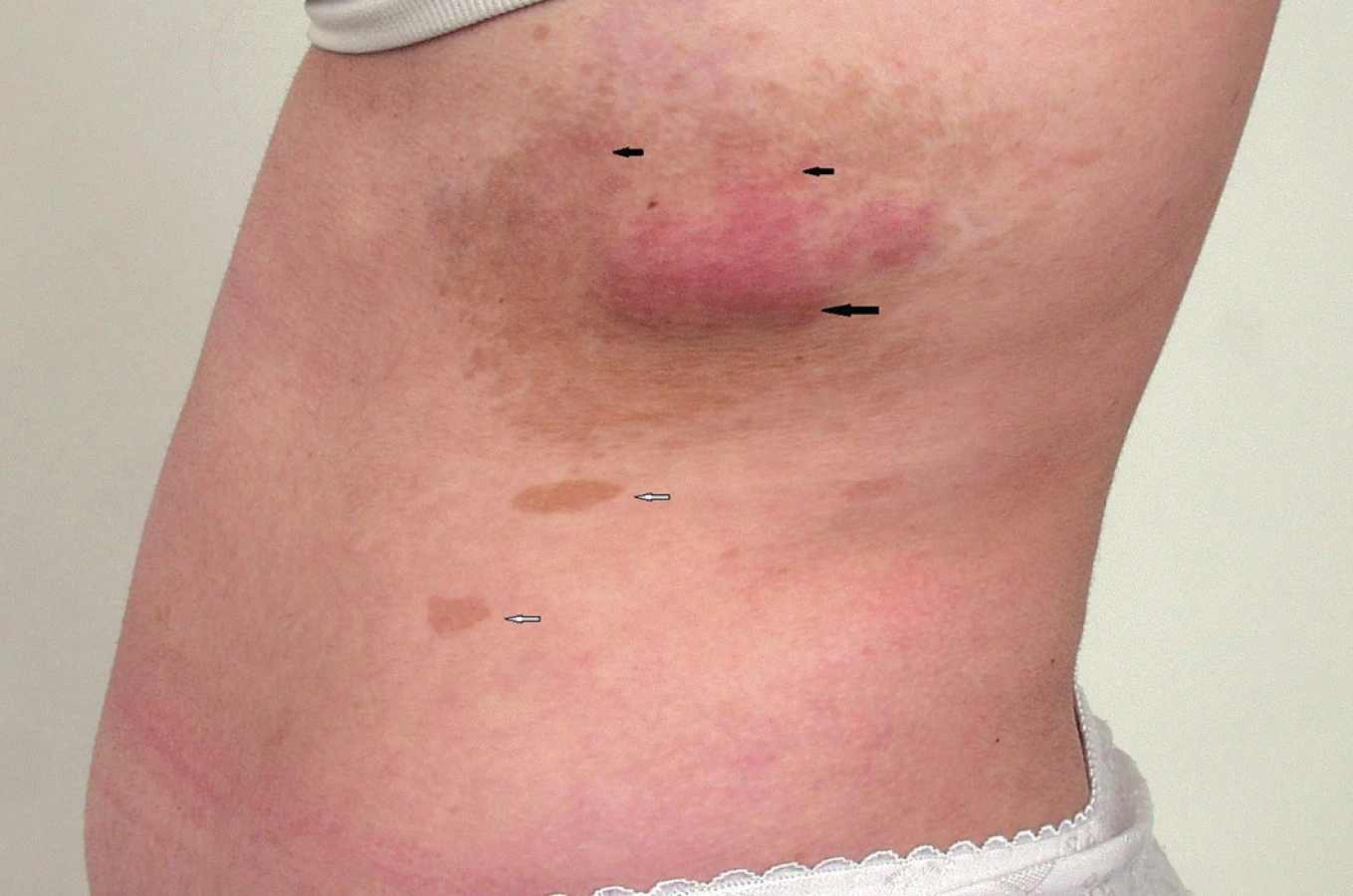
- dva a více neurofibromů nebo jeden plexiformní neurofibrom (obr. 3)
- gliom optického nervu (obr. 4)
- typické kostní léze (dysplázie křídla sfenoidální kosti nebo ztenčení kortikální části dlouhých kostí s nebo bez pseudoartrózy)
- 2 nebo více Lischovy noduly (hamartomy duhovky) oboustranně
- příbuzný 1. stupně (rodič, sourozenec nebo potomek) s prokázanou diagnózou NF1 dle uvedených kriterií (30,31).


Z diferenciálně diagnostických důvodů je nutno zdůraznit, že u NF1 pracujeme s diagnostickými příznaky, které se často vyskytují v celé populaci (např. skvrny barvy bílé kávy) (2). Proto je třeba dodržet diagnostická kriteria. Nesprávně stanovená diagnóza může způsobit významné problémy pacientovi i dalším členům jeho rodiny. Variabilita klinického obrazu bývá výrazná i v rámci jedné rodiny a většina diagnostických i klinických příznaků se manifestuje postupně - v závislosti na věku pacienta (4,32). Klinické projevy NF1 jsou velmi široké, významně přesahují hranice diagnostických kriterií, mají charakter nádorový i nenádorový a vyžadují péči specialistů z různých medicínských oborů.
Skvrny barvy bílé kávy. Nejčastější kožní lézí jsou skvrny barvy bílé kávy (skvrny café au lait, CAL). Nalézají se prakticky u všech pacientů s NF1. Jsou to vícečetné, světle hnědé, hladké, ostře ohraničené okrsky kůže s pravidelnou pigmentací, nejčastěji lokalizované na trupu (ale také na končetinách, na obličeji). Vznikají nahromaděním pigmentovaných melanoblastů (z neurální lišty) na basální vrstvě epidermis a následnou poruchou vývoje melanocytů. Mohou se objevit již u novorozenců a jejich počet přibývá do období puberty. Není žádný vztah mezi počtem skvrn a závažností NF1 (2,31,32) (obr. 2).
Freckling. Častý výskyt má také tzv. freckling, který představuje různý počet drobných okrouhlých makul se zvýrazněnou pigmentací podobného charakteru jako u CAL. Freckling se nachází v podpaží a v tříslech a objevuje se mezi 3. až 5. rokem věku (vzácně dříve) (2,31) (obr. 2).
Lischovy noduly. Mnohočetné hamartomy (noduly) na duhovce lze nalézt pouze u malého procenta dětí ve věku do 6 let, ale téměř u všech (95 %) jedinců s NF1 starších 21 let věku. Oftalmolog je detekuje při vyšetření předního očního segmentu pomocí štěrbinové lampy. Zpočátku jsou tyto melanocytární hamartomy zbarvené světle, ale s věkem tmavnou. Lischovy noduly nezpůsobují poruchu zraku, jsou ale velmi užitečným diagnostickým kriteriem NF1 (31,32).
Anomálie skeletu. Pouze některé anomálie skeletu patří mezi diagnostická kriteria NF1. Mezi diagnostická kriteria patří kongenitální dysplazie dlouhých kostí - nejčastěji jsou nalézány v tibiích, ale mohou být postiženy také ostatní dlouhé kosti. Dysplastické změny kostí sice bývají založeny kongenitálně, ale manifestace v novorozeneckém nebo kojeneckém věku je pouze u závažných pseudoartróz, ostatní změny se projeví později nebo bývají často klinicky němé.
Mezi diagnostická kriteria NF1 nepatří (přes velmi častý výskyt) skolióza. Skolióza může rychle progredovat s nutností rehabilitační terapie, terapie korsetem nebo stabilizačního operačního výkonu (4,32). S věkem pacienta incidence skoliózy u NF1 vzrůstá a často se kombinuje s dysplazií obratlů a/nebo s kostními změnami od přilehlých plexiformních neurofibromů, případně s nálezem imprese zadní hraně obratlového těla při durální ektasii. Nadměrný růst některých kostí je přibližně u 10 % pacientů s NF1 (32).
Neurofibrom a plexiformní neurofibrom. Neurofibrom se může objevit kdekoliv v průběhu periferního nervu a v počtu větším než 2 představují jedno z diagnostických kriterií NF1. Neurofibromy se manifestují v době před začátkem puberty či během ní. Nejtypičtější jsou pendulující a podkožní neurofibrom. U dětí je výskyt neurofibromu velmi variabilní. Neurofibromy jsou ale nalézány také u osob bez diagnózy NF1 (2,31,32) (obr. 2).
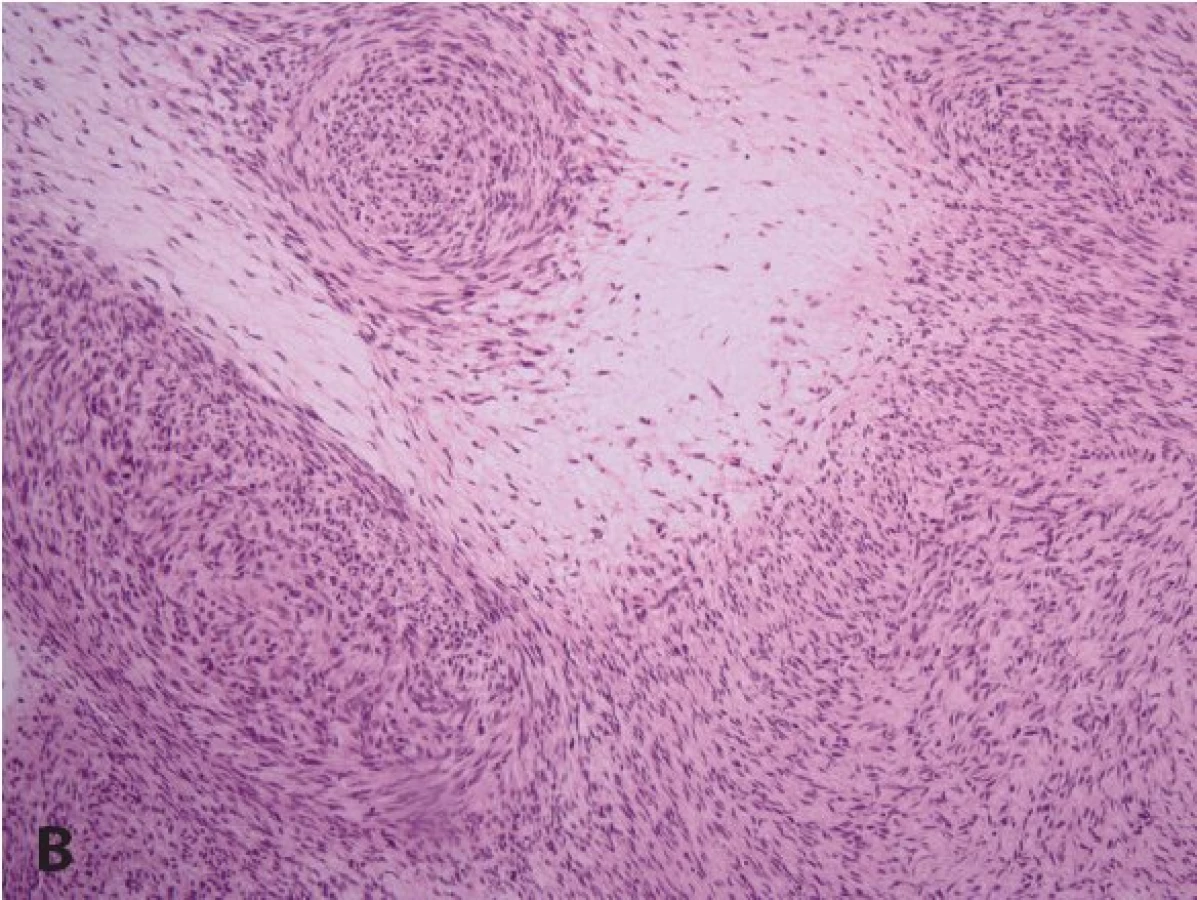
Častý je plexiformní neurofibrom (obr. 5A). Může se objevit na všech částech těla, kde jsou periferní nervy. Plexiformní neurofibromy mají někdy na povrchu hyperpigmentace a/nebo s hypertrichózu a mohou být i velmi rozsáhlé a často se chovají infiltrativně (obr. 3). Celoživotní riziko malignizace plexiformního neurofibromu s rozvojem maligního nádoru pochvy periferního nervu (malignant peripheral nerve sheath tumor, MPNST) (obr. 5B) je v současné době hodnoceno na 8 - 13 % (33). Jeho příznakem může být přetrvávající bolest v oblasti plexiformního neurofibromu trvající déle než měsíc (především klidová bolest a bolest narušující spánek), změna konsistence plexiformního neurofibromu z měkkého na tvrdý a jeho rychlý růst a zvětšení (2,33). Pětileté přežití od stanovení diagnózy MPNST bylo 21 % u pacientů se základní dg. NF1 a u 42 % sporadických případů MPNST bez diagnózy NF1 (33). MPNST je nejčastější příčinou úmrtí dospělých s diagnózou NF1 (33).

Gliomy optické dráhy a gliomy lokalizované jinde v centrálním nervovém systému. Z tumorů centrálního nervového systému (CNS) je pro NF1 charakteristický gliom, nejčastěji pilocytární astrocytom, grade I (obr. 4). U diagnózy NF1 je především riziko rozvoje gliomů zrakové dráhy, mozkového kmene, ale také jiných oblastí mozku. Zraková dráha bývá nejčastěji a zároveň nejdříve postiženou oblastí – s maximem manifestace změn v místech obou optiků a optického chiasmatu. Gliomy zrakové dráhy se vyskytují u 15 až 27 % jedinců s NF1 (34) a manifestují se převážně v období do 6 až 10 let věku (2,31,32). Jsou častější u děvčat s poměrem výskytu u žen a mužů 2 : 1 a oboustranné gliomy optiků lze prakticky nalézt pouze u diagnózy NF1 (34). Až polovina gliomů optiku je asymptomatická, dlouhodobě stacionární a bez růstové dynamiky (2,4,31). Jen malá část gliomů lokalizovaných intraorbitálně se zvětšuje. Naproti tomu nádory postihující optické chiasma progredují častěji (2,32). U druhé poloviny dětí s gliomy se nádory mohou zvětšovat – s tlakem na okolí a okolní struktury a s rozvojem klinických příznaků (zhoršená kvalita zraku, porucha zorného pole, snížení kvality barevného vidění, proptóza, strabismus, edém papily, atrofie optiku a nablednutí optického disku). Při gliomu chiasmatu s tlakem a/nebo propagací do hypothalamu a zásahem do hypothalamo-hypofyzárně-gonadální osy se často rozvíjí předčasná puberta. Diencefalický syndrom je méně častý, vykytuje se podstatně častěji u nádorů hypothalamu bez diagnózy NF1 (2,31,32). Gliomy zrakové dráhy se u NF1 obvykle nechovají agresivně. Proto je vždy třeba pečlivě zvažovat rizika a prospěch každého plánovaného terapeutického postupu a výkonu. Dokud je zrak alespoň částečně funkční, je třeba pokusit se uchránit ho před dalším poškozením. Proto je v současné době při volbě terapeutického postupu na prvním místě chemoterapie - protokol SIOP pro nádory nízkého gradu – carboplatina a vinkristin (4).
Gliomy mozkového kmene. Také výskyt gliomů mozkového kmene je u NF1 častější než v ostatní populaci. Gliomy mozkového kmene u pacientů s NF1 (převážně low grade astrocytomy) většinou způsobují neurologické příznaky (bolesti hlavy, hydrocefalus a postižení mozkových nervů) a jen malá část je klinicky němá. Gliomy mozkového kmene mají (na rozdíl od difúzních gliomů mozkového kmene bez NF1) méně závažný klinický průběh (32).
Intraspinální nádory. Manifestují se pomaleji než nádory nitrolební a v dětském věku se často jedná o náhodný nález při MR zobrazení. Přibližně u jedné poloviny nádorů se jedná o mnohočetný výskyt a náhodně je doprovází další změny - například syringomyelie (31,32). Familiární spinální neurofibromatóza, kterou v dospělosti charakterizují mnohočetné nádory míchy, je variantou NF1 (2,5).
Genetika – příbuzný prvního stupně
Významnou pomocí pro časné stanovení diagnózy NF1 u dětí do 3 let věku je rodinný výskyt NF1 u příbuzného 1. stupně (2,4,31,32). Oba rodiče by měli být pro potřeby genetika vyšetřeni dermatologem (z hlediska výskytu skvrn café au lait a freckling, podkožních neurofibromů) a oftalmologem (kvalita zraku, oční pozadí a přední segment oční na Lischovy noduly)(4).
Diferenciální diagnóza NF1
Některé diagnózy jsou svými příznaky podobné NF1 a je třeba diferenciálně diagnosticky zvážit Legius syndrom (AD dědičnost, mutace SPRED1 genu, chromozóm 15), který je charakterizován nálezem skvrn café au lait a freckling na kůži (jako u NF1), ale nejsou přítomny nádorové procesy (35). Některými autory je uváděn syndrom autozomálně dominantního výskytu skvrn café au lait a syndromy jako je Watson syndrom (charakterizovaný AD dědičnou pulmonální stenózou, skvrnami café au lait na kůži, mentální retardací) a Neurofibromatosis-Noonan syndrom (NFNS) (36), které jsou s NF1 alelické.
Další klinické projevy mimo diagnostická kriteria
- Častý je malý vzrůst (kolem 30% jedinců s NF1), na kterém se podílí řada faktorů jako jsou nízké hodnoty růstového hormonu, suprasellární léze nebo deformity skeletu (32). Makrocefalie je běžným nálezem - u 16 - 45 % dětí je obvod hlavy na 98. nebo vyšším percentilu (2,5,32).
- Hydrocefalus je významnou neurologickou komplikací u dětí s NF1. Většinou je následkem expansivního procesu mozku, vzácnější je jeho vznik následkem stenózy distální části mokovodu (37).
- Bolest hlavy je častá a je popisována u 20 - 25 % dětí s NF1. Obvykle se rozvíjí bez přítomnosti strukturální léze. Vzhledem k riziku nitrolební expanze nebo hypertenze je zde třeba provést MR zobrazení mozku (2,4,32).
- Systémová arteriální hypertenze je nejčastěji renovaskulární při stenóze (stenózách) renální arterie. Je to nejčastější cévní anomálie u NF1 na podkladě fibromuskulární dysplazie (38). Cévní změny mohou mít i jinou lokalizaci. Velmi vzácně je popisovaný moya-moya syndrom s následným rizikem ischemické cévní mozkové poruchy. Vznik moya-moya syndromu je velmi často vázán na předchozí radioterapii nádoru mozku. Arteriální hypertenze může také být následek feochromocytomu, který je popisován u 1 - 4 % dospělých jedinců s NF1 (32). U pacientů s NF1 se také popisují vrozené vady srdce a/nebo velkých cév, kde nejčastější je stenóza plicnice, ale vzácně lze nalézt také stenózy různých částí aorty (38).
- Hypersignální ložiska v T2 vážených obrazech na MRI zobrazení mozku/míchy (nazývaná FASI = Foci of Abnormal Signal Intensity nebo UBO = Unidentified Bright Object) má až 85 % dětí s NF1 (39). Ložiska se nacházejí typicky v oblasti basálních ganglií, globus pallidus, mozečku, v thalamu, mozkovém kmeni a subkortikálně především v temporální oblasti. Nesytí se po podání kontrastní látky (jen výjimečně v oblasti basálních ganglií), jsou isointenzní v T1 vážených obrazech a na CT vyšetření, není přítomen mass-effect (nezpůsobují tlak na okolní struktury) (39). Nejspíše se jedná o ložiska aberantní myelinizace s vakuolární změnou myelinu (40). Z hlediska ložiskového neurologického nálezu jsou FASI asymptomatická, ale jsou dávána do souvislosti s kognitivními poruchami u NF1. V dospělosti mají tendenci se zmenšovat nebo mizet - u jedinců starších 30 let jsou pak spíše vzácné. Od low-grade gliomů se odlišují absencí mass-effektu a sycení po podání kontrastní látky při MR vyšetření mozku. Tyto léze se nesmí zaměnit za nádory. FASI/UBO nebyla v současné době zařazena mezi diagnostická kriteria NF1 (32,39,41).
- Vzácná leukémie dětského věku - juvenilní myelomonocytární leukemie (JMML) se rozvíjí u pacientů s nálezem somatické bodové mutace v RAS genu (resp. mutací v genech NRAS a KRAS2), genu PTPN11 pro Noonan syndrom a v NF1 genu. Mutace v NF1 genu je příčinou JMML ve 14 % případů (42).
- U dospělých pacientů s NF1 je při gastrointestinálních obtížích třeba diferenciálně diagnosticky zvažovat otázku gastrointestinálního stromálního tumoru (GIST), při hypertenzi a vegetativních obtížích je třeba vyloučit možnost feochromocytomu (2,32).
ZÁVĚR
Třebaže NF1 patří svou incidencí mezi vzácná onemocnění, jedná se zároveň o diagnózu, se kterou se můžeme v průběhu delšího času setkat. Je proto třeba znát charakteristické rysy tohoto onemocnění a zároveň počítat s výraznou variabilitou klinického obrazu, který má rozsah od kosmetických změn až po velmi těžký průběh nemoci. Je třeba pochopit obecné rysy průběhu této diagnózy, aby pak bylo možné kvalitně zhodnotit závažnost NF1 individuálně u každého nemocného a dle toho vybrat odpovídající postup při terapii a dlouhodobém sledování – tak aby tento postup svou důrazností pacienta nepoškodil a naopak svou „šetrností“ rizika nemoci nepodcenil.
PODĚKOVÁNÍ
Práce byla podpořena projektem koncepčního rozvoje výzkumné organizace 00064203.
Adresa pro korespondenci:
MUDr. Bořivoj Petrák, CSc.
Klinika dětské neurologie UK 2.LF a FN Motol
V Úvalu 84, 150 06 Praha 5
tel.: 224433301, fax: 224433322
e-mail: borivoj.petrak@post.cz
Sources
1. Von Recklinghausen FD. Ueber die multiplen Fibrome der Haut und ihre Beziehnung zu den multiplen Neuronomen. Berlin, Hirschwald; 1882.
2. Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007; 44 : 81-88.
3. Evans DGR. Neurofibromatosis type 2. In: Roach ES, MillerVS, editors. Neurocutaneous Disorders. Cambridge University Press,U.K; 2004 : 50-59.
4. Petrák B, Plevová P, Novotný J, Foretová L. Neurofibromatosis von Recklinghausen. Klin Onkol 2009; 22(Suppl): 38–44.
5. Riccardi VM. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. 2nd ed. Baltimore: The Johns Hopkins University Press, 1992.
6. Tinschert S, Naumann I, Stegmann E, Buske A, Kaufmann D, Thiel G, Jeanne DE. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet 2000; 8(6): 455-459.
7. Ilenčíková D, Čižmárová M, Krajčiová A, Požgayová S, Rybárová A, Kovács L. Klinické dysmorfické syndromy s tumorigenézou. Klin Onkol 2012; 25(Suppl): 39–48.
8. Huson SM. The neurofibromatoses: classification, clinical featrures and genetic counselling. Kaufmann(ed): Neurofibromatoses, Monogr Hum Genet, Basel, Karger. Volume 16; 2008 : 21-31.
9. Šnajderová M, Riccardi VM, Petrák B, et. al. The importance of advanced parental age in the origin of neurofibromatosis type1. Am J Med Genet Part A 2012; 158A: 519-523.
10. Upadhyaya M. NF1 Gene Structure and NF1 Genotype/Phenotype Correlations. Kaufmann(ed): Neurofibromatoses, Monogr Hum Genet, Basel, Karger. Vol. 16; 2008 : 46-62.
11. Trovo-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet 2006; 70(1): 1-13.
12. Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene 2007; 26(32): 4609-4616.
13. McClatchey AI, Cichowski K. Mouse models of neurofibromatosis. Biochim Biophys Acta 2001; 1471(2): 73-80.
14. Viskochil D, White R, Cawthon R. The neurofibromatosis type 1 gene. Annu Rev Neurosci 1993; 16 : 183-205.
15. Zhu Y, Romero MI, Ghosh P, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 2001; 15(7): 859-876.
16. Welti S. Structure and Function of Neurofibromin. Kaufmann(ed): Neurofibromatoses, Monogr Hum Genet, Basel, Karger. Volume 16; 2008 : 113-128.
17. Lau N, Feldkamp MM, Roncari L, et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol 2000; 59(9): 759-767.
18. Serra E, Rosenbaum T, Nadal M, et al. Mitotic recombination effects homozygosity for NF1 germline mutations in neurofibromas. Nat Genet 2001; 28(3): 294-296.
19. Cooper DN, Ball EV, Stenson PD, Phillips AD, Shaw K, Mort ME. 2012: www.hgmd.org.
20. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet 1996; 33(1): 2-17.
21. Fahsold R, Hoffmeyer S, Mischung C, et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 2000; 66(3): 790-818.
22. Ainsworth P, Rodenhiser D, Stuart A, Jung J. Characterization of an intron 31 splice junction mutation in the neurofibromatosis type 1 (NF1) gene. Hum Mol Genet 1994; 3(7): 1179-1181.
23. Stark M, Assum G, Krone W. A small deletion and an adjacent base exchange in a potential stem-loop region of the neurofibromatosis 1 gene. Hum Genet 1991; 87(6): 685-687.
24. Shen MH, Harper PS, Upadhyaya M. Neurofibromatosis type 1 (NF1): the search for mutations by PCR-heteroduplex analysis on Hydrolink gels. Hum Mol Genet 1993; 2(11): 1861-1864.
25. Harder A, Titze S, Herbst L, et al. Monozygotic twins with neurofibromatosis type 1 (NF1) display differences in methylation of NF1 gene promoter elements, 5‘ untranslated region, exon and intron 1. Twin Res Hum Genet 2010; 13(6): 582-594.
26. Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet 2007; 80(1): 140-151.
27. De Raedt T, Brems H, Lopez-Correa C, Vermeesch JR, Marynen P, Legius E. Genomic organization and evolution of the NF1 microdeletion region. Genomics 2004; 84(2): 346-360.
28. Lazaro C, Gaona A, Estivill X. Two CA/GT repeat polymorphisms in intron 27 of the human neurofibromatosis (NF1) gene. Hum Genet 1994; 93(3): 351-352.
29. Wimmer K, Roca X, Beiglbock H, et al. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5‘ splice-site disruption. Hum Mutat 2007; 28(6): 599-612.
30. Natiomal Institute of Health Consensus Development Conference. Neurofibromatosis: Conference Statement. Arch Neurol Chicago 1988; 45 : 575-578.
31. Goldstein J, Gutmann D. Neurofibromatosis type 1. In: Roach ES, MillerVS, ed. Neurocutaneous Disorders. Cambridge University Press, U.K.; 2004, pp. 42-49.
32. Maria BL, Menkes JH. Neurocutaneous Syndromes. In: Menkes JH, Sarnat HB, Maria BL eds. Child Neurology. 7th ed. Lippincott Williams Wilkins, Philadelphia; 2005, pp. 803-828.
33. Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002; 39 : 311-314.
34. Leisti EL. Radiologic findings of the head and spine in neurofibromatosis 1 (NF1) in Northern Finland. Academic Dissertation. Oulu, Finland: University of Oulu and Oulu Univestity Hospital 2003.
35. Brems, H., Pasmant, E., Van Minkelen, R., Wimmer, K., Upadhyaya, M., Legius, E., Messiaen, L. Review and update of SPRED1 mutations causing Legius syndrome. Hum Mutat 2012; 33 : 1538-1546.
36. De Luca A, Bottillo I, Sarkozy A, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet 2005; 77 : 1092-1101.
37. Kalužová M, Petrák B., Lisý J., Vaculík M., Bendová Š., Komárek V. Idiopatická stenóza akveduktu a porucha vývoje řeči u dětí s neurofibromatosis von Recklinghausen typ 1 – dvě kazuistiky. Cesk Slov Neurol N 2012; 75/108(5): 633-636.
38. Petrák B, Bendová S, Seeman T, Klein T, Lisý J, Zatrapa T, Maříková T. Mid-aortic syndrome with renovascular hypertension and multisystem involvement in a girl with familiar neurofibromatosis von Recklinghausen type 1. Neuroendocrinol Lett 2007; 28(6): 734-738.
39. Petrák B, Lisý J, Kalužová M, Kraus J. Význam hypersignálních ložisek v T2 vážených obrazech na MRI vyšetření mozku pro stanovení diagnosy neurofibromatosis von Recklinghausen typ 1. Cesk-slov pediat 2010; 65(5): 326-327.
40. DiPaolo DP, Zimmerman RA, Rorke LB, Zackai EH, Bilaniuk LT, Yachnis AT. Neurofibromatosis type 1: pathologic substrate of high‑signal - intensity foci in the brain. Radiology 1995; 195(3): 721–724.
41. Ferraz-Filho JR, José da Rocha A, Muniz MP, Souza AS, Goloni-Bertollo EM, Pavarino-Bertelli EC. Unidentified bright objects in neurofibromatosis type 1: conventional MRI in the follow-up and correlation of microstructural lesions on diffusion tensor images. Eur J Paediatr Neurol 2012; 16(1): 42-47.
42. Matsuda K, Shimada A, Yoshida N, et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood 2007; 109(12): 5477-5480.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2015 Issue 1
Most read in this issue
- Neurofibromatosis von Recklinghausen typ 1 (NF1) – klinický obraz a molekulárně-genetická diagnostika
- Patologické hodnocení vzorků kolorektálního karcinomu: pokročilé a časné léze
- Fibroadenom prsu s pleomorfními stromálními buňkami
- Malobuněčná varianta světlobuněčného sarkomu měkkých tkání napodobující Ewingův sarkom - kazuistika