
Obrovskobuněčné léze kostí a jejich diferenciální diagnostika
Giant cell-rich lesions of bone and their differential diagnosis
Giant cell-rich lesions form a heterogeneous group of reactive and truly neoplastic processes with diverse clinical presentation and biological behavior. Common to all of them are variably numerous multinucleated osteoclast-like giant cells and the presence of mononuclear stroma. Based on the histological picture alone it is sometimes impossible to reliably distinguish certain tumors from each other. The pathologist has to know the patient´s age, the exact localization, tumor growth dynamics and its radiographic characteristics. Secondary reactive changes occur frequently and these can completely alter the morphology of the lesion and thus overshadow the underlying neoplasm. Reparative changes in a pathological fracture may histologically mimic primary bone malignancy. Immunohistochemistry helps only in select cases and molecular genetic methods still have very limited utility for the diagnosis of giant cell-rich tumors. It is necessary to correlate the microscopic features of the lesion with clinical and radiological findings. A correct diagnosis is of paramount importance for proper treatment and prognosis.
Keywords:
giant cell tumor – non-ossifying fibroma – chondroblastoma – aneurysmal bone cyst – giant cell reparative granuloma – giant cell-rich osteosarcoma
:
Iva Zambo 1; Lukáš Pazourek 2
:
I. patologicko-anatomický ústav, FN u sv. Anny a LF MU, Brno
1; I. ortopedická klinika, FN u sv. Anny a LF MU, Brno
2
:
Čes.-slov. Patol., 53, 2017, No. 2, p. 61-70
:
Reviews Article
Obrovskobuněčné léze kostí tvoří heterogenní skupinu reaktivních i nádorových procesů s odlišným klinickým projevem a biologickým chováním. Společným znakem pro všechny jsou různě početné mnohojaderné buňky typu osteoklastů a přítomnost stromatu s jednojadernými buňkami. Pouze na základě histologického obrazu není občas možné od sebe některé jednotky spolehlivě odlišit. Patolog musí znát věk pacienta, přesnou lokalizaci a dynamiku růstu tumoru a jeho radiografické charakteristiky. Časté jsou sekundární reaktivní změny, které mohou kompletně alterovat morfologii léze a tím zastřít původní tumor. Reparativní změny v patologické fraktuře mohou mikroskopicky napodobit primární kostní malignitu. Imunohistochemické vyšetření pomáhá jen v některých případech a molekulárně genetické metody mají v diagnostice obrovskobuněčných tumorů dosud jen velmi omezené využití. V mnoha případech stanovení správné diagnózy vyžaduje úzkou mezioborovou spolupráci. Určení přesné diagnózy má zásadní význam pro správnou léčbu a prognózu.
Klíčová slova:
obrovskobuněčný tumor kostí – neosifikující fibrom – chondroblastom – aneuryzmatická kostní cysta – obrovskobuněčný reparativní granulom – osteosarkom bohatý na mnohojaderné buňky
Obrovskobuněčné léze kostí zahrnují skupinu morfologicky a biologicky odlišných reaktivních procesů a pravých nádorů primárně postihujících skelet. Společným znakem pro všechny tyto léze je přítomnost četných nenádorových, osteoklastům podobných obrovských mnohojaderných buněk. Pro správné zařazení obrovskobuněčných tumorů je nezbytná korelace klinického a radiologického nálezu s histologickým obrazem, ve kterém zásadní roli hraje zhodnocení morfologie, distribuce a způsobu růstu všech zastižených buněčných typů.
Diagnostika některých lézí je obtížná a do značné míry závislá na kvalitě a reprezentativnosti dodaného materiálu. Histologický obraz pravých obrovskobuněčných neoplázií může být změněn nebo z velké části zastřen sekundární, reaktivní obrovskobuněčnou proliferací, která může imitovat maligní transformaci nebo naopak vést k nerozpoznání preexistujícího tumoru. K velmi závažným omylům může dojít při nadhodnocení reparativních změn způsobených patologickou zlomeninou. Reaktivní osteoplázie či osteochondroplázie, která je obklopená buněčným, mitoticky aktivním tumorem, napodobuje primární kostní malignitu. Zejména u lokálně pokročilých tumorů a/nebo objemných lézí osového skeletu je vždy nutno počítat s možnou přítomností patologických fraktur, přičemž tyto mohou být vcelku nenápadné, někdy charakteru mikroskopických zlomenin.
Nomenklatura obrovskobuněčných lézí je v mnoha případech zavádějící. V závislosti na klinických projevech existují různé názvy pro tentýž morfologický obraz. Tak například proliferace histologicky odpovídající obrovskobuněčnému reparativnímu granulomu může být v netypické lokalizaci označena za solidní aneuryzmatickou kostní cystu (AKC) a u pacientů s hyperparathyroidismem za hnědý tumor. Pojmenování některých jednotek neodpovídá biologické povaze léze. Bylo prokázáno, že část aneuryzmatických kostních cyst a obrovskobuněčných reparativních granulomů je cytogeneticky charakterizována klonální chromozomální přestavbou, a jedná se tedy o pravé tumory, v případě aneuryzmatické kostní cysty navíc se značně destruktivním typem růstu a vysokým potenciálem pro lokální recidivy (1,2).
V následujícím textu budou diskutovány nejčastější primární kostní obrovskobuněčné léze se zaměřením na jejich diagnostické znaky v korelaci s klinickým nálezem a rentgenologickým (RTG) obrazem. Nejdůležitější charakteristiky vybraných tumorů jsou uvedeny v tabulce 1.

OBROVSKOBUNĚČNÝ NÁDOR KOSTÍ
Obrovskobuněčný nádor kostí (OBN) je považován za prototyp neoplázie bohaté na obrovské mnohojaderné buňky typu osteoklastů. Reprezentuje přibližně 4 % všech primárních kostních tumorů a postihuje dospělé pacienty v 3. – 5. dekádě, nejčastěji mezi 20. a 40. rokem života. Typicky vyrůstá v epifýzách dlouhých kostí, zejména v distálním konci femuru, proximálním konci tibie, distálním konci radia a proximálním konci humeru. Vzácně postihuje krátké kosti nohy a ruky a osový skelet, kde se nejčastěji vyskytuje v kosti křížové. V souvislosti s Pagetovou chorobou kostí může být OBN zastižen v žebrech, plochých kostech či kraniofaciálním skeletu, primárně se však v těchto lokalizacích prakticky nevyskytuje (1,2). Dle recentní WHO klasifikace mezenchymálních tumorů je OBN řazen mezi lokálně agresivní, vzácně metastazující tumory (2). Terminologie vzdáleného rozsevu OBN není ustálená. Někteří autoři preferují označení sekundárních plicních ložisek za implantační noduly, protože tato mívají tendenci chovat se indolentně. U části případů však může dojít k miliárnímu rozsevu či progresi plicních ložisek a dokonce i úmrtí pacienta. Díky pokrokům v molekulární biologii byl objasněn vztah mezi nenádorovými mnohojadernými buňkami typu osteoklastů a jednojadernými neoplastickými buňkami, které v hojné míře exprimují ligand pro aktivační receptor nukleárního faktoru kB (tzv. RANKL). Ten tvoří vazebné místo pro RANK lokalizovaný v membráně mnohojaderných buněk. Interakce RANK-RANKL je zodpovědná za indukci osteoklastogeneze z mononukleárních prekurzorů osteoklastů, a tím také agresivní osteolýzu postižené kosti (3).
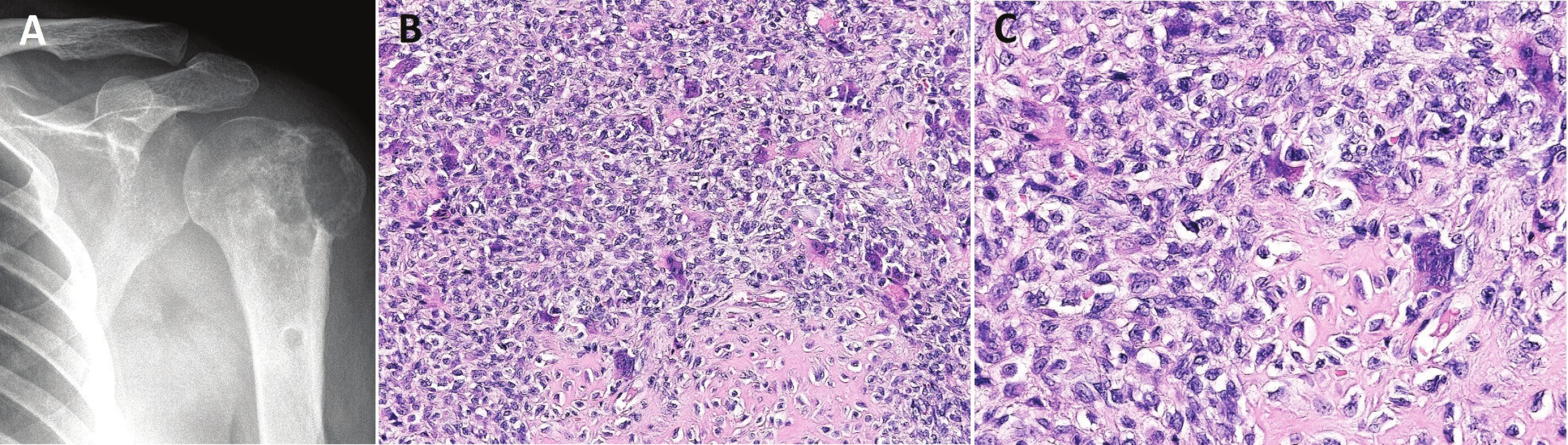
Radiograficky OBN imponuje jako excentricky lokalizované, subchondrální, osteolytické ložisko v epifýze, v době diagnózy však často zasahuje do metafýzy. U pokročilých nálezů je přítomna extraoseální propagace (RTG stádium 3 dle Campanacciho), která ovšem v OBN není známkou maligního zvratu. Zobrazovací vyšetření neumožňují bezpečně rozpoznat vzácně se vyskytující malignitu v OBN. Varovnou známkou je však nezvykle rychlá lokální progrese nálezu. V konvenčním OBN sklerotický lem většinou chybí a není přítomna periostální reakce (obr. 1A). Radiologický obraz může být modifikován druhotnými změnami, např. reaktivní fibrohistiocytární proliferací či sekundární aneuryzmatickou kostní cystou (1).
Histologicky nalézáme uniformní populaci oválných jednojaderných, tzv. stromálních buněk, a četné, rovnoměrně roztroušené obrovské vícejaderné elementy typu osteoklastů, které upoutávají pozornost mnohdy excesivním počtem jader (obr. 1B). Důležitým diagnostickým znakem je identický vzhled jader stromálních a multinukleárních buněk, jádra jsou oválná, se světlým, vezikulárním chromatinem a centrálním nápadným nukleolem (obr. 1C). Někdy mohou být jádra pyknotická. Cytoplazma mnohojaderných buněk je sytě eozinofilní. Mitotická aktivita stromálních elementů bývá nápadná, jednojaderné buňky navíc mohou v různě rozsáhlých oblastech vykazovat poměrně výrazné cytonukleární atypie, které však souvisí s předchozím krvácením nebo jsou vázány na okraje nekrózy či depozita fibrinu. V rámci druhotných změn lze zachytit okrsky se storiformně rostoucími a křížícími se krátkými snopci vřetenitých buněk vzhledu fibroblastů s příměsí méně početných, osteoklastům podobných elementů. Tyto oblasti připomínají neosifikující fibrom a spolu s depozity hemosiderinu, agregáty pěnitých makrofágů a cystickou transformací patří do obrazu reaktivních, sekundárních změn, které mohou být zdrojem diagnostických omylů. V konvenčním OBN chybí pleomorfní buňky, atypické mitózy a produkce chrupavčité či kostní matrix vlastními nádorovými buňkami. Pozor však na interpretaci přítomnosti nezralé pletivové kosti, která je často zastižena na periferii tumoru. Jedná se o reaktivní osifikaci, která bývá zachycena také v okraji rekurentních tumorů a v implantovaných ložiscích OBN v plicích. Na základě histologického obrazu nelze predikovat míru agresivního chování OBN, proto není v současné době grading OBN doporučován.

Konvenční OBN může progredovat do high-grade malignity, která vzniká sekundárně po opakovaných recidivách tumoru nebo za 5 i více let po předchozím ozáření. Primární maligní OBN je extrémně vzácný. Histologicky mohou oblasti s maligní transformací připomínat fibrosarkom, osteosarkom či nediferencovaný sarkom (obr. 2). Klíčem pro správnou diagnózu je zachycení okrsků konvenčního OBN a úseků s atypickými mitózami, jaderným polymorfismem, případně s nádorovým osteoidem. Primární maligní OBN nabývá vzhled nediferencovaného pleomorfního sarkomu s četnými atypickými obrovskými buňkami typu osteoklastů, které také vykazují nápadnou anizonukleózu i mitotickou aktivitu.

Imunohistochemicky lze ve většině konvenčních OBN prokázat nukleární expresi p63, která je vázána na jednojaderné stromální buňky (4-6). V kombinaci s negativitou S100 proteinu a znalostí klinického a RTG obrazu lze tento marker považovat za užitečný v diagnostice OBN. Nutno však mít na paměti, že exprese p63 byla prokázána také v nezanedbatelném procentu chondroblastomů, hnědých tumorů a neosifikujících fibromů (5,6). Obrovské mnohojaderné buňky podle očekávání exprimují histiocytární markery, např. CD68 či CD163. Cytogeneticky lze u přibližně 90 % OBN prokázat mutaci genu H3F3A (7).
Mezi nejdůležitější diferenciálně diagnostické jednotky patří obrovskobuněčný reparativní granulom či hnědý tumor, neosifikující fibrom, chondroblastom, solidní úseky AKC, osteosarkom bohatý na obrovské buňky a z okolních měkkých tkání do kosti prorůstající tenosynoviální obrovskobuněčný tumor.
Obrovskobuněčný reparativní granulom typicky postihuje čelistní kosti, kde OBN primárně vyrůstá vzácně a většinou pouze v terénu Pagetovy choroby (1). Histologicky je distribuce mnohojaderných buněk u reparativního granulomu nepravidelná, navíc bývá přítomna reaktivní novotvorba kosti uvnitř léze a kolagenizace extracelulární matrix. Sekundární změny v OBN ale mohou výše jmenované histologické znaky věrně napodobit. V takových případech je nezbytná klinicko-radiologická korelace. I tak ale nelze v některých případech (zejména u lézí lokalizovaných v krátkých kostech ruky či nohy) obě jednotky bezpečně odlišit. Údaje o hyperkalcémii, hypofosfatémií a zvýšených hodnotách parathormonu jsou důležité pro diagnostiku hnědého tumoru. Věk pacienta, RTG znaky a metafyzární lokalizace pomohou odlišit neosifikující fibrom, ve kterém navíc bývá nápadnější fibroblastické stroma s nepravidelně rozmístěnými mnohojadernými buňkami, mnohdy kompresivně deformovanými. Chondroblastom je charakterizován alespoň fokálně přítomnou chrupavčitou matrix, tzv. drátěnými kalcifikacemi a dužnatými oválnými či krátce vřetenitými buňkami s patrným podélným zářezem v jaderné membráně. Pro odlišení primární AKC od sekundárních změn v konvenčním OBN je důležitý zejména RTG obraz léze. Diagnóza osteosarkomu bohatého na obrovské mnohojaderné buňky je vymezena produkcí nádorového osteoidu neoplastickými buňkami a okrsky histologicky méně nápadnými, napodobujícími konvenční OBN. Problematika tenosynoviálního obrovskobuněčného tumoru prorůstajícího do obratlového těla C6 byla podrobně zdokumentována v kazuistice publikované ve čtvrtém čísle časopisu Česko-slovenská patologie z roku 2016 (8).
Základem léčby je chirurgické odstranění tumoru. V případě intraoseální lokalizace bez významnější destrukce kortikální kosti a propagace do měkkých tkání je preferován intralezionální výkon, tedy kyretáž. S ohledem na vyšší riziko lokální recidivy je výhodné kyretáž doplnit lokální kryoterapií či fenolizací. Vzniklý defekt je vyplněn kostními štěpy či kostním cementem. Mezi výhody cementové výplně patří její termický efekt coby lokálního adjuvans, a také usnadňuje detekci případné lokální recidivy, která se v terénu přestavby kostních štěpů obtížně odhaluje. Lokální recidiva je po kyretáži poměrně častá. Vyskytuje se v 19–50 % případů (9-12), ale na druhou stranu tento výkon výrazně méně zatěžuje pacienta rizikem dalších komplikací spojených s radikální resekcí a implantací vhodné náhrady. U lokálně pokročilých nádorů s destrukcí kortikální kosti a prorůstáním do měkkých tkání (stádium 3 dle RTG) je indikována široká en block resekce a náhrada endoprotézou či masivním kostním homoštěpem. Chirurgická léčba je doporučována také u resekabilních plicních metastáz/implantačních nodulů. Vzhledem k riziku maligní transformace, která bývá asociována s předchozím ozářením, není již radioterapie v základním léčebném algoritmu obecně doporučována. Mezi nové a velmi slibné terapeutické možnosti patří cílená léčba lidskou monoklonální protilátkou, denosumabem, který blokuje interakci RANK-RANKL, a tím potlačuje vyzrávání osteoklastů (13-15). V nádorové tkáni dochází k signifikantnímu úbytku mnohojaderných buněk typu osteoklastů a částečné redukci stromálních buněk (obr. 3B), čímž je umožněna novotvorba kosti (16). Dochází ke sklerotizací tumoru, jeho lepšímu ohraničení, mnohdy také zmenšení obejmu nádoru, čímž je umožněn méně radikální chirurgický výkon (obr. 3A). Terapie denosumabem je doporučována u inoperabilních OBN a u pacientů s neresekabilními plicními metastázami. Mezi nežádoucí účinky dlouhodobé aplikace RANKL inhibitoru patří těžká hypokalcémie, nekróza čelistní kosti a atypické únavové zlomeniny. Dosud však nebyl přesněji specifikován vztah mezi počtem dávek denosumabu, délkou léčby, věkem pacienta, lokalizací a velikostí OBN a rizikem výskytu výše popsaných komplikací (17). Obecně je akceptována dlouhodobá aplikace, ohledně maximální délky léčby však nepanuje shoda. Vzhledem k vysoké ekonomické náročnosti této cílené terapie podléhá v České republice její úhrada schválení revizním lékařem pojišťovny.

NEOSIFIKUJÍCÍ FIBROM / METAFYZÁRNÍ KOSTNÍ DEFEKT
Neosifikující fibrom, metafyzární fibrózní defekt a fibrózní kortikální defekt představují histologicky totožnou fibrohistiocytární proliferaci, která typicky vyrůstá v metafýze dlouhých kostí u mladých jedinců s neuzavřenou růstovou ploténkou. Nomenklatura odráží přesnější lokalizaci léze: při postižení kortexu lze preferovat označení fibrózní kortikální defekt, zatímco více centrálně uložené ložisko s přesahem do dřeňové dutiny je častěji označováno za neosifikující fibrom či metafyzární kostní defekt. Pravděpodobně se ve většině případů jedná o nenádorový proces, který zřejmě souvisí s porušenou osifikací a má tendenci ke spontánní rezoluci (1,2). Vzhledem k asymptomatickému průběhu u většiny pacientů lze incidenci těchto benigních tumorózních lézí jen těžko odhadnout. Dle údajů radiologů je neosifikující fibrom detekován u 30-40 % skeletálně nezralých pacientů, přibližně dvakrát častěji u chlapců, a to nejčastěji v 2. dekádě, typicky v distálním femuru, v proximální či distální tibii, méně často v dlouhých kostech horní končetiny (18). Pokud se neosifikující fibrom klinicky projeví, pak bolestí v postiženém místě, vzácněji může být příčinou patologické fraktury. Raritně lze zachytit vícečetná symetrická ložiska neosifikujícího fibromu, která mohou být asociovaná s některými klinickými syndromy. Výskyt neosifikujících fibromů tak například patří mezi méně známé projevy neurofibromatózy I. typu. Při nálezu mnohočetných metafyzárních kostních defektů v nezvyklých lokalizacích (v kostech kraniofaciálního skeletu, obratlových tělech atp.) je nutno pomýšlet na možnou souvislost s Jaffeho-Campanacciho syndromem, který na sebe ovšem upoutává pozornost svými nápadnějšími klinickými projevy, jako jsou mentální retardace, pigmentace kůže typu bílé kávy, kryptorchismus, malformace kardiovaskulárního systému a další (19).
Zobrazovací metody odhalí excentricky lokalizované osteolytické ložisko vázané na kortex, který bývá ztenčen, ale neporušen. Lem tumoru je sklerotický (obr. 4A). V souvislosti s růstem skeletu dochází postupně k posunu léze směrem k diafýze, v případě spontánní regrese se ložisko skleroticky mění. Radiologický obraz bývá natolik příznačný, že biopsie ve většině případů není nutná.
Mikroskopicky je léze v typických oblastech tvořená buněčnou proliferací storiformně uspořádaných krátce vřetenitých stromálních buněk vzhledu fibroblastů. Mnohojaderné buňky jsou nepočetné, disperzně, a spíše nepravidelně roztroušené (obr. 4B, C). Většinou lze zastihnout okrsky xantomatózní reakce, drobné fokusy hemoragií či hemosiderinové pigmentace. Pseudocystická transformace typu sekundární AKC je možná a bývá příčinou rychlého zvětšení léze. Nekrózy ani příležitostné mitózy nejsou v neosifikujících fibromech známkou malignity. Atypické mitózy však chybí.

Imunohistochemicky jsou stromální buňky difúzně CD68 pozitivní, patrně tedy mají původ blíže histiocytům než fibroblastům. V části neosifikujících fibromů byly prokázány různé chromozomální aberace, nejčastěji translokace t(1;4)(p31;q34). Tyto však nejsou smysluplně využitelné v rutinní diagnostice (1).
Diagnóza neosifikujícího fibromu by v korelaci s klinickým a RTG nálezem neměla činit potíže. Nemá-li však patolog dostupné adekvátní informace (týkající se zejména přesné lokalizace léze), pak může být metafyzární kostní defekt zaměněn za OBN. Vřetenobuněčnou proliferaci lze považovat za reaktivní změny v konvenčním OBN. Na rozdíl od neosifikujícího fibromu však OBN typicky roste v epifýze a postihuje dospělé pacienty s uzavřenou růstovou ploténkou. V případě kortikálních mikroskopických fraktur a následné reaktivní osteoplázii může mikroskopický obraz připomínat fibrózní dysplázii. Ve fibrózní dysplázii ale nejsou zřetelné lemy osteoblastů kolem trabekul pletivové kosti a většinou chybí obrovské mnohojaderné buňky. Také excentrická lokalizace osteolýzy podporuje diagnózu neosifikujícího fibromu. Benigní fibrózní histiocytom má totožnou histologickou stavbu jako metafyzární kostní defekt, od kterého se liší „nezvyklou“ lokalizací v plochých kostech, žebrech či obratlových tělech.
Při náhodném nálezu nekomplikovaného ložiska neosifikujícího fibromu není terapie nutná a je doporučováno pouze sledování pacienta. Kyretáž s následným vyplněním defektu kostními štěpy je upřednostňována u objemnějších lézí, zejména v případech s hrozící nebo již přítomnou patologickou frakturou.
CHONDROBLASTOM
Chondroblastom je charakterizovaný proliferací nezralých chondrocytů a alespoň fokální produkcí chrupavčité matrix. Ve většině případů se chová benigně, ojediněle však roste lokálně agresivně a zcela raritně může metastazovat do plic. Proto je v současné WHO klasifikaci řazen k tumorům s nejistým biologickým chováním (2). V porovnání s konvenčním OBN se jedná o vzácný nádor, který představuje necelé 1 % všech kostních neoplázií. Většinou je diagnostikován ve 2. – 3. dekádě, přibližně dvakrát častěji u mužů (1,2). Chondroblastom je téměř vždy lokalizován v epifýze či apofýze, typicky v oblasti kolene a proximálních konců pažní a stehenní kosti. Vzácností není ani postižení pánve, lopatky, páteře, žeber, čéšky a zánártních kostí, zejména kosti patní. Vyskytovat se však může i v kostech kraniofaciálního skeletu (20). Hlavním klinickým příznakem bývá bolest lokalizovaná do oblasti kloubu, která může být spojena s poruchou jeho funkce a vzácněji, při perforaci kloubní chrupavky, s reaktivní synovialitdou a nitrokloubním výpotkem.
Radiograficky se chondroblastom projevuje jako excentricky uložené, oválné osteolytické ložisko, dobře ohraničené tenkým lemem sklerotické kosti (obr 5A). Roste v epifýze nebo apofýze s možným přesahem do metafýzy. V centru mohou být patrné jemné bodové kalcifikace. Na rozdíl od OBN je typická tendence k prorůstání až do těsného sousedství kloubní plochy s možným zborcením kloubu a následnou těžkou deformující artrózou. V diagnostice chondroblastomu se s výhodou uplatňuje magnetická rezonance, která navíc pomůže identifikovat sekundární AKC, která komplikuje až 20 % případů chondroblastomu, nejčastěji při lokalizaci tumoru v tarzálních kostech (1).
V mikroskopickém obraze většinou dominují buněčné oblasti tvořené nahloučenými chondroblasty a různě početnými mnohojadernými buňkami typu osteoklastů. V rozvolněnějších úsecích je patrná produkce hyalinní chrupavčité matrix. Chondroblasty jsou polygonální a mají okrouhlá či oválná jádra, mnohdy s podélným zářezem (obr 5B, C). Atypické mitózy chybí. Mezi diagnosticky cenný histologický znak patří nález tzv. drátěných kalcifikací. Jde o lineárně mineralizovanou extracelulární matrix, která v podobě sítě obkružuje jednotlivé chondroblasty a je nejlépe patrná ve vzorcích, které nebyly odvápňovány. Výrazné kalcifikace jsou vázány na oblasti s vyzrálejší chrupavčitou matrix. Do obrazu sekundárních změn v chondroblastomu patří okrsky koagulační nekrózy či cystoidní degenerace, která může napodobit AKC či prostou cystu. Často se vyskytující vřetenobuněčná transformace výrazně znesnadňuje stanovení správné diagnózy, zejména v případech, ve kterých v lézi převládá a maskuje tak charakteristické znaky chondroblastomu. Část těchto vřetenitých buněk představují protáhlé chondroblasty, některé odpovídají reaktivním fibroblastům (1).
Chondroblasty exprimují S100 protein, SOX9 a vimentin. Exprese p63 kolísá případ od případu (4-6). V rámci diagnostického algoritmu je tedy nutno vzít možnou jadernou pozitivitu p63 v úvahu. Nekonstantně může být v chondroblastech prokázána exprese cytokeratinu, EMA a hladkosvalového aktinu (21). Cytogenetické analýzy prokázaly heterogenní aberace nejčastěji zahrnující chromozomy 5 a 8 (1). Absence specifické chromozomální přestavby však neumožňuje využití těchto poznatků v diagnostice.
V malých vzorcích může být v případě rozsáhlých sekundárních změn histologické odlišení chondroblastomu od konvenčního OBN nemožné. Diagnostické rozpaky většinou pomůže vyřešit vyšetření exprese p63 a S100 proteinu. Zatímco v OBN jsou stromální buňky typicky p63 pozitivní a S100 negativní, chondroblasty by měly zřetelně exprimovat S100 protein při variabilní expresi p63. Chondromyxoidní fibrom se od chondroblastomu liší postižením metafýz, absencí kalcifikací a přítomností úseků s myxoidními lobuly. Při výskytu AKC v lokalizaci typické pro chondroblastomy a mladém věku pacienta je nutno zaslaný materiál extenzivně (mnohdy kompletně) zpracovat a velmi pečlivě vyšetřit, včetně imunohistochemické exprese S100 proteinu. Sekundární AKC totiž dokáže téměř kompletně zastřít původní tumor, takže na zobrazovacích vyšetřeních tento imponuje jako prokrvácená multilokulární cysta. Mikroskopické odlišení solitární kostní cysty od chondroblastomu, ve kterém sekundárně vznikla cysta vyplněná serózní tekutinou, lze bez větších obtíží pomocí klinicko-radiologické korelace. Solitární kostní cysta bývá lokalizována v metafáze či diafýze dlouhých kostí. Podobnost světlobuněčného chondrosarkomu s chondroblastomem je dána podobnými lokalizacemi obou tumorů i obdobným radiologickým obrazem. Chondrosarkom však lze rozpoznat histologicky na základě identifikace maligních chondrocytů a charakteristických objemných buněk se světlou cytoplazmou.Chondroblastom bývá většinou léčen kyretáží. Lokální recidiva se vyskytuje přibližně v 10 % případů, u kterých je mírně zvýšeno riziko zakládání implantačních uzlíků do plic (1).
ANEURYZMATICKÁ KOSTNÍ CYSTA (AKC)
AKC je benigní, avšak destruktivní a expanzivní multilokulární cysta, resp. pseudocysta, vyplněná krví. Jedná se o poměrně vzácnou lézi, která představuje necelá 3 % kostních tumorů (1). V naprosté většině vyrůstá u mladých pacientů, nejčastěji ve druhé dekádě. AKC může postihnout kteroukoli kost a dokonce se primárně vyskytovat i v měkkých tkáních (2). Absence charakteristické lokalizace je mezi ostatními kostními neopláziemi nezvyklá a snadno se může stát zdrojem diagnostických pochybností, zejména při odlišení primární a sekundární AKC. O něco častěji se AKC vyskytuje v metafýzách dlouhých kostí končetin, v zadních segmentech obratlů, kraniofaciálním skeletu a krátkých kostech horních i dolních končetin. Klinicky se AKC projevuje nápadně. Mezi typické příznaky patří bolest a mnohdy rychlé zduření postižené oblasti. Při lokalizaci v obratlích může kompresí nervů způsobit neurologické symptomy. AKC byla dlouhou dobu považována za reaktivní proces a část případů skutečně vzniká v důsledku intraoseální hemoragie, na kterou může okolní tkáň zareagovat buď tvorbou cysty nebo vznikem obrovskobuněčného reparativního granulomu. Z tohoto předpokladu vychází pozoruhodný termín solidní AKC, což je označení pro lézi vzhledu obrovskobuněčného reparativního granulomu v netypické lokalizaci. Cytogenetická analýza prokázala, že alespoň část primárních AKC je charakterizována chromozomálními přestavbami, nejčastěji translokací t(16;17)(q22;p13), v důsledku které vzniká chimérický gen CDH11-USP6 (2). Některé AKC tedy patří mezi pravé nádory.
Radiograficky se většinou jedná o osteolytickou expanzivní lézi s dobře ohraničenými okraji, periferně často s úzkým sklerotickým lemem a někdy patrnou přítomností sept. RTG obraz připomíná nafouklý balónek, který v dlouhých kostech expanduje jejich konturu (obr. 6A), jindy se obraz připodobňuje k mýdlovým bublinám. Výpočetní tomografie a magnetická rezonance pomohou dobře ozřejmit vnitřní strukturu léze a přítomnost hladinek (tzv. fluid-fluid levels). AKC může kontinuálně postihnout i více kostí spojených kloubem, což bývá nejlépe patrné v oblasti páteře, kde mnohdy dochází k prorůstání do několika sousedních obratlů a jednostrannému, asymetrickému postižení paravertebrálních tkání. Případné solidní oblasti jsou suspektní z sekundární AKC a měly by být biopticky verifikovány.
Histologický obraz AKC závisí na velikosti odebraného materiálu. Fragmenty získané kyretáží většinou zachytí kolabovaná septa a okrsky hemoragií. V resekátech a někdy i probatorních excizních je dobře patrná struktura krví vyplněné multilokulární pseudocysty. Široká septa svojí stavbou připomínají mladou granulační tkáň. Jsou tvořena edematózní fibrózní tkání s bohatou kapilární sítí, extravazací erytrocytů, zánětlivým infiltrátem a různě početnými obrovskými buňkami typu osteoklastů, které mají tendenci shromažďovat se v úsecích s výraznějším krácením. Luminální povrch sept postrádá výstelku, jen místy mohou být patrny oploštělé buňky připomínající endotelie. Reaktivní osteoplázie většinou bývá zřetelná alespoň fokálně, svým rozsahem však může vystupovat do popředí histologického obrazu (obr. 6). Nezralý osteoid lemovaný dužnatými osteoblasty může připomínat nádorový osteoid, nicméně v AKC tvoří pruhovitá depozita paralelní s dlouhou osou sept a někdy je částečně mineralizovaný. V takovém případě získává osteoid, případně navazující pletivová kost, namodralou barvu (tinkčními vlastnostmi připomíná chrupavčitou matrix) a patří do diagnostických znaků AKC pod označením „modrá kost“ (1,2,21). Dle zkušeností z našeho pracoviště však je tento mikroskopický znak zastižen poměrně zřídka a pouze ve velmi drobných fokusech, takže může snadno uniknout pozornosti. Přítomnost „modré kosti“ patrně souvisí s délkou dekalcifikace. V dlouhé ose sept kromě osteoidu probíhají také novotvořené kapiláry. Na periferii léze jsou patrné přestavbové změny kosti s vystupňovanou osteoklastickou resorpcí. Výčet preexistujících tumorů, které mohou tvořit podklad pro sekundární AKC, je poměrně široký. Kromě v tomto článku diskutovaných lézí mezi ně patří také osteoblastom, fibrózní dysplázie, benigní fibrózní histiocytom, chondromyxoidní fibrom, eozinofilní granulom, myositis ossificans, high-grade konvenční osteosarkom, nediferencovaný pleomorfní sarkom kostí či metastatický karcinom.

K největším diferenciálně diagnostickým úskalím pro AKC patří odlišení teleangiektatického osteosarkomu, který se s AKC překrývá svým klinickým a radiografickým obrazem, pročež veškerá tíha konečného rozhodnutí spočívá na hodnotícím patologovi. Pouze nález zřetelně pleomorfních nádorových osteoblastů produkujících krajkový osteoid a/nebo atypických mitóz opravňuje k diagnóze malignity. V malých vzorcích však tyto diagnostické partie mohou chybět. Teleangiektatický osteosarkom se velmi vzácně vyskytuje v páteři, kraniofaciálním skeletu a malých kostech ruky a nohy, kde je naopak AKC poměrně běžná. Tuto informaci však lze vnímat pouze jako pomocnou. Rozhodujícím je histologický obraz. V průběhu vyšetřovacího procesu je vždy nutno pátrat po případné existenci původní léze, na jejímž podkladě AKC vznikla, a tím vyloučit sekundární AKC. Biologickou povahou nebezpečným (prokrváceným a cysticky změněným) tumorem je konvenční osteosarkom, u kterého je však patologicko-radiologická korelace výrazně přínosnější než v předchozím případě. I tady pro správnou diagnózu platí nezbytná přítomnost maligních osteoblastů tvořících nádorový osteoid. Klinický a RTG nález pomáhají odlišit AKC od sekundárně změněného OBN či chondroblastomu. Lokalizace osteolytického ložiska v epifýze u dospělého pacienta upřednostňuje diagnózu OBN, zatímco podobná léze u skeletálně nezralého jedince svědčí spíše pro chondroblastom. Obrovskobuněčný reparativní granulom se v mnohých znacích s AKC překrývá a je dokonce synonymem pro solidní variantu AKC. Dokud nebude vyjasněna nomenklatura těchto jednotek, zůstává nejdůležitějším vodítkem pro správné pojmenování histologicky identických lézí radiografický obraz. Obrovskobuněčný reparativní granulom je převážně solidní, chybí zřetelné multilokulární projasnění. Reparativní změny patologické fraktury v terénu solitární kostní cysty mohou velmi napodobit AKC, včetně fokusů reaktivní osteoplázie a přítomnosti osteoklastů. Korelace s RTG obrazem je v takových případech nezbytná (1).
Způsob léčby AKC odvisí od lokalizace léze, její velikosti a míry agresivního chování. U nekomplikovaného lokálního nálezu se nejčastěji provádí kyretáž s následným vyplněním defektu kostním štěpy či kostním cementem. Riziko lokální recidivy lze snížit aplikací fenolu, tekutého dusíku či fotokoagulací argonovým laserem během operačního výkonu (1). U agresivní AKC je indikována resekce s adekvátní náhradou. Jako doplňkovou léčebnou metodu lze použít arteriální embolizaci, s opatrností v indikovaných případech také radioterapii.
OBROVSKOBUNĚČNÝ REPARATIVNÍ GRANULOM
Obrovskobuněčný reparativní granulom je heterogenní jednotkou, která zahrnuje patogeneticky různorodé léze s identickým histologickým obrazem, ale odlišnou klinickou manifestací. Pod toto označení patří čistě reaktivní procesy v čelistních kostech při hyperparathyroidismu (tzv. hnědé tumory), ale i pravé tumory s prokázanou přestavbou USP6 genu, které se překrývají se solidní AKC a typicky bývají lokalizované v distálních oblastech apendikulárního skeletu. Hnědý tumor častěji postihuje dolní čelist dětí a adolescentů (v 1. a 2. dekádě), kde typicky vyrůstá před prvními stálými moláry. Vzácně může postihnout i dlouhé kosti. Obrovskobuněčný reparativní granulom, obvykle lokalizovaný v krátkých kostech ruky a nohy, bývá nejčastěji diagnostikován v 2. – 3. dekádě. Ženy jsou postiženy dvakrát častěji než muži (1). Obrovskobuněčné reparativní granulomy rostou většinou solitárně. Jejich vícečetný výskyt patří do obrazu cherubismu, vzácněji také Noonanova syndromu. První zmíněná jednotka je charakterizována symetrickou, až bizarní remodelací čelistních kostí, nápadněji mandibuly, následovanou proliferací mnohočetných lézí vzhledu obrovskobuněčného reparativního granulomu (2). Pacienti s Noonanovým syndromem trpí kromě výskytu multifokálních obrovskobuněčných reparativních granulomů různě závažnými vývojovými defekty jakými jsou vrozené srdeční vady, krvácivá diatéza, krátká postava s krátkým krkem či hypertelorismus. Vícečetné obrovskobuněčné reparativní granulomy mohou být asociovány také s Pagetovou chorobou kostí, multifokální hnědé tumory bývají poměrně běžné u pacientů s hyperparathyroidismem (1).
Zobrazovací metody odhalí oválné, dobře ohraničené, projasněné ložisko. Kontura kosti může být rozšířená, avšak kortex je neporušen a chybí periostální reakce. Při lokalizaci v čelistech může dojít k dislokaci zubů, tyto však nebývají resorbovány.
Mikroskopická stavba hnědého tumoru a obrovskobuněčného reparativního granulomu je totožná. Název jednotky „obrovsko-buněčný reparativní granulom“ postrádá oporu v patogenezi i mikroskopickém obraze. Struktury pravých granulomů v této lézi nenajdeme a o reparativní proces se většinou jedná při lokalizaci v čelistních kostech, kde jsou ovšem tyto léze mnohdy asociovány se zvýšenou funkcí příštítných tělísek a měly by tedy být označeny za hnědý tumor. Histologicky převažuje vřetenobuněčná proliferace fibroblastů či myofibroblastů, která místy přechází do fibrotizovaných úseků. Obrovské mnohojaderné buňky typu osteoklastů mají tendenci shlukovat se v místech s čerstvým i starším krvácením (obr. 7). Mnohdy bývá extracelulární matrix prostoupena málo početnými lymfocyty a často je přítomna reaktivní osteoplázie. V oblastech s výrazným krvácením mnohdy dochází k cystické transformaci. Tyto úseky připomínají AKC. Sporadické mitózy mohou být zastiženy, ne však atypické.
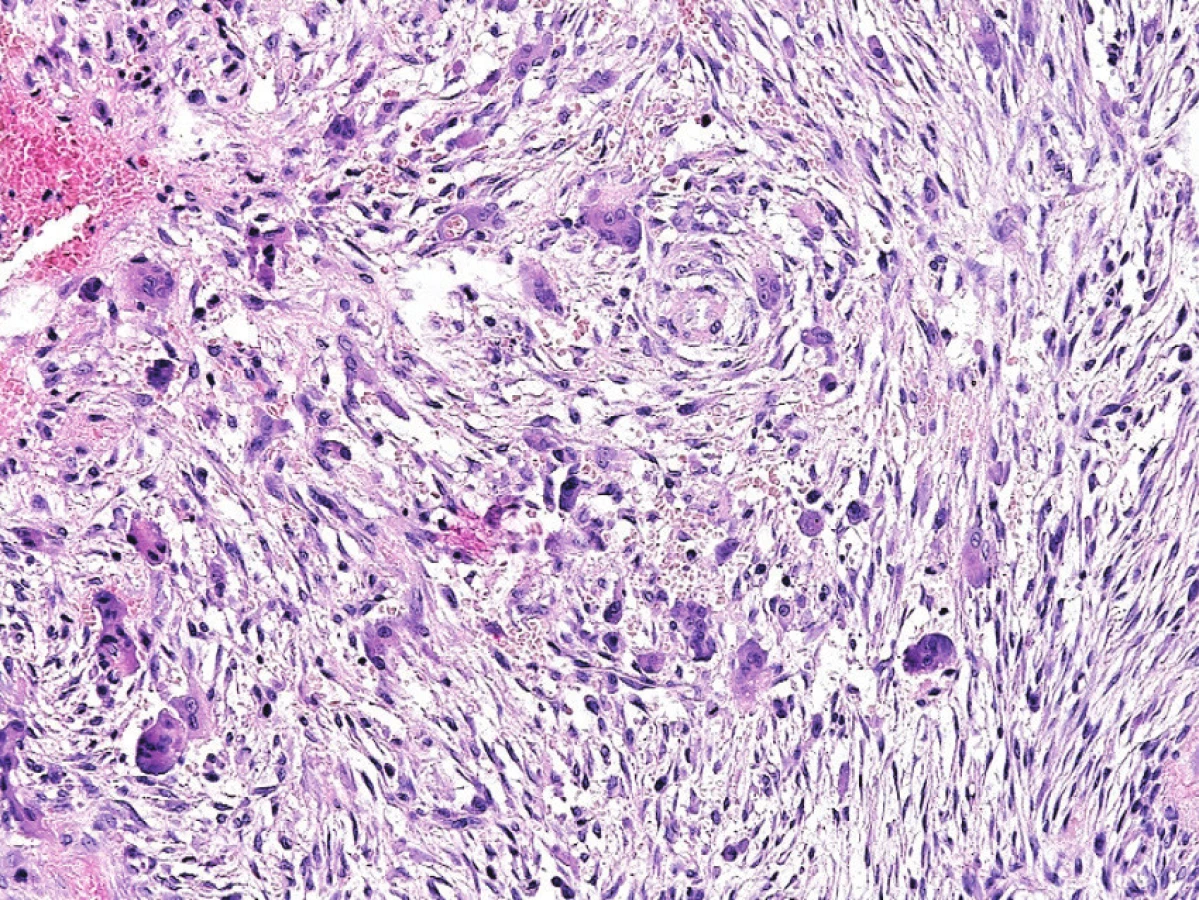
V rámci diferenciálně diagnostické rozvahy je vždy nutno pomýšlet na možnou souvislost léze typu obrovskobuněčného reparativního granulomu s hyperparathyroidismem, a to zejména při lokalizaci v čelistních kostech či multifokálním výskytu. K záměně za konvenční OBN by mohlo dojít zejména při lokalizaci v krátkých kostech ruky a nohy anebo v souvislosti s Pagetovou chorobou kostí. V těchto situacích může pomoci klinicko-radiologická korelace, i přesto však někdy nelze jednoznačnou diagnózu stanovit.
Solitární neosifikující fibrom se od obrovskobuněčného reparativního granulomu liší lokalizací i detaily radiografického obrazu. Obtížným se ale stává jeho odlišení u pacientů s Jaffeho-Campanacciho syndromem, při kterém se neosifikující fibromy mohou vyskytovat také v čelistních kostech (1).
Terapeuticky je při postižení čelistních kostí metodou volby kyretáž, při primárním hyperparathyroidismu je základem odstranění příštítných tělísek. Obrovskobuněčné reparativní granulomy maxily mohou vykazovat dobrou odpověď na steroidy, kalcitonin a interferon alfa-2b. Při lokalizaci v krátkých kostech apendikulárního skeletu a rozsáhlém nálezu je někdy nezbytné amputovat prst či celý paprsek.
OSTEOSARKOM BOHATÝ NA OBROVSKÉ MNOHOJADERNÉ BUŇKY
Osteosarkom je nejčastější primární nehematoonkologickou malignitou kostí (2). Tvoří heterogenní skupinu zhoubných tumorů s různým biologickým potenciálem a variabilní mikroskopickou stavbou. Osteosarkom bohatý na obrovské mnohojaderné buňky je velmi vzácným subtypem konvenčního (intramedulárního) high-grade osteosarkomu, představuje méně než 3 % všech osteosarkomů (1,21,22). Křivka incidence konvenčního osteosarkomu má bimodální charakter s nejvyšším vrcholem mezi 15. a 20. rokem života a druhým, nižším ve 4. až 5. dekádě (2). Častěji se vyskytuje u mužů a typicky postihuje metafýzy dlouhých kostí, zejména v blízkosti kolenního kloubu (1,2,21).
Radiograficky se manifestuje sklerotickou, osteolytickou či kombinovanou destruktivní lézí s neostrými okraji, většinou porušeným kortexem a periostální reakcí spojenou s různě zřetelnou infiltrací do okolních měkkých tkání (obr. 8).
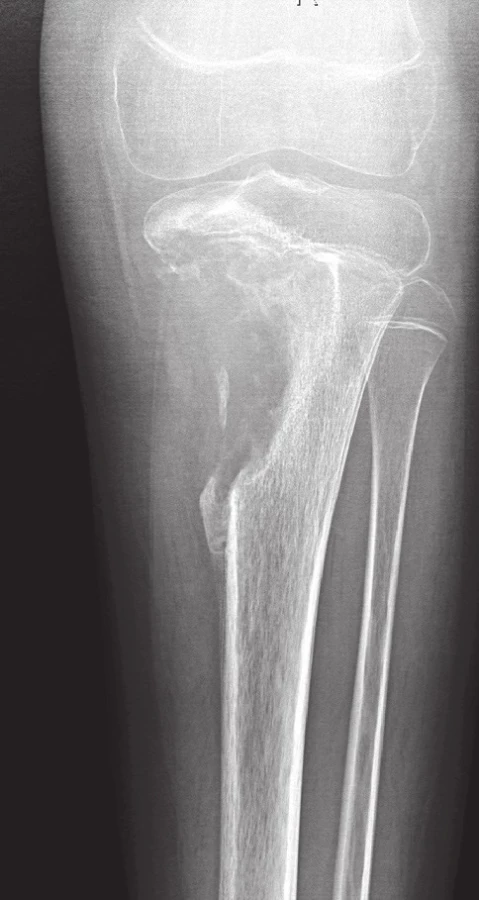
Pro stanovení diagnózy osteosarkomu bohatého na obrovské mnohojaderné buňky musí být v nádorové tkáni v nadpoloviční většině zastoupeny benigně vyhlížející vícejaderné buňky typu osteoklastů (1,22). Oblasti typické pro konvenční osteosarkom bývají méně nápadné, takže hrozí záměna za OBN, což sebou přináší závažné následky pro pacienta. Je tedy důrazně doporučováno, aby byly všechny obrovskobuněčné léze u adolescentů, které zasahují metafýzu dlouhých kostí, důkladně histologicky zpracovány a vyšetřeny. Pečlivá korelace s klinickým obrazem a nálezem na zobrazovacích vyšetřeních je samozřejmě nezbytná (22). V archívu bioptických případů I. patologicko-anatomického ústavu FN u sv. Anny není tato varianta konvenčního osteosarkomu zastoupena.
Standardní léčebný protokol zahrnuje neoadjuvantní chemoterapii, s následnou resekcí, přičemž je preferován končetinu šetřící výkon a náhrada endoprotézou. Následuje histologické zhodnocení efektu předoperační chemoterapie. Za dobrou léčebnou odpověď je považována nekróza nádorové tkáně v 90 a více procentech objemu tumoru. Na efektu iniciální léčby je závislý výběr chemoterapeutik v pooperační fázi. Zavedením této agresivní multimodální léčby je dosaženo dlouhodobého přežití až v 70 % pacientů s lokalizovaným high-grade konvenčním osteosarkomem (23).
ZÁVĚR
Kostní léze s bohatým zastoupením obrovských buněk typu osteoklastů zahrnují heterogenní skupinu reaktivních i nádorových procesů. Většina obrovskobuněčných lézí se vyznačuje benigním biologickým chováním. Typickým zástupcem tumorů bohatých na buňky podobné osteoklastům je OBN. Stanovení správné diagnózy je někdy obtížné a v mnoha případech nemožné bez pomoci korelace nálezu klinického, radiologického a histologického. Reparativní změny v patologické fraktuře zásadním způsobem ovlivňují histologický obraz. Bez upozornění klinika na zlomeninu v oblasti vyšetřovaného tumoru může snadno dojít k záměně benigní léze za primární kostní malignitu (za osteosarkom či chondrosarkom) se závažnými důsledky pro pacienta. V případě nálezu AKC je vždy nutno pomýšlet na možnou preexistující lézi, na jejímž podkladě došlo k druhotným hemoragickým změnám s následnou cystickou přestavbou tumoru. Zejména u mladých pacientů či při klinicky netypickém obrazu je důležité bioptický materiál extenzivně zpracovat. Při pochybnostech o správném zařazení obrovskobuněčného tumoru lze v diagnostickém algoritmu vyšetřit imunohistochemickou expresi p63 a S-100 proteinu. Molekulárně genetické metody mají pro rutinní diagnostiku ve většině případů jen okrajový význam. V současnosti lze využít detekce přestavby USP6 genu u části lézí typu AKC či obrovskobuněčného reparativního granulomu, u OBN lze prokázat mutaci genu H3F3A. Validní diagnostika obrovskobuněčných kostních lézí není možná bez úzké mezioborové spolupráce. Stanovení správné diagnózy je nezbytné pro volbu adekvátní terapie a má tak zásadní vliv na morbiditu i mortalitu pacienta.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
MUDr. Iva Zambo, Ph.D.
I. PAÚ FN u sv. Anny v Brně
Pekařská 53,
656 91 Brno
Tel.: +420 543 183 220
email: iva.zambo@fnusa.cz
Sources
1. Czerniak B. Dorfman and Czerniak´s bone tumors (2nd edn). Philadelphia: Elsevier; 2016 : 246-1085.
2. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. WHO classification of tumours of soft tissue and bone (4th edn). Lyon: IARC; 2013 : 262-374.
3. Liao TS, Yurgelun MB, Chang SS et al. Recruitment of osteoklast precursors by stromal cell derived factor 1 (SDF-1) in giant cell tumor of bone. J Orthop Res 2005; 23(1): 203-209.
4. Shooshtarizadeh T, Rahimi M, Movahedinia S. P63 expression as a biomarker discrimiating giant cell tumor of bone from other giant cell-rich bone lesions. Patlogy – Research and Practise 2016; 212(10): 876-879.
5. Hammas N, Laila C, Youssef ALM et al. Can p63 serve as a biomarker for giant cell tumor of bone? A Morroccan experience. Diagnostic Pathology 2012; 7 : 130.
6. Lee CH, Espinosa I, Jensen KC et al. Gene expression profilig identificates p63 as a diagnostic marker for giant cell tumor of bone. Mod Pathol 2008; 21(5): 531-539.
7. Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45(12): 1479-1482.
8. Kinkor Z, Svoboda T, Grossman P, Bludovský D, Heidenreich F, Švec A, Mečiarová I. Difúzní obrovskobuněčný tumor šlachových pochev krční páteře s destrukcí obratle C6 – kazuistika. Cesk Patol 2016; 52(4): 218-221.
9. Bakle M, Schremper L, Gebert C et al. Giant cell tumor of bone: treatment and outcome of 214 cases. J Cancer Res Clin Oncol 2008; 134(9): 969-978.
10. Gaston CL, Bhumbra R, Watanuki M et al. Does the addition of cement improve the rate of local recurrence after curettage of giant cell tumours in bone? J Bone Joint Surg Br 2011; 93(12): 1665–1669.
11. Kivioja AH, Blomqvist C, Hietaniemi K et al. Cement is recommended in intralesional surgery of giant cell tumors: a scandinavian sarcoma group study of 294 patients followed for a median time of 5 years. Acta Orthop 2008; 79(1): 86–93.
12. Klenke FM, Wenger DE, Inwards CY et al. Giant cell tumor of bone: risk factors for recurrence. Clin Orthop Relat Res 2011; 489(2): 591–599.
13. López-Pousa A, Martín Broto J, Garrido T and Vázquez J. Giant cell tumour of bone: New treatments in development. Clin Transl Oncol 215; 17(6): 419-430.
14. Chawla S, Hnshaw R, Seeger L et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol 2013; 14(9): 901-908.
15. Thomas D, Henshaw R, Skubitz K et al. Denosumab in patients with giant-cell tumour of bone: An open-label, phase 2 study. Lancet Oncol 2010; 11(3): 275-280.
16. Branstetter DG, Nelson SD, Manivel JC et al. Denosumab induces tumor reduction and bone formativ in patiens with giant-cell tumor of bone. Clin Cancer Res 2012; 18(16): 4415-4424.
17. Gaston CL, Grimer RJ, Parry M et al. Current status and unanswered questions on the use of Denosumab in giant cell tumor of bone. Clin Sarcoma Res 2016; 6(1): 15.
18. Mallet JF, Rigault P, Padovani JP et al. Non-ossifying fibroma in children: a surgical condition? Chir Pediatr 1980; 21(3): 179-189.
19. Colby RS, Saul RA. Is Jaffe-Campanacci syndrome just a manifestation of neurofibromatosis type 1? Am J Med Genet 2003; 123A(1): 60-63.
20. Kurt AM, Unni KK, Sim FH, McLeod RA. Chondroblastoma of bone. Hum Pathol 1989; 20(10): 965-976.
21. Rosenberg AE and Nielsen GP. Giant cell containing lesions of bone and their differential diagnosis. Curr Diagn Pathol 2001; 7(4): 235-246.
22. Bertoni F, Bacchini P, EL Staals EL. Giant cell-rich osteosarcoma. Orthopedics 2003; 26(2): 179-181.
23. Ta HT, Dass CR, Choong PF, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metastasis Rev 2009; 28(1-2): 247-263.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2017 Issue 2
Most read in this issue
- Myxoid tumors of soft tissues
- Giant cell-rich lesions of bone and their differential diagnosis
-
Hybrid peripheral nerve sheath tumors:
A review - Mature teratoma of the uterine corpus: A case report