
Výsledky morfologické depistáže Lynchova syndromu v období 2013-2016
Results of morphological screening for Lynch syndrome during the period 2013-2016
The introduction of a screening system for Lynch syndrome in pathology laboratories in Plzen yielded 24 diagnoses of Lynch syndrome during the period of 2013-2016, 20 of them presenting with colorectal cancer. In 8 of those 24 cases germline mutations of MMR genes, previously not recognized as pathogenic with certainty, were detected. Although the frequency of Lynch syndrome in patients with colorectal cancer was only 0.34 % in total, following introduction of the universal immunohistochemical investigation of MMR (mismatch repair) proteins expression in all colorectal cancers examined in Sikl´s Institute of Pathology the frequency per year in this department reached 2.4 %. The results favor universal immunohistochemical screening for Lynch syndrome in colorectal and endometrial cancer cases over a selective approach based on a combination of clinical and morphological criteria. Increased effectiveness of the universal approach is not brought about only by higher sensitivity of the immunohistochemical examination per se, but also by the possibility of automation of the process leading to increased adherence even of pathologists not directly engaged in Lynch syndrome management. However, the introduction of a nation-wide universal screening system requires support from the government and health insurance companies.
Keywords:
colorectal cancer – endometrial cancer – immunohistochemistry – Lynch syndrome – MMR – screening
:
Martin Dušek 1,2; Ladislav Hadravský 3; Jan Stehlík 2; Kateřina Černá 2; Radmila Čurčíková 2,4; Marián Švajdler 1,2; Bohuslava Šašková 1,2; Magdaléna Dubová 1,2; Michal Michal 1
![]() ; Tomáš Jirásek 3,4; Ondřej Daum 1,2
; Tomáš Jirásek 3,4; Ondřej Daum 1,2
![]()
:
Šiklův ústav patologie LF UK v Plzni a FN Plzeň
1; Bioptická laboratoř, s. r. o., Plzeň
2; Ústav patologie 3. LF UK a FN Královské Vinohrady Praha
3; Oddělení patologie Krajské nemocnice Liberec, a. s.
4
:
Čes.-slov. Patol., 54, 2018, No. 2, p. 86-92
:
Original Article
Zavedení systému depistáže Lynchova syndromu na pracovištích patologie v Plzni vedlo v letech 2013-2016 k diagnóze 24 případů, z toho 20 prezentujících se kolorektálním karcinomem. V 8 z těchto 24 případů byly detekovány germinální mutace MMR genů, které předtím nebyly v databázích evidovány jako patogenní. V celkovém souhrnu byla sice četnost Lynchova syndromu u pacientů s kolorektálním karcinomem pouze 0,34 %, po zavedení systému univerzálního imunohistochemického vyšetřování exprese MMR (mismatch repair) proteinů ve všech kolorektálních karcinomech diagnostikovaných v Šiklově ústavu patologie však četnost případů Lynchova syndromu za rok na tomto pracovišti dosáhla až 2,4 %. Naše výsledky svědčí ve prospěch univerzálního imunohistochemického screeningu Lynchova syndromu v případech kolorektálního a endometriálního karcinomu oproti výběrovým depistážním metodám založeným především na klinických, méně i morfologických znacích. Vyšší efektivita univerzálního screeningu nespočívá pouze ve vyšší senzitivitě imunohistochemického vyšetření, ale i v možné automatizaci procesu a tím zvýšení adherence k depistáži i patologů přímo nezainteresovaných v managementu Lynchova syndromu. Plošné zavedení národního univerzálního depistážního systému však vyžaduje podporu ze strany státní správy a zdravotních pojišťoven.
Klíčová slova:
depistáž – endometriální karcinom – imunohistochemie – kolorektální karcinom – Lynchův syndrom – MMR
Lynchův syndrom (LS) je autozomálně dominantně dědičný familiární karcinomový syndrom v současné době definovaný průkazem germinální inaktivační mutace některého z MMR genů, genu EPCAM nebo germinální metylace promotoru genu MLH1 (tab. 1)(1). Nejčastěji postiženými MMR geny při LS jsou MLH1 a MSH2 (dohromady více než 80 %)(2), dále následuje MSH6 (10 %). Vzácněji mohou dysfunkci MMR proteinů, a tím pádem i LS, způsobovat zárodečná hypermetylace promotoru genu MLH1 vedoucí k jeho epigenetické inaktivaci (3,4) nebo zárodečné delece 3´ konce genu EPCAM, které zase vedou k epigenetické inaktivaci MSH2 (5,6).
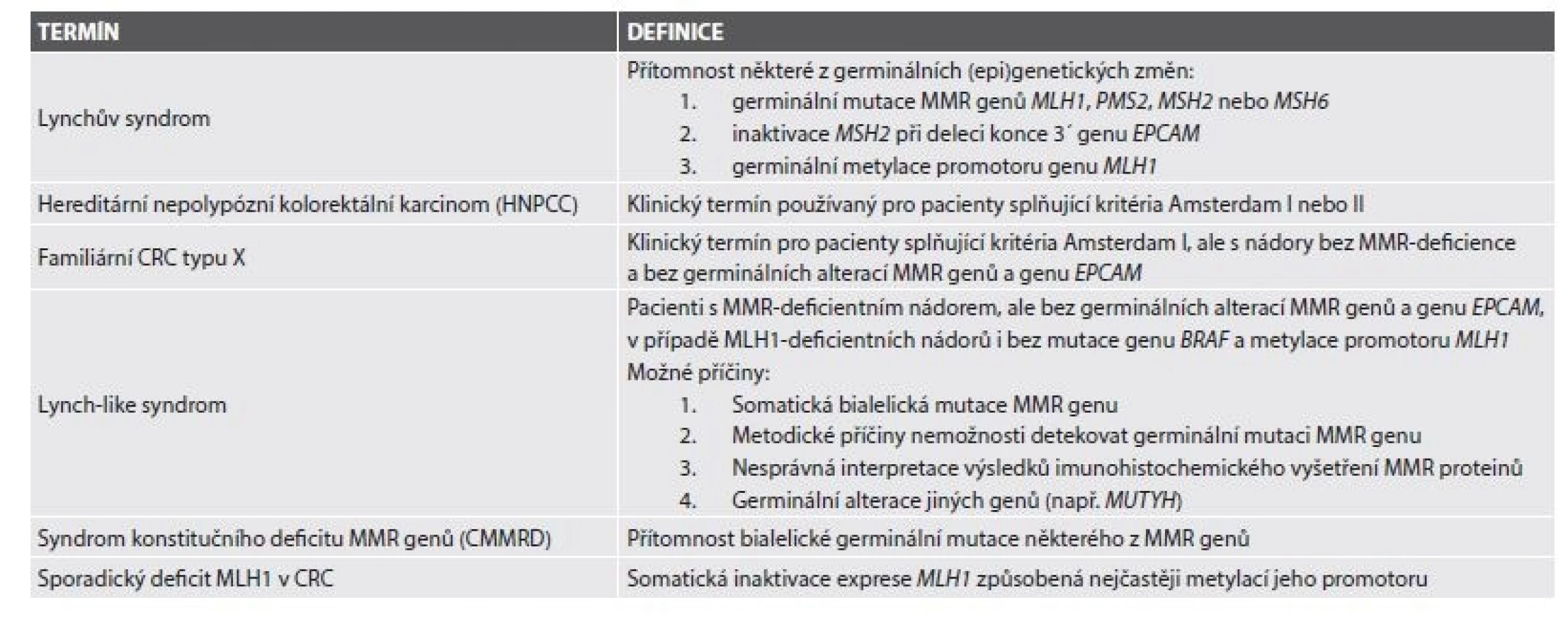
Nejčastějším karcinomem vznikajícím při LS je kolorektální karcinom (CRC), je však zvýšené riziko vzniku i dalších malignit, zejména karcinomů endometria, tenkého střeva, ovaria, ledvinné pánvičky a močovodu, nádorů mozku a kůže. Na podkladě LS vzniká podle současných dat asi 2,8 % CRC (1). Na rozdíl od familiárních karcinomových syndromů s premorbidním fenotypem (např. familiární adenomatózní polypózy, FAP) může být LS diagnostikován prakticky až při nálezu maligního nádoru, případně při genetickém vyšetření rodinných příslušníků již diagnostikovaného probanda. Výjimkou z tohoto pravidla je fenotypická varianta LS projevující se vznikem kožních sebaceózních nádorů, označovaná jako Muir-Torreho syndrom (MTS)(7).
Argumentů pro zavedení co nejsenzitivnějšího systému depistáže je několik. V prvé řadě je to četnost germinálních mutací MMR genů (a tedy LS) v populaci, která se podle odhadů pohybuje v rozmezí 1/370 – 1/400 (8,9). Dále je to vysoké riziko vzniku maligních nádorů u těchto pacientů, které v případě CRC u obou pohlaví a endometriálního karcinomu u žen s LS dosahuje v závislosti na typu mutace až 80 % (10-14). K progresi z adenomu do CRC navíc údajně dochází již během 2-3 let, na rozdíl od 8-10 let u sporadických případů (15,16). V neposlední řadě je důležitým faktorem i vznik maligních nádorů v poměrně nízkém věku, podle literárních údajů je průměrný věk v době diagnózy CRC 44-61 let, tedy až o 20 let méně než u sporadického CRC, a samozřejmě i zvýšené riziko vzniku synchronních a metachronních maligních nádorů (15,17).
Původně byl LS, tehdy dosud nazývaný hereditární nepolypózní kolorektální karcinom (HNPCC) diagnostikován na podkladě klinických Amsterdamských kritérií (18), respektive pro zohlednění případné extrakolonické prezentace Amsterdamských kritérií II (19). K záchytu pacientů suspektních z molekulárně geneticky definovaného LS slouží na klinické úrovni revidovaná Bethesda guidelines (RBG)(20), která však také nezachytí všechny případy LS (21), zejména v případě postižení MSH6 a PMS2 (22-26). Podle současných odhadů až 25 % pacientů s LS není při aplikaci těchto guidelines zachyceno. Z toho důvodu se v posledních letech obrací pozornost k možnostem morfologické diagnostiky LS spočívající ve vyšetření nádorů statisticky významně asociovaných s LS, zejména CRC, metodami moderní patologie, které byly detailně popsány v přehledových článcích v českém písemnictví (27-29). Tyto morfologické metody lze využít v depistáži buď výběrově, anebo univerzálně. V druhém případě je záchytnost signifikantně vyšší (30). I studie poměru nákladů a efektivity prokázala nejen sociálně-zdravotní, ale i ekonomickou výhodnost univerzálního sytému vyšetřování LS (31). V letech 2013-2016 jsme se proto pokusili v Šiklově ústavu patologie (ŠÚP) a Bioptické laboratoři s.r.o. (BL) postupně zavést model univerzální depistáže směřující k plošnému vyšetřování imunoexprese MMR proteinů ve všech CRC a následně i v endometriálních adenokarcinomech, protože bylo prokázáno, že endometriální prezentace u žen s LS může dosahovat i 3-5 % a často předchází kolorektální prezentaci (12,32,33).
MATERIÁL A METODIKA
Algoritmus výběru pacientů
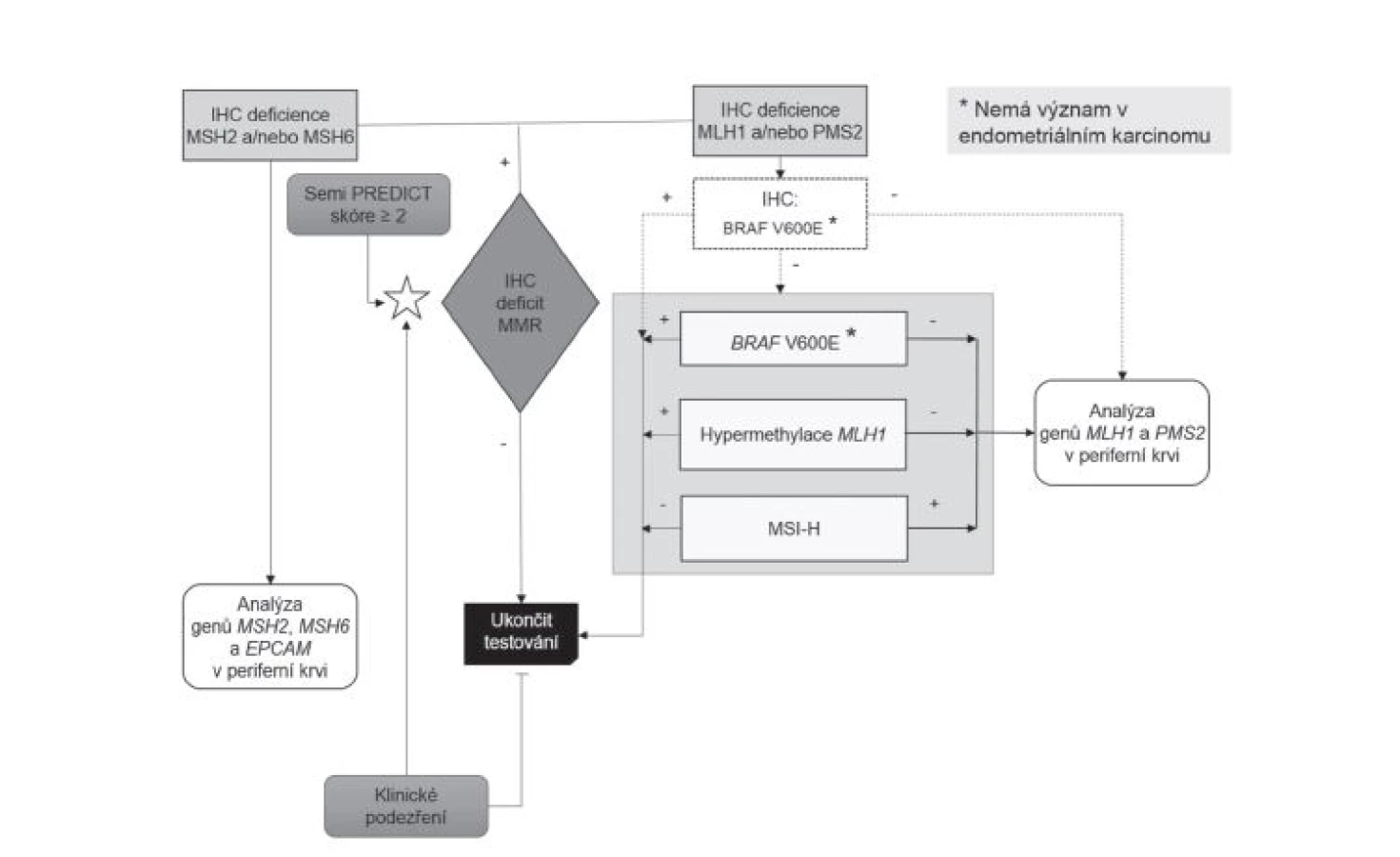
Metodika výběru pacientů pro genetické vyšetření se v průběhu sledovaného období s ohledem na vývoj poznatků a technologických možností měnila, jak dokumentují průběžně publikované přehledové články (27-29). Až do poloviny roku 2014 byla vstupní metodou na obou pracovištích analýza morfologie asociované s nestabilitou mikrosatelitů (MSI), takzvaná MSI-H morfologie, všech CRC prostřednictvím Semi PREDICT skóre (27,34), při jejímž pozitivním výsledku byl indikován komplex vyšetření somatického genomu sestávající z molekulárně genetické analýzy MSI, hypermetylace promotoru genu MLH1 a mutace V600E genu BRAF. Byla-li takto v nádoru potvrzena MSI a zároveň nebyla vyloučena možnost LS průkazem hypermetylace promotoru MLH1 a/nebo mutace BRAF, byl pacient, po bližším určení dysfunkčního MMR proteinu na základě imunohistochemického vyšetření, indikován k analýze germinálních mutací MMR genů (27,28). Od poloviny roku 2014 začaly být v ŠÚP všechny CRC vyšetřovány imunohistochemicky (v případě resekátů se zjevným nádorem byl automaticky jeden řez pro imunohistochemické vyšetření MMR proteinů zabločkován již při přikrojení, v případě endoskopických vzorků bylo vyšetření indikováno až na základě histologického nálezu), navíc začaly být imunohistochemicky vyšetřovány i všechny endometriální karcinomy (na základě histologického nálezu). V téže době však naopak musela být, s výjimkou konzultačních biopsií, prakticky ukončena depistáž LS v BL z důvodu finančního zatížení klientů na podkladě indukované péče. Vyšetřovány imunohistochemicky tak byly pouze vzorky zaslané do BL z jiného pracoviště na základě pozitivního Semi PREDICT skóre. Po zhodnocení výsledků imunohistochemického vyšetření bylo poté indikováno vyšetření somatického genomu nádorových buněk jako výše, případně analýza germinálních mutací MMR genů (obr. 1)(29).
Histologické a imunohistochemické vyšetření
Vzorky tkáně fixované v 10% formolu a zpracované klasickou parafinovou technikou byly obarveny hematoxylinem a eozinem při tloušťce řezů 4 μm. Pro imunohistochemické vyšetření byly řezy odparafínovány a rehydratovány v jednom kroku na přístroji BenchMark ULTRA (Ventana). Následně byla skla pokryta Tris-EDTA pufrem (pH 8,6) a zahřáta na 95°C po dobu 64 minut pro odmaskování antigenu. Skla byla poté inkubována při 37°C po dobu 40 minut se 100 μl primární protilátky proti MLH1 (MutL Protein Homolog 1, M1, RTU, Ventana), MSH2 (MutS Protein Homolog 2, G219-1129, RTU, Cell Marque), MSH6 (MutS Protein Homolog 6, 44, RTU, Ventana) a PMS2 (Postmeiotic Segregation Increased 2, EPR3947, RTU, Cell Marque). Poté byla skla omyta TBS (Tris-buffered saline) pro odstranění reagentů. Detekce byla zprostředkována alkalickou fosfatázou s amplifikací (ultraView Universal Alkaline Phosphatase Red detection Kit, Ventana) nebo inkubací s 200 μl DAB (3,3 - diaminobenzidine) substrate/chromogen po dobu 3 minut. Kvalita barvení byla verifikována jak použitím adekvátní vnější kontroly, tak zejména vnitřní kontrolou, kterou představují nenádorové lymfocyty. Jako ztráta exprese byla označena absence barvení jader nádorových buněk při zachované barvitelnosti vnitřní kontroly.
Analýza somatického genomu
Nejprve byly z bloků nakrájeny 20 μm silné řezy do mikrozkumavky a po odparafinování byla extrahována DNA dle manuálu výrobce (QIAsymphony DSP DNA kit). Na spektrofotometru Nanodrop byla změřena koncentrace a čistota DNA a za použití PCR s primery k vybraným „housekeeping“ genům i její kvalita.
Pro vyšetření nestability mikrosatelitních markerů (MSI) ve tkáni byla využita multiplexní PCR s následnou fragmentační analýzou pěti monukleotidových (BAT-25, BAT-26, NR-21, NR-24, MONO-27) a dvou pentanukleotidových repeticí (Penta C and Penta D) kitem Promega MSI Analysis System.
Exon 15 genu BRAF se zaměřením na detekci mutací v kodonech V600 a K601 byl analyzován metodou PCR a reverzní hybridizace kitem BRAF 600/601 StripAssay (Viennalab), nebo metodou real-time PCR kitem cobas 4800 BRAF V600 Mutation Test (Roche).
Analýza metylace promotoru genu MLH1 byla prováděna pomocí metody bisulfidické konverze a metylačně specifické PCR (35).
Analýza germinálních mutací
DNA a RNA z krve odebrané do EDTA zkumavky, respektive PAX zkumavky (PAXgene blood RNA tubes IVD) byla extrahována dle manuálu výrobce (QIAsymphony DSP DNA kit a PAXgene blood RNA kit IVD).
Detekce mutací celé kódující oblasti genu MLH1 byla provedena pomocí metody PCR (s primery amplifikujícími všech 19 exonů, včetně exon-intronových spojů) a přímého sekvenování. Analýza strukturálních aberací byla provedena pomocí MLPA-mP003 kitu.
Detekce případné germinální metylace promotoru genu MLH1 byla provedena pomocí MLPA-ME011 kitu.
Detekce mutací celé kódující oblasti genu PMS2 byla provedena pomocí metody RT-PCR (mRNA byla přepsána na cDNA, a ta použita jako templát pro PCR) a přímého sekvenování všech 15 exonů. Analýza strukturálních aberací byla provedena pomocí MLPA-mP008 kitu.
Detekce mutací celé kódující oblasti genu MSH2 byla provedena pomocí metody PCR (s primery amplifikujícími všech 16 exonů, včetně exon-intronových spojů) a přímého sekvenování. Analýza strukturálních aberací byla provedena pomocí MLPA-mP003 kitu.
Detekce mutací celé kódující oblasti genu MSH6 byla provedena pomocí metody PCR (s primery amplifikujícími všech 10 exonů, včetně exon-intronových spojů) a přímého sekvenování. Analýza strukturálních aberací byla provedena pomocí MLPA-mP072 kitu.
Hodnocení významu detekovaných genetických změn
Patogenní význam variant MMR genů zjištěných molekulárně genetickou analýzou byl stanoven srovnáním s databázemi HGMD (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php)(36), MMRGVD (Mismatch Repair Genes Variant Database, http://www.med.mun.ca/mmrvariants)(37), InSIGHT (https://www.insight-group.org/variants/databases)(38), dbSNP NCBI (Database of Single Nucleotide Polymorphisms, National Cancer for Biotechnology Information, https://www.ncbi.nlm.nih.gov/snp) a IGSR (International Genome Sample Resource, http://www.internationalgenome.org) vycházející z dat získaných v rámci projektu sekvenování „1000 Genomes Project“ (39). V případě neznámé patogenicity byl také využit predikční program Provean (Protein Variation Effect Analyzer, http://provean.jcvi.org)(40).
VÝSLEDKY
V roce 2013 bylo na pracovištích ŠÚP a BL vyšetřeno celkem 1382 pacientů (unikátních rodných čísel) s diagnózou C18-20 (z toho 1006 případů v BL a 376 v ŠÚP). Na základě Semi PREDICT skóre bylo u 84 pacientů (6,1 %) vysloveno podezření na MSI a byla provedena další analýza, na jejímž základě bylo pro analýzu germinálních mutací MMR genů vybráno 20 případů. U 7 pacientů (0,5 %) byla prokázána zárodečná mutace některého z MMR genů, u 6 pacientů byla vyloučena. Do konce roku 2016 se však nezdařilo provést analýzu germinálních mutací u 7 pacientů (0,5 %) suspektních z diagnózy LS. Navíc byl jednou diagnostikován Lynch-like syndrom v materiálu z endometriálního karcinomu (somatická frameshift duplikace c.1597_1600dupCTTC v exonu 10 genu MSH2). Z pacientů s CRC diagnostikovaných v ŠÚP se ve 4 (1,06 %) případech jednalo o LS, v 10 (2,66 %) o MSI-H sporadický karcinom a v 1 (0,27 %) o nedovyšetřený suspektní LS.
V roce 2014 bylo vyšetřeno celkem 1568 pacientů s diagnózou C18-20 (z toho 1181 v BL a 387 v ŠÚP). Pro další analýzu bylo na základě morfologie a IHC vyšetření určeno 67 pacientů (4,3 %), z toho 39 (10,1 %) diagnostikovaných v ŠÚP. U 3 pacientů (0,2 %) byl molekulárně geneticky potvrzen LS, ve 36 případech (2,3 %) se jednalo o sporadický MSI-H karcinom, u 2 pacientů (0,1 %) se jednalo o Lynch-like syndrom v důsledku somatické jednobázové substituce c.942+3A>T třetí hraniční base intronu 5 v genu MSH2, a v důsledku somatické mutace c.1252delA v exonu 7 genu MSH2. 4 případy (0,3 %) se suspektním LS zůstávají dosud nevyšetřeny. Navíc byl LS diagnostikován u 2 pacientek s endometriálním karcinomem a u 1 pacientky s adenokarcinomem duodena. Tyto 3 případy LS s extrakolonickou prezentací byly diagnostikovány v ŠÚP. Naopak u žádného pacienta s CRC diagnostikovaným v ŠÚP nebyl prokázán LS.
V roce 2015 mělo diagnózu C18-20 celkem 1441 pacientů (z toho 1118 v BL a 323 v ŠÚP). 32 (2,2 %) nádorů bylo MMR-deficientních, z toho 20 (6,19 %) diagnostikovaných v ŠÚP. Molekulárně genetická analýza prokázala LS u 5 pacientů (0,35 %). Ve 20 případech (1,4 %) se jednalo o sporadický MSI-H karcinom, 7 pacientů (0,49 %) se suspektním LS nebylo dosud geneticky vyšetřeno. Z toho u pacientů diagnostikovaných v ŠÚP se ve 2 (0,61 %) případech jednalo o LS, ve 14 (4,33 %) o MSI-H sporadický karcinom a v 5 (1,54 %) o nedovyšetřený suspektní LS.
V roce 2016 bylo celkem vyšetřeno 1423 pacientů s diagnózou C18-20 (v BL 1092 a v ŠÚP 331). Jako MMR-deficientní bylo určeno 42 případů (3 %), z toho v ŠÚP 34 (10,3 %). LS byl molekulárně geneticky prokázán u 5 pacientů (0,35 %), ve 27 případech (1,9 %) šlo o sporadický MSI-H tumor. U 1 pacienta (0,1 %) byl diagnostikován Lynch-like syndrom se somatickou frameshift delecí c.1670_1677delAAGAACTG v exonu 15 genu MLH1. Dále byl LS diagnostikován u jedné pacientky s endometriálním karcinomem. Osm pacientů (0,56 %) velmi suspektních z diagnózy LS však dosud nemá provedenu analýzu germinálních mutací. Z pacientů s CRC diagnostikovaných v ŠÚP se ve 4 (1,2 %) případech jednalo o LS, ve 24 (7,25 %) o MSI-H sporadický karcinom a ve 4 (1,2 %) o nedovyšetřený suspektní LS.
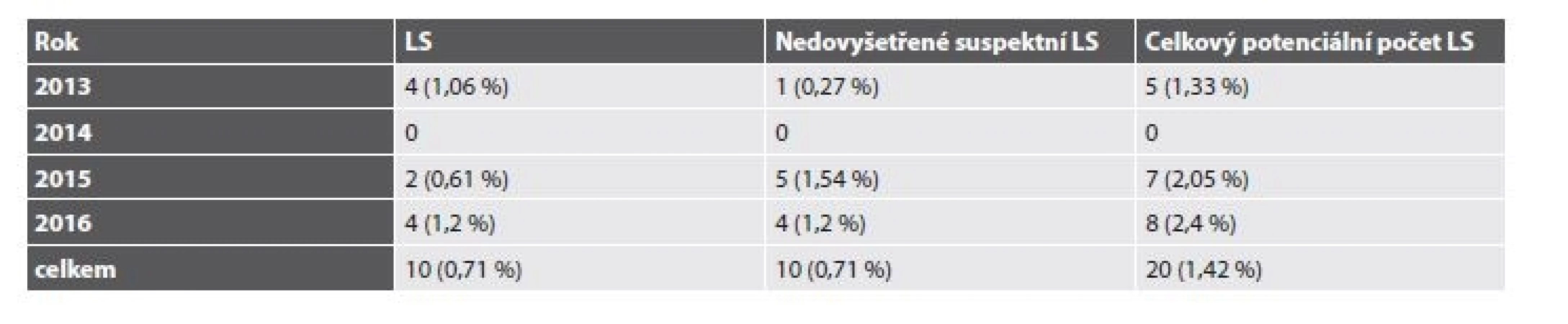
Celkem tedy bylo v letech 2013-2016 vyšetřeno 5814 pacientů s diagnózou C18-20 (4397 pacientů v BL a 1417 v ŠÚP). Suspektní MSI-H morfologie a/nebo IHC deficit MMR proteinů byl popsán u 225 případů. LS byl molekulárně geneticky potvrzen u 20 pacientů (0,34 %) s CRC (tab. 2). Z toho v BL bylo diagnostikováno 10 případů (0,22 %), v ŠÚP také 10 případů (0,71 %). Navíc byl u 3 pacientek LS diagnostikován na podkladě endometriálního karcinomu a u jedné pacientky prezentující se adenokarcinomem duodena (tab. 3). U 4 pacientů (0,07 %) byl diagnostikován Lynch-like syndrom. U 26 pacientů (0,45 %) se dosud nepodařilo provést analýzu germinálních mutací. Celkem by tedy mohl být LS teoreticky podkladem až 0,79 % případů CRC. Při omezení statistického zhodnocení na CRC vyšetřované v ŠÚP, které představují skupinu s kontrolovaným vstupem a poměrně homogenním složením, dosahuje potenciální frekvence LS u pacientů s CRC za celé sledované období až 1,42 %, v roce 2016 až 2,4 % (tab. 4).


Celkový poměr M/Ž byl 10/14, v případě CRC asociovaného s LS byl tento poměr 10/10, zatímco všichni 4 pacienti s extrakolonickou prezentací byly ženy. Věkové rozpětí bylo 26-80 let (průměrný věk: 55,9 roku, při omezení na CRC 56,6 roku, v případě extrakolonických malignit 52,5 roku). 5 pacientům (21,7 %) bylo v době diagnózy více než 70 let. Germinální mutace (nebo epimutace) v genu MLH1 byla zaznamenána v 6 případech CRC (25 %), v genu PMS2 ve 3 případech CRC (12,5 %), v genu MSH2 v 5 případech CRC a 1 endometriálního karcinomu (celkem 25 %) a v genu MSH6 v 6 případech CRC, 1 duodenálního adenokarcinomu a 1 endometriálního karcinomu (celkem 37,5 %).
DISKUZE
Během let 2013-2016 byl postupně vypracován algoritmus morfologické diagnostiky Lynchova syndromu a zaveden do rutinní praxe. V současné době jsou v ŠÚP vyšetřovány všechny CRC a endometriální karcinomy, v BL vzhledem ke způsobu financování zdravotnictví pouze případy zaslané ke konzultaci cíleně na diagnostiku LS. V uvedeném intervalu bylo na našich pracovištích diagnostikováno 24 případů LS, z toho 20 s CRC. Četnost LS ve skupině pacientů s CRC za celé sledované období souhrnně činí 0,34 %, z toho v BL 0,22 %, v ŠÚP 0,71 %. Relativně nízký počet případů a změna metodiky v průběhu roku 2014 znemožňuje validní souhrnné statistické zhodnocení. S určitou rezervou lze hodnotit výsledky za roky 2015 a 2016, během kterých probíhala na obou pracovištích depistáž konstantním, byť vzájemně odlišným způsobem. V obou těchto letech byla frekvence LS u pacientů s CRC detekovaná metodou plošného imunohistochemického vyšetřování dvakrát vyšší než celková frekvence v obou souborech. Frekvence LS v souboru CRC diagnostikovaných v ŠÚP dosáhla až 1,54 %, při společném hodnocení s dosud nedovyšetřenými případy suspektního LS až 2,4 %. Fakt, že jsou výsledky stále nižší než podle literárních údajů, může mít několik vysvětlení.
V prvé řadě může mít vliv „problém malých čísel“, protože vzhledem k náhodné distribuci patogenních alel v populaci mohou být tyto v malých souborech nehomogenně rozděleny. Tento faktor se mohl (v asociaci s dalšími vlivy) uplatnit zejména v anomálním roce 2014, kdy v ŠÚP nebyl detekován žádný LS u pacienta s CRC, přestože v témže roce zde byly diagnostikovány 2 případy LS s endometriálním karcinomem a 1 s duodenálním adenokarcinomem. Druhou možností je vliv zevních faktorů, které by mohly zvyšovat výskyt sporadických CRC, čímž by došlo k relativnímu snížení četnosti LS v populaci pacientů s CRC. V souvislosti se statistickými údaji o signifikantně vyšší incidenci CRC v Plzeňském kraji nelze vyloučit působení neznámého exogenního faktoru zvyšujícího výskyt sporadického CRC (http://www.svod.cz/analyse.php?modul=regionprehled#, ÚZIS ČR). I nižší frekvence MMR-deficientních nádorů ve skupině CRC diagnostikovaných v ŠÚP v roce 2015 a 2016 (6,19 % a 10,3 %) v porovnání s literárními údaji by mohla svědčit pro hypotézu o regionálním působení exogenního faktoru zvyšujícího incidenci sporadického MSS-CRC (30,41,42). Na druhou stranu, nízká frekvence MSI-H (respektive MMR-deficientních) karcinomů také může být vysvětlena falešnou pozitivitou exprese MMR proteinů. Tato diskordance mezi výsledkem hodnocení exprese MMR proteinů na základě imunohistochemického vyšetření a jejich funkcí může být dána jednak charakterem mutace umožňující syntézu proteinu zachovávajícího antigenicitu, ale postrádajícího funkci, jednak chybným hodnocením imunohistochemické reakce. Druhá možnost přichází v úvahu zejména v případě komplexu MLH1/PMS2. Je totiž známo, že tyto proteiny jsou velmi citlivé na autolýzu, a proto zejména ve větších resekátech je jejich antigenicita často snížena v důsledku pomalého a/nebo nedostatečného průniku fixativa do tkáně. Z toho vyplývající adaptivní snížení nároků na intenzitu hodnocené imunohistochemické reakce pak může mít za následek mylnou interpretaci slabého zbarvení v důsledku abnormální struktury proteinu jako následek neadekvátní fixace. Pro tuto možnost by mohl svědčit fakt, že v našem souboru převažovaly případy LS asociované s germinální mutací MSH6 nad případy způsobenými germinální mutací MLH1, které dle literatury představují největší skupinu pacientů s LS. Ani v tomto ohledu však současný nedostatečný stav poznatků o výskytu patogenních variant MMR genů v české populaci neumožňuje vyloučit, že námi detekovaný vysoký výskyt germinálních mutací MSH6 neodráží skutečnou regionální anomálii relativně geneticky homogenní populace ČR. Konečně nelze ani vyloučit, že literární údaje o četnosti LS jsou nadhodnocené. Ne všechny literární zdroje udávající incidenci, prevalenci nebo četnost LS v rámci CRC mají totiž jasně definována kritéria diagnózy LS, tedy podle dnešní definice průkaz germinálních mutací MMR genů, germinálních mutací genu EPCAM nebo germinální metylace promotoru genu MLH1. Některé zdroje tedy mohou zahrnovat i případy Lynch-like syndromu, případně, byla-li použita pouze klinická kritéria, i jiné familiární syndromy asociované s CRC.
Ačkoli je problematika nízké frekvence LS v našem souboru zajímavá a důležitá a vyžaduje hlubší analýzu většího souboru v delším časovém období, aktuálně se jako zásadnější jeví vysoký počet nedovyšetřených případů suspektních z LS. Vzhledem k tomu, že analýzu germinálních mutací může dle platné legislativy indikovat pouze lékařský genetik, k němuž může pacienta odeslat pouze jiný klinický lékař, je evidentní, že mezioborová spolupráce s aktivní účastí zainteresovaných klinických lékařů je v současnosti jedním z nejkritičtějších momentů v diagnostice LS, stejně jako v následném genetickém vyšetření příbuzných pacientů s LS a v organizaci navazujících screeningových metod. Několik let snahy o zavedení plošného screeningového systému nás přesvědčilo i o tom, že výše uvedené nebude možné bez politické podpory a bez finanční účasti zdravotních pojišťoven.
Zajímavým vedlejším výsledkem depistáže LS byla identifikace 8 variant MMR genů, které předtím v genetických databázích nebyly evidovány jako patogenní. Jmenovitě jde o jednobázovou substituci c.366+1G>A první hraniční báze intronu 2 v genu MSH2 (dosud hodnocena pouze jako pravděpodobně patogenní), in-frame deleci dvanácti bází c.170_181del v exonu 2 genu MLH1 (dosud hodnocena jako nejasná), inframe deleci c.1153_1155delAGG v exonu 4 genu MSH6 (dosud hodnocena pouze jako pravděpodobně patogenní), frameshift deleci c.3573delT v exonu 7 genu MSH6 (dosud hodnocena pouze jako pravděpodobně patogenní), frameshift duplikaci c.2252_2253dupAA v exonu 19 genu MLH1 (dosud hodnocena jako nejasná), jednobázovou nonsense substituci c.856G>T v exonu 4 genu MSH6 (dosud hodnocena pouze jako pravděpodobně patogenní), jednobázovou nonsense substituci c.1691C>A v exonu 4 genu MSH6 (dosud hodnocena pouze jako pravděpodobně patogenní) a frameshift duplikaci c.1862dupT v exonu 4 genu MSH6 (dosud hodnocena pouze jako pravděpodobně patogenní)(tab. 2 a 3).
V neposlední řadě je zajímavým zjištěním, že 5 pacientům s LS (21,7 %) bylo v době diagnózy více než 70 let, což ve shodě s jinými studiemi zpochybňuje efektivitu těch systémů depistáže, mezi jejichž vstupní kritéria patří věk pacienta (30). Ačkoli by se mohlo zdát, že v této věkové kategorii nemá smysl rozlišovat mezi LS a sporadickým CRC, tak vzhledem ke skutečnosti, že LS jako autozomálně dominantně dědičné onemocnění je charakterizován variabilní expresivitou (tedy i různým věkem vzniku maligního nádoru) v rámci rodiny, na osud dosud zdravých rodinných příslušníků může mít výrazný dopad i diagnóza LS v netypických věkových kategoriích.
ZÁVĚR
Naše zkušenosti jednoznačně potvrzují prospěšnost univerzálního imunohistochemického vyšetřování MMR proteinů všech CRC a endometriálních karcinomů v rámci depistáže LS. Zvýšení efektivity záchytu pacientů s LS lze v rámci specializovaných center dále dosáhnout soustředěním interpretace imunohistochemického barvení a následné molekulárně genetické analýzy na omezený počet patologů obeznámených s jejími úskalími. V současné době se perspektivně také jeví možnost imunohistochemické analýzy exprese MMR proteinů ve všech endometrioidních a světlobuněčných karcinomech ovaria, které mohou být s LS dle literatury asociovány až ve 20 % případů (12,32,33,43). Špatně ovlivnitelným prvkem depistážního systému je účast klinických lékařů a lékařských genetiků a vzájemná koordinace nezbytné preventivní péče, která v současné době není dostatečně podporována aktuálním nastavením systému zdravotnictví v ČR.
SEZNAM ZKRATEK
BL: Bioptická laboratoř s.r.o., Plzeň
CMMRD: constitutional mismatch repair deficiency, syndrom konstitučního deficitu MMR genů
CRC: colorectal carcinoma, kolorektální karcinom
EPCAM: epithelial cell adhesion molecule
FAP: familiární adenomatózní polypóza
HNPCC: hereditary non-polyposis colorectal cancer, hereditární nepolypózní kolorektální karcinom
LS: Lynchův syndrom
MLH1: mut L homolog 1
MMR: mismatch repair
MSH2: mut S homolog 2
MSH6: mut S homolog 6
MSI: microsatellite instability, nestabilita mikrosatelitů
MSI-H: microsatellite instability – high, vysoký stupeň nestability mikrosatelitů
MSS: microsatellite stable, stabilní mikrosatelity
MTS: Muir – Torreho syndrom
MUTYH: mutY homolog
PMS2: postmeiotic segregation increased 2
RBG: revidovaná Bethesda guidelines
ŠÚP: Šiklův ústav patologie Fakultní nemocnice Plzeň a Lékařské fakulty Univerzity Karlovy v Plzni
PODĚKOVÁNÍ
Tento výstup vznikl v rámci projektu Specifického vysokoškolského výzkumu 2017–260 391.
The work was supported by the grant SVV 2017–260 391.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
Doc. MUDr. Ondřej Daum, Ph.D.
Šiklův ústav patologie LF UK a FN Plzeň
Edvarda Beneše 13, 305 99 Plzeň
tel.: +420377402523
e-mail: DAUM@fnplzen.cz
Sources
1. Pai RK. A practical approach to the evaluation of gastrointestinal tract carcinomas for Lynch syndrome. Am J Surg Pathol 2016; 40(4): e17-34.
2. Nystrom-Lahti M, Wu Y, Moisio AL, et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet 1996; 5(6): 763-769.
3. Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002; 62(14): 3925-3928.
4. Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 2007; 356(7): 697-705.
5. Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet 2009; 41(1): 112-117.
6. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 2009; 30(2): 197-203.
7. Kacerovská D, Kazakov DV, Černá K, et al. Muir-Torre syndrom - fenotypická varianta Lynchova syndromu. Cesk Patol 2010; 46(4): 86-94.
8. Hampel H, de la Chapelle A. The search for unaffected individuals with Lynch syndrome: do the ends justify the means? Cancer Prev Res (Phila) 2011; 4(1): 1-5.
9. Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 2006; 296(12): 1479-1487.
10. Jenkins MA, Baglietto L, Dowty JG, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006; 4(4): 489-498.
11. Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. Journal of Medical Genetics 2005; 42(6): 491-496.
12. Zeimet AG, Mori H, Petru E, et al. AGO Austria recommendation on screening and diagnosis of Lynch syndrome (LS). Arch Gynecol Obstet 2017; 296(1): 123-127.
13. Sehgal R, Sheahan K, O’Connell PR, Hanly AM, Martin ST, Winter DC. Lynch syndrome: an updated review. Genes (Basel) 2014; 5(3): 497-507.
14. Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer 2013; 12(2): 229-240.
15. Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36(11): 801-818.
16. Jass JR, Stewart SM. Evolution of hereditary non-polyposis colorectal cancer. Gut 1992; 33(6): 783-786.
17. Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol 2014; 109(8): 1159-1179.
18. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34(5): 424-425.
19. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116(6): 1453-1456.
20. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96(4): 261-268.
21. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352(18): 1851-1860.
22. Liu T, Yan H, Kuismanen S, et al. The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res 2001; 61(21): 7798-7802.
23. van der Klift H, Wijnen J, Wagner A, et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 2005; 44(2): 123-138.
24. Dovrat S, Figer A, Fidder HH, et al. Mutational analysis of hMsh6 in Israeli HNPCC and HNPCC-like families. Fam Cancer 2005; 4(4): 291-294.
25. Hegde MR, Chong B, Blazo ME, et al. A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res 2005; 11(13): 4689-4693.
26. Hendriks YM, Jagmohan-Changur S, van der Klift HM, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130(2): 312-322.
27. Daum O, Beneš Z, Hadravský L, et al. Lynchův syndrom v rukách patologa. Cesk Patol 2014; 50(1): 18-24.
28. Kokošková B, Daum O, Beneš Z, et al. Moderní diagnostika Lynchova syndromu. Gastroent a Hepatol 2014; 68(2): 157-165.
29. Dušek M, Hadravský L, Černá K, et al. Diagnóza Lynchova syndromu od patologa. Klin Onkol 2016; 29(3): 180-186.
30. Hartman DJ, Brand RE, Hu H, et al. Lynch syndrome-associated colorectal carcinoma: frequent involvement of the left colon and rectum and late-onset presentation supports a universal screening approach. Hum Pathol 2013; 44(11): 2518-2528.
31. Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med 2010; 12(2): 93-104.
32. Mills AM, Liou S, Ford JM, Berek JS, Pai RK, Longacre TA. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol 2014; 38(11): 1501-1509.
33. Mills AM, Longacre TA. Lynch syndrome screening in the gynecologic tract: current state of the art. Am J Surg Pathol 2016; 40(4): e35-44.
34. Hyde A, Fontaine D, Stuckless S, et al. A histology-based model for predicting microsatellite instability in colorectal cancers. Am J Surg Pathol 2010; 34(12): 1820-1829.
35. Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A. CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol 2002; 160(5): 1823-1830.
36. Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017; 136(6): 665-677.
37. Woods MO, Williams P, Careen A, et al. A new variant database for mismatch repair genes associated with Lynch syndrome. Hum Mutat 2007; 28(7): 669-673.
38. Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014; 46(2): 107-115.
39. Birney E, Soranzo N. Human genomics: The end of the start for population sequencing. Nature 2015; 526(7571): 52-53.
40. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015; 31(16): 2745-2747.
41. Mas-Moya J, Dudley B, Brand RE, et al. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum Pathol 2015; 46(11): 1616-1625.
42. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138(6): 2073-2087 e2073.
43. Vierkoetter KR, Ayabe AR, VanDrunen M, Ahn HJ, Shimizu DM, Terada KY. Lynch Syndrome in patients with clear cell and endometrioid cancers of the ovary. Gynecol Oncol 2014; 135(1): 81-84.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2018 Issue 2
Most read in this issue
- Frozen section: history, indications, contraindications and quality assurance
- Frozen section examination of pancreas, gallbladder, extrahepatic biliary tree, liver, and gastrointestinal tract
- Intraoperative diagnosis of the head and neck lesions, thyroid and parathyroid gland, bone and soft tissue, and genitourinary tract
- Results of morphological screening for Lynch syndrome during the period 2013-2016