
Neuronálna ceroidná lipofuscinóza s postihnutím srdca
Neuronal ceroid lipofuscinosis with cardiac involvement
Neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders with clinical presentation predominantly in the childhood. The NCLs represent lysosomal storage disorders characterized by the accumulation of autofluorescent lipopigment storage material. The most common clinical features include development failure, psychomotor regression, seizures, and progressive loss of vision. We present a case of neuronal ceroid lipofuscinosis with cardiac involvement diagnosed post-mortem in a 9,5-year-old boy, whose clinical symptomatology comprised partial epilepsy, psychomotor decline and sinus bradycardia. In contrast to ventricular hypertrophy, being more frequently associated with NCLs, we discovered cardiac atrophy. Histologic examination of the heart revealed not only the lipofuscinosis affecting cardiac conducting cells and cardiomyocytes, but also basophilic degeneration of myocardium.
Keywords:
Neuronal ceroid lipofuscinosis – NCL7/MFSD8 – bradycardia – Basophilic degeneration of myocardium
:
Silvia Farkašová Iannaccone 1; Peter Vasovčák 2; Dorota Sopková 1; Mária Pisarčíková 3; Marián Švajdler 4; Lucia Fröhlichová 5; Lucia Mistríková 6; Daniel Farkaš 7
:
Bioptická laboratoř s. r. o., Plzeň
; Ústav súdneho lekárstva UPJŠ LF, Trieda SNP č. 1, Košice
1; PROGENET s. r. o., Strečnianska 13, Bratislava
2; Klinika pediatrickej anesteziológie a intenzívnej medicíny, Detská fakultná nemocnica Košice, Trieda SNP 1, Košice
3; Šiklův ústav patologie, Univerzita Karlova Praha, Lékařská fakulta Plzeň, Česká Republika
4; Oddelenie patológie, Univerzitná nemocnica Louisa Pasteura, Rastislavova 48, Košice
5; Kardiochirurgické oddelenie, Východoslovenský ústav srdcových chorôb, Ondavská č. 8, Košice
6; Úrad pre dohľad nad zdravotnou starostlivosťou, SLaPA pracovisko, Ipeľská 1, Košice
7
:
Čes.-slov. Patol., 55, 2019, No. 3, p. 176-181
:
Original Articles
Neuronálne ceroidné lipofuscinózy predstavujú skupinu geneticky podmienených neurodegeneratívnych ochorení s klinickými prejavmi prevažne v detskom veku. Jedná sa o lyzozomálne ochorenia sprevádzané intracelulárnou akumuláciou autofluorescentného materiálu charakteru lipofuscínu. Medzi najčastejšie klinické prejavy týchto ochorení patria poruchy vývoja, psychomotorický regres, epileptické záchvaty a poruchy zraku. Popisujeme prípad posmrtne diagnostikovej lipofuscinózy u 9,5-ročného chlapca s postihnutím srdca. Ochorenie sa okrem neurologických prejavov v zmysle parciálnej epilepsie a psychomotorického regresu prejavovalo sínusovou bradykardiou. Makroskopicky sme na rozdiel od častejšie udávanej hypertrofie srdca zistili výraznú atrofiu. Histologickým vyšetrením srdca bola diagnostikovaná nielen lipofuscinóza buniek prevodového systému a kardiomyocytov, ale aj bazofilná degenerácia kardiomyocytov.
Klíčová slova:
Neuronálna ceroidná lipofuscinóza – NCL7/MFSD8 – bradykardia – bazofilná degenerácia kardiomycytov
Neuronálne ceroidné lipofuscinózy sú skupinou geneticky podmienených lyzozonálnych neurodegeneratívnych ochorení vyznačujúcich sa intracelulárnou akumuláciou autofluorescentného materiálu charakteru lipofuscínu. Klinické príznaky sú zväčša podmienené postihnutím centrálnej nervovej sústavy. Medzi najčastejšie klinické prejavy patria poruchy vývoja, psychomotorický regres, záchvaty a poruchy zraku (1), medzi zriedkavejšie diagnostikované sprievodné klinické prejavy patrí postihnutie srdca (2-5). Popisujeme prípad posmrtne mikroskopicky a geneticky diagnostikovej neuronálnej ceroidnej lipofuscinózy u 9,5-ročného chlapca s postihnutím srdca, u ktorého sa ochorenie okrem typických klinických príznakov vyplývajúcich z poškodenia mozgu prejavovalo intermitentnou sínusovou bradykardiou.
POPIS PRÍPADU
V opisovanom prípade sa jednalo o 9,5-ročného rómskeho chlapca (113 cm, 14 kg, BMI 11), ktorý bol pred hospitalizáciou umiestnený v domove sociálnych služieb. Z anamnestických údajov vyplynulo, že mal 6 súrodencov, pričom staršia sestra je sledovaná pre epilepsiu. Narodil sa spontánnym pôrodom v 38. gestačnom týždni, nebol kriesený a popôrodná adaptácia bola primeraná. Od veku 4,5 roka bol sledovaný s diagnózou parciálnej epilepsie so sekundárnou generalizáciou. Diagnostikovaný bol psychomotorický regres s vyslovením podozrenia na metachromatickú leukodystrofiu, alebo iné neurodegeneratívne ochorenie. Pre agenézu pravej obličky bol dispenzarizovaný v nefrologickej ambulancii. Opakovane bol hospitalizovaný pre infekcie dýchacích ciest, pričom počas hospitalizácií bola opakovane zistená intermitentná sínusová bradykardia. Echokardiografické vyšetrenie srdca vykonané dva roky pred úmrtím bolo v medziach normy. Až do úmrtia pacient nechodil a nerozprával. Očným vyšetrením bola diagnostikovaná anoptická amblyopia (amlyopia ex anopsia) a atrofia zrakového nervu. Naposledy bol hospitalizovaný pre prevažne pravostranný hnisavý a abscedujúci zápal pľúc, ktorý po 2. dňoch viedol pri rozvoji septického šoku k smrti. V čase prijatia do nemocnice mal nemerateľnú telesnú teplotu. Od prijatia do nemocnice až do úmrtia mal výraznú bradykardiu od 38/min po 48/min.
Pitva bola vykonaná 8 hodín po smrti. Pri vonkajšej obhliadke bola výrazná celková hypotrofia, flekčné postavenie končatín a mikrocefalická hlava. Z vnútorného nálezu okrem zjavného pravostranného zápalu pľúc boli najvýraznejšie zmeny na mozgu. Markantná bola celková atrofia mozgu (hmotnosť 626 g) s hydropsom mozgových plien (obr. 1). Na rezných plochách mozgu fixovaného vo formole boli ryhy medzi závitmi výrazne rozšírené, samotné závity stenčené, komorový systém stredne rozšírený, biela hmota nevykazovala makroskopicky viditeľné zmeny (obr. 2). Vo všetkých systematicky vyexcidovaných rezoch mozgu bola deplécia neurónov kôry so stratou pôvodného laminárneho usporiadania, v zachovaných neurónoch bolo výrazné prejasnenie a zväčšenie objemu cytoplazmy, ktoré boli sprevádzané reaktívnou hypertrofiou (gemistocytárna premena) astrocytov. Zmeny týkajúce sa poškodenia kôry boli najzreteľnejšie viditeľné v hemisférach mozočka kde bola evidentná deplécia neurónov stratum granulosum (obr. 3). Imunohistochochemickým vyšetrením kôry hemisfér mozgu bola deplécia neurónov sprevádzaná výrazným znížením expresie na prítomnosť neurofilament (NF). V mozočku farbením na NF bola naviac zistená ektopia Purkyňových buniek v molekulárnej vrstve (obr. 4), celkom sporadicky „torpédové“ rozšírenie axónov Purkyňových buniek, abnormálne vetvenie dendritov Purkyňových buniek pripomínajúce „smutnú vŕbu“ (weeping willow), respektíve horizontálne dendritické vetvenie (obr. 5). Farbením na kyslý gliálny fibrilárny proteín (GFAP) bola výrazne zvýšená pozitivita hypertrofických astrocytov v subkortikálnych oblastiach. Farbením na dôkaz makrofágov pomocou CD68 bola zistená ich abnormálne zvýšená prítomnosť prevažne v hlbších vrstvách kôry, pričom ich hustota priamo korelovala s pôvodnou hustotou a laminárnemu rozloženiu neurónov (menej v molekulárnej oblasti a bielej hmote) (obr. 6). Prejasnenie cytoplazmy so zväčšením objemu cytoplazmy neurónov bolo podmienené prítomnosťou granulárneho materiálu, svetložltej a ružovkastej farby, ktorý sa hromadil v neurónoch. Tento pigment bol Luxol fast blue, PAS a PAS s diastázou pozitívny, pričom pri vyšetrení fluorescenčným mikroskopom vykazoval známky intracelulárnej autofluorescencie (obr. 7). Dvojjadrové Purkyňové bunky, ich vakuolizácia, ani prejavy demyelinizácie neboli zistené.


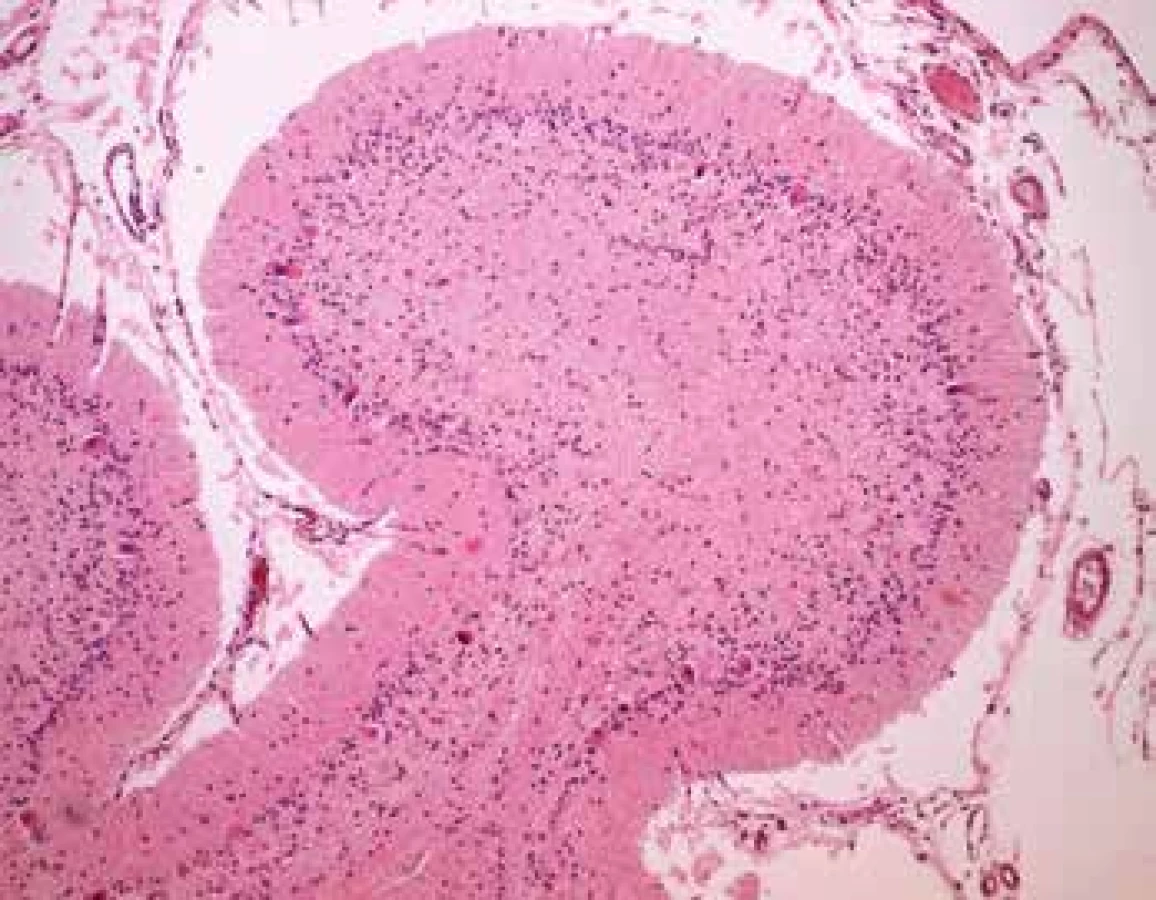




Srdce bolo anatomicky správne konfigurované, tuhšej konzistencie, hmotnosti 74 g. V pravej a ľavej komore myokardu boli difúzne v perinukleárnej oblasti kardiomyocytov nakopeniny hnedožltého pigmentu, ktorý bol slabo PAS a PAS diastáza pozitívny. Následným fluorescenčným vyšetrením bola zistená výrazná autofluorescencia tohoto pigmentu nielen v svalovine myokardu (obr. 8), ale aj v oblasti prevodového systému srdca, ktorá vo farbení hematoxylínom-eozínom nebola zjavná. Naviac bol v kardiomyocytoch sporadicky výskyt slabo bazofilne sfarbených amorfných, miestami jemne granulárnych hmôt, ktoré ojedinele vypĺňali kompletne celú cytoplazmu kardiomyocytov (obr. 9), pričom tieto hmoty vykazovali PAS a PAS diastáza pozitivitu, ale nevykazovali prejavy autofluoresnecie. Z ostatných patologických nálezov bol zistený ascites (250 ml) a agenéza pravej obličky. Na seróznej ploche prvej kľučky jejuna bola prítomná ostro od okolia ohraničená tumoriformná lézia svetložltej farby (1,5 cm x 0,7 cm x 0,8 cm) histologickým vyšetrením diagnostikovaná ako heterotopia tkaniva pankreasu (obr. 10). Po histologickom diagnostikovaní neuronálnej ceroidnej lipofuscinózy (neuronal ceroid lipofuscinosis – NCL) bol genetickým vyšetrením tkaniva mozgu špecifikovaný NCL variant v homozygotnom stave. c.881C>A (p.Thr294Lys) v géne MFSD8.


Na základe histologického nálezu mozgu (výrazná deplécia neurónov, hromadenie sa pigmentu v neurónoch, difúzna hyperplázia reaktívnych astrocytov), klinického obrazu (parciálna epilepsia so sekundárnou generalizáciou, psychomotorický regres, anoptická amblyopia s atrofiou zrakového nervu) a genetického vyšetrenia sme daný prípad uzavreli ako NCL7/MFSD8. Bezprostrednou príčinou smrti bol hnisavý až abscedujúci zápal pľúc.
DISKUSIA
Lyzozomálne ochorenia (LO) tvoria početnú skupinu vzácne sa vyskytujúcich metabolických ochorení. LO sú podmienené genetickým defektom lyzozomálnych enzýmov a klinické prejavy sú viazané prevažne na detský vek (1,6). LO môžu byť klasifikované podľa chemického zloženia ukladajúceho sa materiálu na neuronálne lipidózy, NCL a ochorenia uhľovodíkov (cukrov, carbohydrate disorders) (7). LO nervového systému sú tradične delené do dvoch skupín. Neuronálne tezaurizmózy so zmenami prevažne v sivej hmote a leukodystrofie so zmenami prevažne v bielej hmote. Medzi typické neuronálne tezaurizmózy patria NCL (6). Najčastejšou príčinou neuronálnych tezaurizmóz je hromadenie glykoproteolipidov, ktoré pripomínajú ceroid a lipofuscín. Klinicky sa NCL delili na infantilný, neskorý infantilný (so začiatkom medzi 18. mesiacom až 4. rokom života), juvenilný a adultný typ (7,8), neskôr pribudol kongenitálny typ (1). Termín NCL bol po prvýkrát zavedený v roku 1969 (9). Pokroky v molekulárne genetických technológiach, štúdie rodín a sporadických prípadov umožnili rozdelenie NCL do 14. skupín (NCL1-14) (1,10,11). Incidencia sa uvádza 0,1 až 7/100000 živorodených detí, pričom sa jedná o autozomálne recesívne ochorenia (12) s výnimkou niektorých adultných foriem s autozomálne dominantnou dedičnosťou (1,12). V Čechách je odhad incidencie 1,3/100000 (13). V našom prípade sa jednalo o geneticky diagnostikovanú NCL7/MFSD8, ktorá sa pôvodne uvádzala ako Turecký neskorý infantilný variant, nakoľko bola prvýkrát popísaná v tejto etnickej skupine. V súčasnom období je geneticky rozpoznaných minimálne 38 mutácii MFSD8 (11), pričom už dávnejšie boli popísané aj v bývalom Československu (14). NCL sú ochorenia panetnického charakteru s celosvetovým rozšírením, pričom klinicky je vo všetkých variantach prítomná cerebelárna ataxia, epileptické záchvaty a myoklonické kŕče. Infantilný a neskorý infantilný typ (kam patrí aj nami popisovaný prípad) sa prejavujú psychomotorickým regresom, zlyhávaním zraku progredujúcim do slepoty na podklade makulárnej a retinálnej degenerácie. Klinický priebeh je charakteristický tým, že čím je skorší začiatok, tým je rýchlejšia progresia a kratší priebeh. V infantilnom a neskorom infantilnom type je prežívanie od niekoľkých rokov po 10 rokov (7). V nami popisovanom prípade sme zaznamenali 5-ročné prežívanie od objavenia sa prvotných klinických symptómov. Klinicky by sa malo myslieť na NCL7 aj v prípadoch s prejavmi podobnými Rettovmu syndrómu, ktorý môže imitovať NCL (15).
Makroskopicky je cerebrálna a cerebelárna atrofia prítomná vo všetkých klinických typoch, ale najvýraznejšia je pri infantilnom a neskorom infantilnom type, pričom v týchto typoch NCL sú najvýraznejšie aj mikroskopické nálezy (7,12). V popisovanej kazuistike bola zistená hmotnosť mozgu 626 g (norma pre daný vek je 1275 g) (16). Mikroskopický obraz, vo všetkých miestach vyexcidovaných podľa doporučenia pre pitvy mozgu neurodegeneratívnych ochorení (17), vykazoval obraz straty neurónov kôry, v zachovaných neurónoch zväčšenie ich cytoplazmy nahromadením PAS pozitívneho a autofluorescentného pigmentu, ktorý bol sprevádzaný výrazným zmnožením hypertrofických astrocytov, čo zodpovedá typickému mikroskopickému obrazu NCL (7,12). Zmeny boli najvýraznejšie a nejlepšie detekovateľné v kôre mozočka, kde okrem deplécie buniek stratum granulosum a ektopie Purkyňových buniek boli prítomné nápadné zmeny abnormálnej arborizácie dendritov a sporadického rozšírenia ich axónov, tzv. torpéd. Pojem torpédo sa používa na popis rozšírenia axónov Purkyňových buniek, ktoré sa však môžu vyskytovať pri mnohých iných metabolických a degeneratívnych mozočkových ochoreniach (18,19). Samotná ektopia Purkyňových buniek nie je patognomická pre NCL, nakoľko sa môže vyskytovať aj pri iných chorobných stavoch (granular layer aplasia (19,20). Z tohto pohľadu je pre predbežnú diagnózu NCL najdôležitejšia prítomnosť intracelulárneho autofluorescentného pigmentu, pre záverečnú diagnózu genetické vyšetrenie. Vzhľadom k tomu, že makroskopický a mikroskopický obraz môžu do istej miery imitovať aj iné chorobné alebo poúrazové stavy s následným dlhodobým prežívaním (napr. perinatálna hypoxia, stavy po dusení sa, po ťažkom hemoragickom šoku) považujeme odporúčania na široké a cielené vyšetrenie centrálnej nervovej sústavy za veľmi vhodné (17).
V literatúre sa aspoň v niektorých typoch juvenilnej NCL (NCL3) uvádzajú zmeny činnosti srdca v zmysle zmien T-vĺn, arytmií, bradykardie, hypertrofie svaloviny ľavej komory (2,3), u neskorej infantilnej NCL2 kompletná atrioventrikulárna blokáda (4). Okrem zmien na EKG bola v práci Hofmana u troch pacientov pri klasickom juvenilnom a variante juvenilnej NCL popísaná taktiež ventrikulárna hypertrofia, výrazná bradykardia so sínusovým zastavením srdca a ťažká supraventrikulárna tachykardia počas anestézy. Mikroskopickým vyšetrením vykonaným po pitve bolo v daných prípadoch zistené v kardiomyocytoch a bunkách prevodového systému ukladanie patologického materiálu (5). U nami popisovaného pacienta bola počas opakovaných hospitalizácií zistená intermitentná sínusová bradykardia, pri poslednej dvojdňovej hospitalizácii pretrvávajúca bradykardia od 38/min po 48/min, ktorá však už bola prítomná v stave s laboratórnymi prejavmi sepsy. V danom prípade bola zistená hmotnosť srdca 74 gramov (za normu sa považuje v danom veku hmotnosť 115 gramov) (16), takže na rozdiel od kazuistík popísaných v literárnych zdrojoch sa jednalo o výraznú atrofiu. Mikroskopickým vyšetrením bola v perinukleárnej oblasti kardiomyocytov zistená prítomnosť výrazne väčšieho množstva pigmentu, ktorý vykazoval známky autofluorescencie. Vyšetrením pomocou fluorescencie bola zistená prítomnosť autofluorescentných hmôt aj v bunkách prevodového systému, ktoré v svetelnej mikroskopii neboli zjavné. Domnievame sa, že bradykardia zistená počas opakovaných hospitalizácií bola spôsobená hromadaním sa patologického materiálu v bunkách prevodového systému srdca, čo mohlo negatívne ovplyniť samotnú činnosť srdca. Okrem hromadenia sa lipopigmentu v kardiomyocytoch bola v niektorých typoch NCL popísaná v srdci aj nápadná depozícia kalciových a cholesterolových zložiek spôsobujúca reštriktívny typ srdcového poškodenia (21). V našom prípade bola zistená bazofilná degenerácia kardiomyocytov, s ktorou sme sa pri doposiaľ publikovaných prácach o NCL nestretli. Bazofilná degenerácia (nazývaná aj mukoidná degenerácia, mucinózna degenerácia, alebo kardiálny koloid) (22) sa zväčša nevyskytuje v prvej dekáde života (23,24). Na našom pracovisku sme sa s bazofilnou degeneráciou kardiomyocytov v prvej dekáde života taktiež doposiaľ nestretli.
Hypotermia zistená pri prijatí dieťaťa do nemocnice nebola podmienaná vplyvom vonkajšieho prostredia. Udáva sa, že pacienti s NCL majú nižšiu bazálnu teplotu a počas anestézie sú vo zvýšenom riziku hypotermie, čo si vyžaduje peroperačnú intervenciu v zmysle ohrievania tela (25). Vzhľadom k vyššie uvedenému predpokladáme, že hypotermia mohla byť podmienená postihnutím termoregulačných oblastí mozgu. Úplne však nie je možné vylúčiť to, že hypotermia bola spôsobená celkovým orgánovým zlyhávaním v prebiehajúcom septickom stave.
ZÁVER
V kazuistike sme prezentovali prípad 9,5-ročného chlapca s 5-ročnou symptomatológiou neurodegeneratívneho ochorenia prejavujúceho sa hlavne epileptickými záchvatmi, psychomotorickým regresom a poruchami zraku. Mikroskopickým vyšetrením mozgu a srdca bola zistená prítomnosť intracelulárneho autofluorescentného materiálu. Stanovenie definitívnej diagnózy genetickým vyšetrením (NCL7/MFSD8) bolo vykonané po smrti. Ochorenie sa počas života v dôsledku postihnutia buniek prevodového systému srdca a kardiomyocytov prejavovalo intermitentou sínusovou bradykardiou, čo je podľa našich dostupných možností prvý prípad bradykardie u NCL7/MFSD8.
PREHLÁSENIE
Autor práce prehlasuje, že v súvislosti s témou, vznikom a publikáciou tohto článku nie je v konflikte záujmov a vznik publikácie a článku neboli podporené žiadnou farmaceutickou firmou. Toto prehlásenie sa týka aj všetkých spoluautorov.
Adresa pre korešpondenciu:
MUDr. Daniel Farkaš, PhD.
Úrad pre dohľad nad zdravotnou starostlivosťou
SLaPA pracovisko Košice
Ipeľská 1, 043 74 Košice
tel.: +421552852660
fax: +421552852655
e-mail: farkas.dany@gmail.com
Sources
1. Glykys J, Sims KB. The Neuronal Ceroid Lipofuscinosis Disorders. In: Swaiman FK, Ashwal S, eds. Swaiman´s Pediatric Neurology (6th ed). Edinburg: Elsevier; 2018 : 960-986.
2. Ostergaard JR, Rasmussen TB, Mølgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011; 76(14): 1245-1251.
3. Michielsen P, Martin JJ, Vanagt E, Vrints C, Gillebert T, Snoeck J. Cardiac involvement in juvenile ceroid lipofuscinosis of the Spielmeyer-Vogt-Sjögren type: prospective noninvasive findings in two siblings. Eur Neurol 1984; 23(3): 166-172.
4. Fukumura S, Saito Y, Saito T, et al. Progressive conduction defects and cardiac death in late infantile neuronal ceroid lipofuscinosis. Dev Med Child Neurol 2012; 54(7): 663-666.
5. Hofman IL, Van der Wal AC, Dingemans KP, Beckrer AE. Cardiac pathology in neuronal ceroid lipofuscinoses – a clinicopathologic correlation in three patients. Eur J Pediatr Neurol 2001; 5(supll. A): 213-217.
6. Povyšil C, Šteiner I, et al. Speciální patologie (druhé, doplněné a přepracované vydání). Galén Karolinum 2007 : 306-307.
7. Haberland C. Clinical neuropathology. Text And Color Atlas. New York: Demos; 2007 : 174-175.
8. Armstrong D, Halliday W, Hawkins C, Takashima S. Pediatric Neuropathology: A Text Atlas. Tokyo: Springer; 2007 : 148-151.
9. Zeman W, Dyken P. Neuronal ceroid-lipofuscinosis (Batten’s disease): relationship to amaurotic family idiocy? Pediatrics 1969; 44(4): 570-583.
10. Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta 2013; 1832(11): 1795-1800.
11. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 2015; 1852(10 Pt B): 2237-2241.
12. Gray F, Duyckaerts C, De Girolami U. Escourolle & Poirier Manual of Basic Neuropathology (5th edn.) Oxford: Oxford UIniversity Press; 2014 : 238-240.
13. Elleder M, Franc J, Kraus J, Nevšímalová S, Sixtová K, Zeman J. Neuronal ceroid lipofuscinosis in the Czech Republic: analysis of 57 cases. Report of the ‘Prague NCL group’. Eur J Paediatr Neurol 1997; 1(4): 109-114.
14. Kousi M, Siintola E, Dvorakova L, et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009; 132(3): 810-819.
15. Craiu D, Dragostin O, Dica A, et al. Rett-like onset in late-infantile neuronal ceroid lipofuscinosis (CLN7) caused by compound heterozygous mutation in the MFSD8 gene and review of the literature data on clinical onset signs. Eur J Paediatr Neurol 2015; 19(1): 78-86.
16. Coppoletta JM, Wolbach SB. Body Length and Organ Weights of Infants and Children: A Study of the Body Length and Normal Weights of the More Important Vital Organs of the Body between Birth and Twelve Years of Age. Am J Pathol 1933; 9(1): 55-70.
17. Rohan Z, Matěj R. Pitva mozku a míchy při diagnóze neurodegenerativního onemocnění – praktický postup pro optimalizaci vyšetření. Cesk Patol 2015; 51(4): 199-204.
18. Gray F, Duyckaerts C, De Girolami U. Escourolle & Poirier Manual of Basic Neuropathology (5th edn.) Oxford: Oxford UIniversity Press; 2014 : 10.
19. Imataka G, Yamanouchi H, Hirato J, et al. Autopsy report of a 7-year old patient with the mosaic trisomy 13. Cell Biochem Biophys 2013; 67(2): 813-817.
20. Graham DI, Lantos PL. Greenfield´s Neuropathology, Volume 1 (6th edn). Oxford University Press, Arnold: London; 1997 : 474.
21. Reske-Nielsen E, Baandrup U, Bjerregaard P, Bruun I. Cardiac involvement in juvenile amaurotic idiocy – A specific heart muscular disorder. Acta Path Microbiol Scand 1981; 89 : 357-365.
22. Buja LM, Butany J. Cardiovascular Pathology, Elsevier, Fourth Edition 4th Edition. 2016 : 59-60.
23. Buja LM, Butany J. Cardiovascular Pathology, Elsevier, Fourth Edition 4th Edition. 2016 : 111-112.
24. Rosai J, Lascano EF. Basophilic (mucoid) degeneration of myocardium: a disorder of glycogen metabolism. Am J Pathol 1970; 61(1): 99–116.
25. Miao N, Levin SW, Baker EH, et al. Children with infantile neuronal ceroid lipofuscinosis have an increased risk of hypothermia and bradycardia during anesthesia. Anesth Analg 2009; 109(2): 372-378.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2019 Issue 3
Most read in this issue
- Cytological examination of cerebrospinal fluid
- Neuronal ceroid lipofuscinosis with cardiac involvement
- Pituitary adenomas – practical approach to the diagnosis and the changes in the 2017 WHO classification
- Atypical fibroxanthoma, rare and often unrecognized cutaneous soft tissue tumor – a case report and review of the literature