
Využití imunohistochemie při diagnostice renálních neoplázií
Immunohistochemistry and renal neoplasias
Tento přehledový článek stručně shrnuje možnosti využití imunohistochemie při vyšetřování především renálních karcinomů a základní molekulárně genetické znaky vybraných neoplázií. Článek však v žádném případě nelze brát jako univerzální návod pro diagnostiku renálních tumorů. Renální karcinomy dokážou mít velmi variabilní morfologický vzhled a to i v rámci jedné léze (nádorová heterogenita) a často velmi nepředvídatelný a neuniformní imunohistochemický profil. Některé renální neoplázie jsou diagnostikovány striktně na podkladě molekulárně-genetických vlastností, bez ohledu na morfologický vzhled.
Keywords:
immunohistochemistry – angiomyolipoma – chromophobe renal cell carcinoma – clear cell renal cell carcinoma – renal oncocytoma
Authors:
Kristýna Pivovarčíková; Květoslava Michalová
![]() ; Ondřej Hes
; Ondřej Hes
Authors‘ workplace:
Šiklův ústav patologie LF UK a FN Plzeň
Published in:
Čes.-slov. Patol., 56, 2020, No. 3, p. 130-139
Category:
Reviews Article
Overview
Tento přehledový článek stručně shrnuje možnosti využití imunohistochemie při vyšetřování především renálních karcinomů a základní molekulárně genetické znaky vybraných neoplázií. Článek však v žádném případě nelze brát jako univerzální návod pro diagnostiku renálních tumorů. Renální karcinomy dokážou mít velmi variabilní morfologický vzhled a to i v rámci jedné léze (nádorová heterogenita) a často velmi nepředvídatelný a neuniformní imunohistochemický profil. Některé renální neoplázie jsou diagnostikovány striktně na podkladě molekulárně-genetických vlastností, bez ohledu na morfologický vzhled.
Klíčová slova:
angiomyolipom – imunohistochemie – chromofóbní renální karcinom – světlobuněčný renální karcinom – renální onkocytom
Klasifikace renálních neoplázií doznala v posledních 40 letech významných změn a stále se velmi dynamicky mění. Prakticky neustále jsou publikovány práce popisující nové renální nádorové jednotky, zároveň se však začíná významně měnit i pohled a přístup k některým dlouhodobě uznávaným renálním neopláziím. V současné době platná WHO klasifikace (z roku 2016) odráží významné pokroky na poli morfologie, imunohistochemie, cytogenetiky a molekulární patologie a obsahuje několik desítek nádorových jednotek a čtyři tzv. provizorní jednotky (1).
Není účelem tohoto článku do detailu popsat všechny renální tumory - morfologické spektrum různých jednotek může být opravdu široké. Bez nadsázky lze říct, že typický imunohistochemický profil jednotlivých renálních karcinomů (RCC) neexistuje a stoprocentně se nelze spolehnout ani na molekulárně genetické vyšetření. Pozornost zde tak bude věnována pouze určitým diferenciálně diagnostickým okruhům RCC, které budou stručně komentovány. V žádném případě však nelze brát toto sdělení jako univerzální návod pro diagnostiku renálních neoplázií.
Na prvním místě by v tomto přehledovém článku mělo být zdůrazněno, že u drtivé většiny případů ke správné diagnóze renální neoplázie vystačí obyčejné barvení hematoxylinemeosinem a dostatečný sampling léze. Histologický nález zůstává nejdůležitějším diagnostickým kritériem, které pokud nedovolí určit diagnózu s definitivní platností, alespoň rozhoduje o dalším diagnostickém postupu. Nutné je však mít též na paměti, že některé renální tumory jsou dnes klasifikovány čistě na podkladě molekulárně genetických znaků (bez ohledu na přítomnost či absenci typických morfologických znaků). Molekulárně genetická analýza tak v urogenitální patologii začíná být součástí rutinní diagnostické praxe a je společně s imunohistochemií hojně využívána. Molekulárně genetickou analýzu a imunohistochemická barvení však nelze v běžné praxi užívat jako univerzální diagnostický nástroj a s tímto vědomím by k nim mělo být přistupováno. Tyto metody tak v žádném případě neřeší diagnostické dilema ve 100 % případů. Diagnostika části renálních neoplázií tedy rozhodně patří do rukou zkušeného urogenitálního patologa a laboratoře disponující výše jmenovanými diagnostickými modalitami.
MARKERY RENÁLNÍHO ORIGA
Patology je obecně používáno různé spektrum imunohistochemických markerů, ale asi mezi ty pro renální origo úplně nejčastější patří PAX2, PAX8, RCC, CD10 a vimentin. Při indikaci těchto a jiných barvení je třeba vždy myslet na jejich nízkou specificitu pouze pro renální origo. Též bývá pravidlem, že neobvyklé varianty a „podivné“ RCC obvykle mívají stejně tak neobvyklý a podivný imunohistochemický profil, a že imunohistochemický profil metastázy RCC může být zcela odlišný od tumoru primárního.
PAX2 kromě renálního origa (senzitivita 56 % u primárních RCC a 79 % u metastáz RCC (2)) může vykazovat též pozitivitu např. u tumorů i nenádorových lézí Mülleriánského origa (3), karcinomu příštítného tělíska či světlobuněčného karcinomu ovaria (4). PAX8 je podle doporučení ISUP považován za patrně nejlepší marker renálního origa (5), se sensitivitou až 95 % u primárních RCC (6), ale ani toto barvení se nevyznačuje specificitou. Nukleární pozitivita PAX8 je též často využívána k potvrzení origa ovariálního karcinomu či karcinomu štítné žlázy (7). S PAX8 pozitivitou též nutno počítat u B-lymfocytů a B-buněčných lymfomů (zkřížená pozitivita s PAX5) a velkého množství dalších (viz tab. 1) (8). Imunohistochemický marker RCC (Renal Cell Carcinoma marker) vykazuje membránovou a cytoplasmatickou pozitivitu a v publikacích je uváděna sensitivita přes 79 % u primárních RCC a 56 % u metastáz RCC (9). I přes velmi sugestivní název však nutno počítat s pozitivitou tohoto markeru nejen u RCC (pozitivita popsána u karcinomů prsu, embryonálního karcinomu a jiných lézí (9)). CD10 je další oblíbený marker renálního origa, reaguje membránově a kromě RCC vykazuje pozitivitu i u velkého množství různých jiných malignit i nenádorových lézí (ve velmi zkráceném výčtu – různé typy lymfomů, leukémií, uroteliální karcinom, hepatocelulární karcinom, adenokarcinom prostaty atd.). Stejně tak velkou nespecifitou je znám i vimentin.
RENÁLNÍ KARCINOMY SPOJENÉ S PAPILÁRNÍ MORFOLOGIÍ
Renální karcinomy rostoucí pod obrazem papilární léze mají sice překryvné charakteristiky morfologické (tj. papilární růst), avšak odlišné vlastnosti imunohistochemické, molekulárně genetický profil a různé klinické chování. Mezi tyto léze lze ve zkráceném výčtu zařadit papilární renální karcinom (PRCC), renální karcinom asociovaný se syndromem familiární leiomyomatózy a karcinomem ledviny (HLRCC)/fumaráthydratáza deficientní renální karcinom (FHRCC), světlobuněčný papilární renální karcinom (CCPRCC), Xp11.2 translokační renální karcinom (Xp11 TRCC) a metanefrický adenom (MA).
U PRCC je dnes již oproti původnímu smýšlení a dělení (tradičně typ 1 a 2 (10)) evidentní, že PRCC představuje velmi heterogenní skupinu, s neustále expandujícím morfologickým spektrem. PRCC typicky exprimuje cytokeratiny (AE1/AE3, CAM5.2, HMWK), membránový epitelový antigen (EMA), α-methylacyl-CoA-racemasu (AMACR), vimentin a CD10 (1). Variabilní je reaktivita pro CK7, kdy pozitivita CK7 je častější u PRCC typ 1, u PRCC typ 2 se pozitivita pohybuje okolo 50 % případů (11). Za typické molekulárně genetické znaky PRCC jsou tradičně považovány trisomie/polysomie chromosomů 7 a 17 a ztráta gonosomu Y u mužských pacientů (1). Avšak v současné době je zřejmé, že toto všeobecně uznávané dogma je nepravdivé a zůstává v platnosti snad pouze u PRCC typu 1 (12-19).
Renální karcinom asociovaný se syndromem familiární leiomyomatózy a karcinomem ledviny (HLRCC)/fumaráthydratáza deficientní renální karcinom (FHRCC) je nádor s širokým morfologickým spektrem zahrnujícím tumory predominantně s papilární architektonikou, typicky smíšené s jiným růstovým typem (cystický, tubulární, tubulolopapilární, solidní) (20,21), či nádory tubulocystické (22,23). Velká část tumorů v literatuře publikovaných jako HLRCC/FHRCC byla původně klasifikována jako renální karcinomy blíže nespecifikované (RCC NOS dle WHO) (24). V minulosti byl u těchto nádorů popisován jako typický morfologický znak přítomnost eosinofilních makrojadérek s perinukleolárním projasněním. Dnes je jasné, že tento znak není při diagnostice nikterak nápomocný, neboť nemusí být přítomen (25), či je naopak často přítomen i u jiných RCC (21, 26). Pro nádory je tak typická a definující pouze inaktivační mutace genu pro FH (1,27). Tato mutace vede ke kompletní ztrátě či redukci aktivity enzymu fumarát hydratasy, která je za normálních okolností zodpovědná za konverzi fumarátu na malát během Krebsova cyklu (28-30). Z toho resultující zvýšené množství fumarátu vede k modifikaci buněčných enzymatických a látkových pochodů, kde jedním z důsledků je i zvýšená produkce S-(2-sukcino)-cysteinu (2SC). Akumulace 2SC vede k pozitivním výsledkům imunohistochemických barvení na 2SC (31,32), naopak ztráta enzymatické aktivity FH způsobuje negativitu imunohistochemického barvení na FH (imunohistochemický profil 2SC+/FH-) (24,31). Imunohistochemická protilátka proti 2SC je komerčně nedostupná, vzhledem k raritě těchto lézí a tedy ne příliš častému užití protilátky je obtížná i interpretace výsledku vyšetření, její využití v rutinní praxi je tedy limitováno. Protilátka proti FH je sice běžně dostupná, avšak četné práce jasně dokazují její významnou nespolehlivost a to ve smyslu jak falešně negativních tak falešně pozitivních výsledků (21,26). Definitivní diagnóza musí být potvrzena molekulárně genetickým vyšetřením s průkazem mutace/ztráty heterozygozity genu FH a to i u případů s nespecifickým imunoprofilem a sugestivní morfologií a klinickým průběhem (mladý pacient s agresivním tumorem).
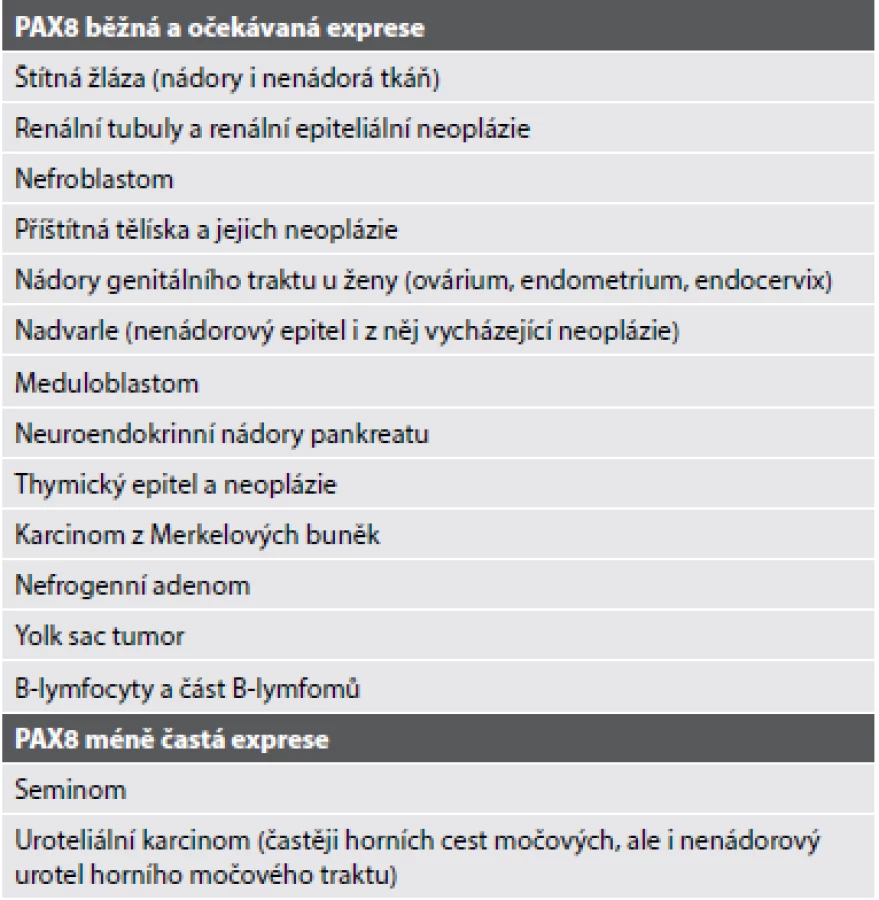
Světlobuněčný papilární renální karcinom (CCPRCC) se skládá z blandně vyhlížejících cylindrických buněk s objemnou „clear“ cytoplasmou a jádrem s low-grade morfologií, lehce nadzdviženým od bazální membrány. Tumor roste pod obrazem papilární, tubulární (Obr. 1) či tubulopapilární léze. Imunohistochemicky tyto nádory vykazují difúzní pozitivitu v CK7, HMWK a vimentinu. Karboanhydráza IX (CA-IX) je pozitivní v typické v tzv. cup-like (bazolaterální) distribuci. Naopak negativní jsou nádorové buňky v racemáze (AMACR) a TFE3. Variabilní reaktivitu lze prokázat v CD10 (1,33,34). CCPRCC byl dlouhou dobu považován za tumor bez molekulárně genetických aberací typických ať už pro CCRCC, či pro PRCC (33). U ojedinělých v literatuře popsaných případů však byly detekovány abnormality genu VHL (34,35). Podle našeho názoru by však léze s nejednoznačnou morfologií, ne příliš typickým imunohistochemickým profilem a se současně prokázanou abnormalitou VHL genu neměly být klasifikovány jako CCPRCC (36).
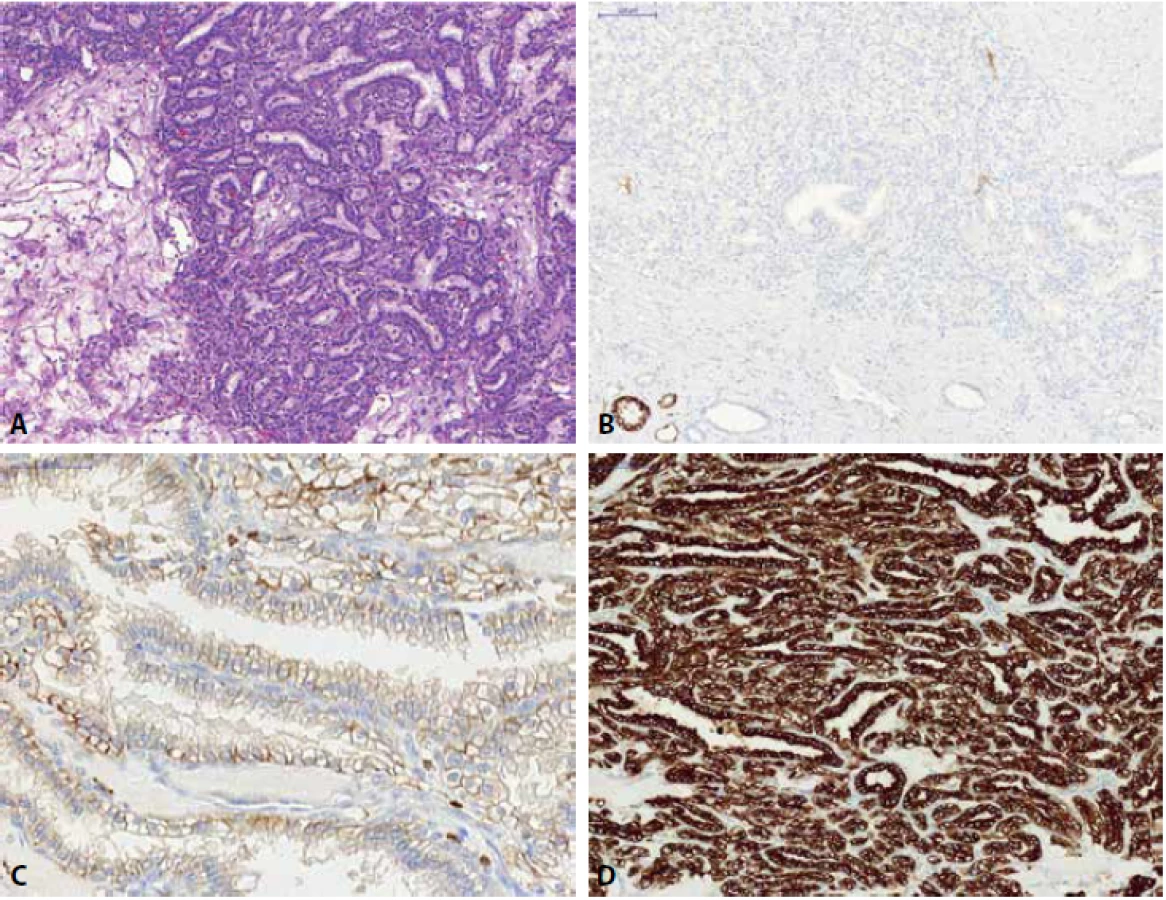
Xp11.2 translokační renální karcinom (Xp11 TRCC) je renální neoplázií, kde přítomnost translokace transkripčního faktoru TFE3 (se širokým spektrem genů partnerských za vzniku různých fúzních genů) je charakteristikou tuto nádorovou jednotku definující (37-40). Pro nádory je typická velká variabilita morfologického vzhledu, klasicky je však popisována papilární architektonika, buňky s jasnou a eozinofilní objemnou cytoplasmou, psammomatózní tělíska, hyalinní globule, krevní lakuny. Imunohistochemický profil není konstantní, nádory obyčejně neexprimují cytokeratinové markery, jsou pozitivní v PAX8, někdy jsou reaktivní v průkazu melanomových markerů (Melan A, HMB45), část nádorů je cathepsin K pozitivní. Imunohistochemický průkaz proteinu TFE3 (jaderná reaktivita) je problematický, nevykazuje konstantní pozitivitu (ovlivněn fixací materiálu, vykazuje falešně pozitivní i negativní výsledky) a je tedy nutno k jeho interpretaci přistupovat obezřetně a finální diagnózu konfirmovat molekulárně genetickým vyšetřením zlomu TFE3 pomocí vyšetření FISH (41).
Metanefrický adenom (MA) se typicky skládá s těsně nahloučených malých uniformních acinů, zhruba polovina případů však může obsahovat papilární okrsky (1) a nádor tak morfologicky může významně připomínat PRCC typ 1 se solidním způsobem růstu (5). MA je však oproti PRCC difúzně pozitivní v barvení WT1 a většinou CD57. Cytokeratin CK7 může být u MA fokálně pozitivní, avšak negativní je nádor v barvení racemázou a EMA (1). Nedávno byla též prokázána užitečnost imunohistochemického barvení s BRAF V600E (88% senzitivita a 100% specificita v barvení MA (42)). Pro MA též není typická polysomie chromozomů 7 a 17 a ztráta chromozomu Y (5,43).
RENÁLNÍ KARCINOMY SE SVĚTLOBUNĚČNOU MORFOLOGIÍ
Na prvním místě je zde potřeba zmínit nejčastější renální tumor vůbec – světlobuněčný renální karcinom (CCRCC). Do této skupiny spadá i nádor, který má s CCRCC překryvný genetický profil – multilokulární cystická renální neoplázie nízkého maligního potenciálu a lze zde zařadit i provizorní jednotku současné WHO – renální karcinom s leiomyomatózním stromatem (RCCLS). Další jednotky v této diferenciálně diagnostické skupině se překrývají s neopláziemi uvedenými výše – mezi ně patří jak CCPRCC, Xp11 TRCC tak i PRCC, který též může vykazovat výrazné světlobuněčné změny.
Světlobuněčný renální karcinom (CCRCC) má typicky acinární či alveolární uspořádání, buňky se světlou cytoplazmou a na pozadí relativně pravidelnou jemnou síť kapilár. Morfologie těchto RCC však může být i velmi různorodá, mohou obsahovat low-grade vřetenité buňky, “syncitiální” obrovské buňky, produkci mucínu, high-grade okrsky s emperipoézou, buňky podobné buňkám Panethovým, mohou být tvořeny buňkami s eosinofilní cytoplazmou (granulární varianta CCRCC) či vzácně mohou vykazovat i papilární způsob růstu. Hlavním klíčem pro správnou diagnózu je u CCRCC dostatečný sampling léze, neboť typické morfologické znaky mohou být v tumorózní mase vyjádřeny pouze fokálně. CCRCC je typicky pozitivní v barvení vimentinem a CA-IX. CK 7 je často vnímán jako marker, který je u CCRCC negativní, není tomu ovšem tak. Recentní práce ukazuje, že CK7 může být pozitivní až u 66 % low-grade CCRCC (Obr. 2) (u CCRCC s high-grade morfologií a cysticky změněných CCRCC je pozitivita CK7 udávána s nižší frekvencí – u 6 %, resp. 25 % takových lézí) (44). Mokelulárně genetické znaky typické pro CCRCC jsou abnormality VHL genu (mutace, hypermethylace, LOH3p).
Multilokulární cystická renální neoplázie nízkého maligního potenciálu (MCRNLMP) je novou jednotkou WHO 2016, kompletně sestávající z cystických prostor lemovaných low-grade buňkami se světlou cytoplazmou, bez přítomnosti solidních okrsků. Tyto léze vykazují pozitivitu CK7 (44) až v 92 % případů (45). Nádor je též pozitivní v CD10 (63 %), racemáze - AMACR (21 %), vimentinu (58 %), CA-IX (100 %) (45). Genetický profil je stejně jako ten imunohistochemický překryvný s CCRCC, diagnóza (a odlišení od CCRCC) je tak plně závislá pouze na morfologickém nálezu – absenci solidních nádorových okrsků a vyžaduje tak důkladný sampling léze.
Relativně blízko k dříve zmíněné skupině CCPRCC stojí tzv. renální karcinom s objemným leiomyomatózním stromatem (RCCLS), v současné WHO uváděný jako „provizorní“ jednotka. Nádory mají epitelovou komponentou prakticky totožnou se světlobuněčným renálním karcinomem a typicky obsahují objemné hladkosvalové stroma. Renální karcinomy s objemným leiomyomatózním stromatem jsou poměrně heterogenní skupinou nádorů, kde většinu tumorů představuje klasický CCRCC, který pouze produkuje objemné stroma. Malá část tumorů této skupiny je tvořena nádory, které reagují pozitivně s CK7 a nemají abnormality ve VHL genu. Ještě menší část představují morfologicky identické nádory, které vykazují mutaci v TCEB1 genu. Pouze malá část těchto lézí nemá prokázány abnormality ani v VHL, ani v TCEB1 genu (46). Shrnout lze vše následujícím způsobem: máme-li nádor se světlobuněčnými elementy a objemným hladkosvalovým stromatem, je vhodné barvit CK7. Je-li léze negativní, popř. slabě či fokálně pozitivní, statisticky pravděpodobněji jde o CCRCC. Jde-li o lézi difuzně CK7 pozitivní, spíše bychom měli myslet na TCEB1 mutovaný RCC či skutečný RCC s objemným leiomyomatózním stromatem (47). Je nutno říci, že jde o okrajovou část renální patologie.
PRCC se světlobuněčnými změnami je subtyp PRCC s typickou papilární architektonikou, kde papilární struktury jsou lemovány buňkami se světlou cytoplasmou. Některé ze studií věnujících se těmto tumorům připisují světlobuněčný vzhled degenerativním změnám v tumoru, krvácení a/nebo nekróze (48,49). Většina prací popisujících tuto jednotku však po provedení molekulárně genetických vyšetření shledala většinu popsaných tumorů i přes často excesivně vyjádřenou papilární architektoniku za CCRCC (49-52). Jen ojedinělé případy RCC s papilární architektonikou a buňkami se světlou cytoplazmou tak patrně budou opravdovými PRCC. Studie přesně definující diagnostické modality a shrnující biologické chování nejsou v současné době v literatuře k dispozici.
RENÁLNÍ KARCINOMY S ONKOCYTÁRNÍ MORFOLOGIÍ
Do této skupiny lze zařadit onkocytický papilární renální karcinom, renální onkocytom (RO), chromofobní renální karcinom (ChRCC), „granulární“ variantu světlobuněčného renálního karcinomu (CCRCC), eozinofilní solidní a cystický renální karcinom (ESC RCC), sukcinátdehydrogenáza deficientní renální karcinom (SDHB-RCC), t(6;11) translokační renální karcinom (t(6;11)TRCC), tzv. hybridní onkocytom-chromofóbní tumory (HOCT), ale morfologickým spektrem do této skupiny může spadat i HLRCC/FHRCC. Recentně jsou pak v literatuře popisovány i neoplázie jako low-grade onkocytický tumor (LOT) (53), či high-grade onkocytický tumor (HOT) (54,55), které si též rozhodně zaslouží být zmíněny. Ač se to může zdát nepravděpodobné, v této skupině bude zmíněn i epiteloidní angiomyolipom (eAML), který svou morfologií může někdy připomínat RCC.
Onkocytický PRCC (OPRCC) je dobře známá morfologická varianta PRCC, u nějž jsou papily lemovány buňkami s hojnou granulární a výrazně eozinofilní cytoplasmou a obyčejně nízkým nukleárním gradem. Imunohistochemický profil těchto lézí vykazuje typicky pozitivitu vimentinu, AMACR a v barvení protilátkou proti mitochondriálnímu antigenu (MIA). CK7 je v pozitivitě variabilní. Zisk chromosomů 7 a 17 je dle dostupných studií nejčastěji detekovanou molekulárně genetickou změnou (56-61), i když významná část OPRCC (dle některých studií až 43,5 % OPRCC (62)) vykazuje disomický status chromosomů 7 a 17. Mezi další chromozomální aberace patří zisk chromosomů 3 a 11, ztráty chromosomů 1, 4, 11, 14 a gonosomu X a Y (56-61). U části tumorů pak byla explicitně detekována delece chromosomu 14, změny v genu CCND1, delece 1p (locus 1p36) a ztráta chromosomu Y (62, 63), přičemž tyto změny jsou typicky popisovány i u renálního onkocytomu (RO). Je tedy zřejmé, že OPRCC a RO mají z části překryvný chromozomálně aberační genotyp a molekulárně genetická analýza má v diferenciální diagnostice těchto lézí jen velmi omezený význam.
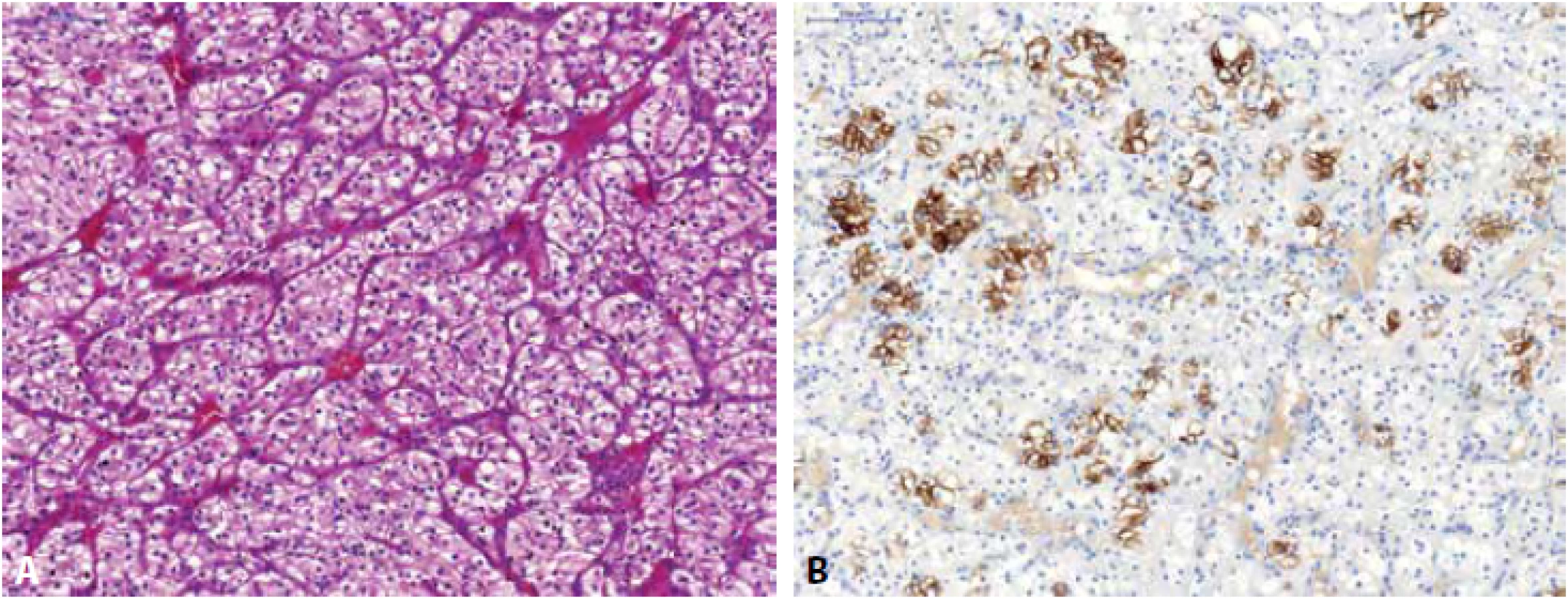
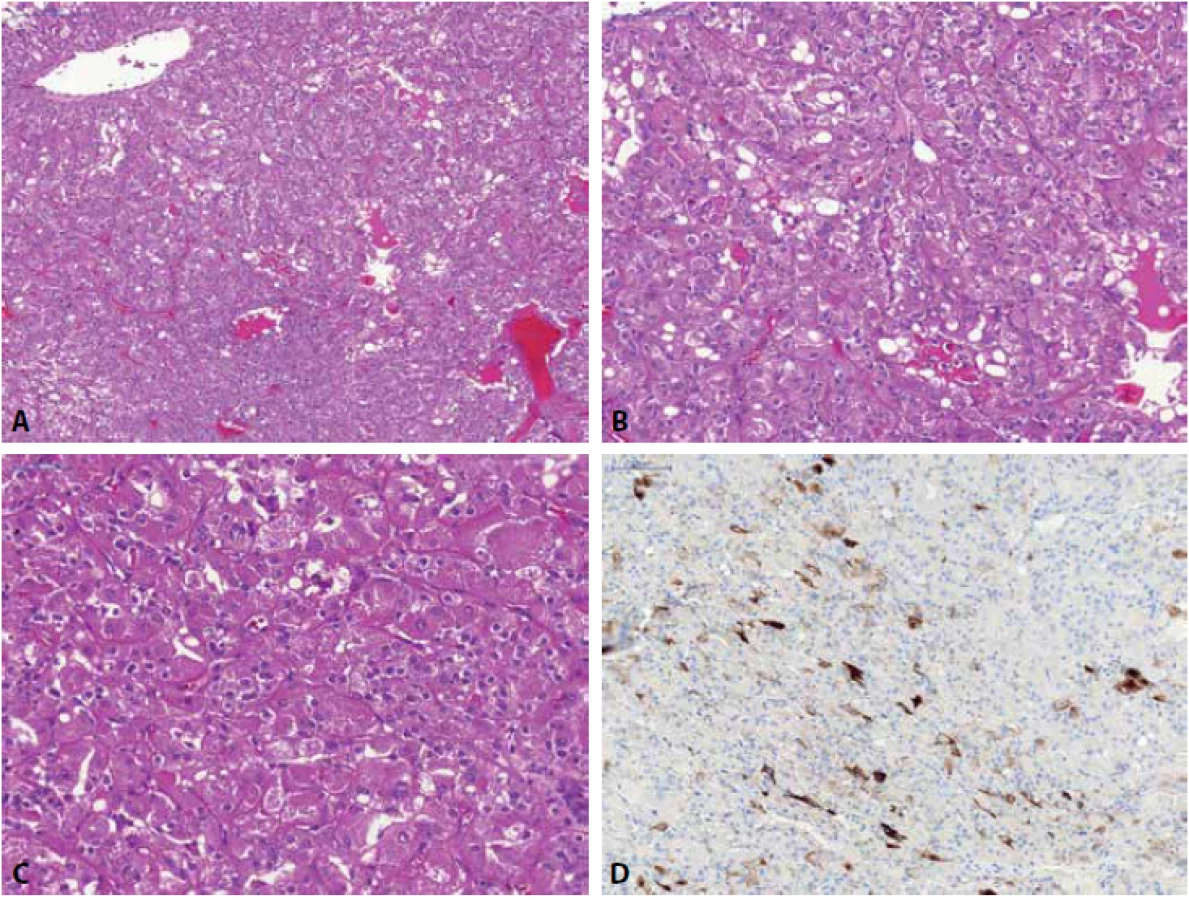
Renální onkocytom (RO) může mít variabilní morfologický vzhled, nejčastěji je však popisován jako nádor složený z tzv. onkocytů (eozinofilní okrouhlé buňky s denzně granulární cytoplazmou), které jsou uspořádány v solidní hnízda, typické jsou okrsky, kde onkocytické buňky rostou v řídké hypocelulární pojivové tkáni (Obr. 3) (1). RO je difúzně silně pozitivní v barvení MIA (64), toto však není pravidlem (např. malobuněčná varianta RO může reagovat velmi chabě (65) či být negativní (66)). Stejně tak i pozitivita S-100A1 je udávána ve velkém procentu případů RO a je tak některými autory doporučována k odlišení od ChRCC (ChRCC je obvykle S-100A1 negativní) (67), avšak malobuněčná varianta RO bývá popisována též jako S-100A1 negativní (66). Cytokeratin CK7 a vimentin jsou v RO obvykle negativní, ale není ani výjimkou, že tumor může fokálně reagovat pozitivně (68) (s CK7 obvykle reagují roztroušené individuální buňky či skupiny buněk a to silnou pozitivitou (69), též centrální jizevnaté okrsky RO mohou vykazovat reaktivitu jak v CK7, tak i ve vimentinu (70,71)). CD117 je u RO membránově pozitivní (5). Molekulárně genetický profil rozděluje RO do různých cytogenetických kategorií – RO může mít normální karyotyp, některé RO mají aneuploidický chromozomální status se ztrátou chromozomu 1 (celý chromozom, nebo jen jeho část), kombinovaný se ztrátou chromosomu Y či X a/nebo chromosomu 14 a 21 a u některých RO lze detekovat translokaci lokusu 11q13 s alterací CCND1 a overexpresí cyclinu D1 (72-74).
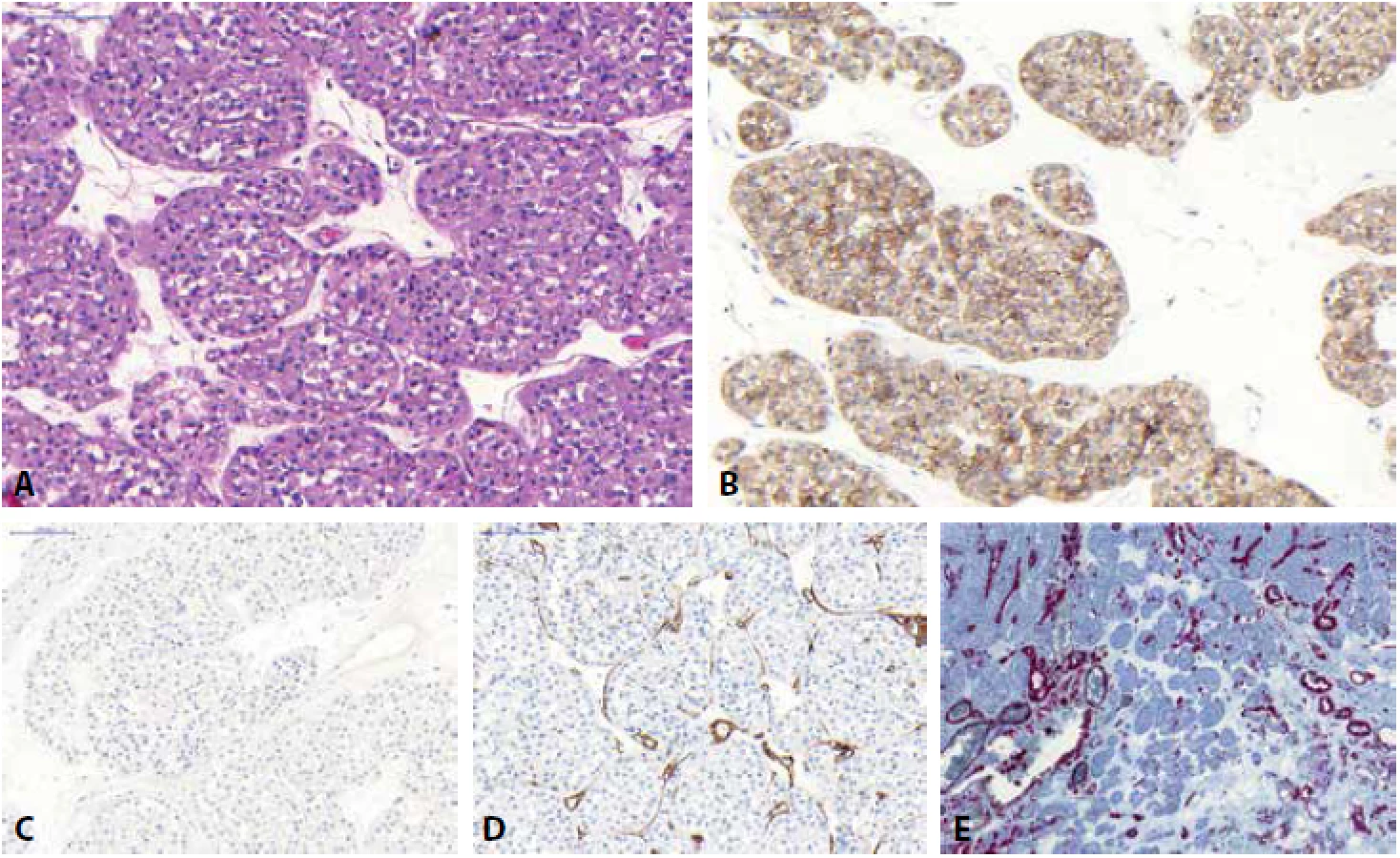
U chromofóbního renálního karcinomu (ChRCC) jsou klasicky rozeznávány dvě hlavní morfologické varianty (klasická a eozinofilní), morfologické spektrum této neoplázie je ale mnohem širší (v literatuře jsou dokumentovány případy adenomatoidního mikrocystického pigmentovaného ChRCC, tzv. onkocytické varianty ChRCC, ChRCC s neuroendokrinní diferenciací či multicystický ChRCC (75-83)). ChRCC tak může být predominantně tvořený velkými slabě eosinofilními buňkami, které jsou typické pro klasickou variantu ChRCC, naopak menší oxyfilní buňky s granulární cytoplazmou bývají u eosinofilní varianty ChRCC (1). Jádra jsou obvykle velká s nerovnými okraji (tzv. raisinoidní jádra), perinukleárním projasněním (halo efekt) a časté jsou i binukleární buňky (Obr. 4). ChRCC je pozitivní v CD117, EMA, CK8, CK18 a obvykle v parvalbuminu. Cytokeratin CK7 je difúzně pozitivní, nikoliv však ve 100 % případů a na toto pak nelze spoléhat především u eosinofilní varianty ChRCC, kde CK7 bývá často pozitivní jen fokálně, nebo může být i zcela kompletně negativní (5, 84). Difúzní reaktivita CK7 však podle většiny autorů favorizuje diagnózu ChRCC (84). Vimentin je obecně negativní, vzácně však může vykazovat ojedinělou reaktivitu (stejně jako u RO) (68). Obecně lze říci, že ChRCC morfologie je podmíněna řadou fixačních artefaktů (raisinoidní jádra, perinukleární haló) a je tedy nutno počítat s promítnutím tohoto faktu i do výsledků imunohistochemických vyšetření. Chromozomálně aberační status u ChRCC vykazuje mnohočetné ztráty postihující chromozomy 1, 2, 6, 10, 13, 17, 21 či Y (85-87), ale byly dokumentovány i případy se ziskem chromosomů 4, 7, 15, 19, 20 (87), přičemž tyto „gainy“ bývají častěji detekovány u ChRCC se sarkomatoidní dediferenciací (změny s větší frekvencí detekovány právě v sarkomatoidní nádorové komponentě) (87,88). Popsány byly i případy ChRCC s disomickým profilem (87).
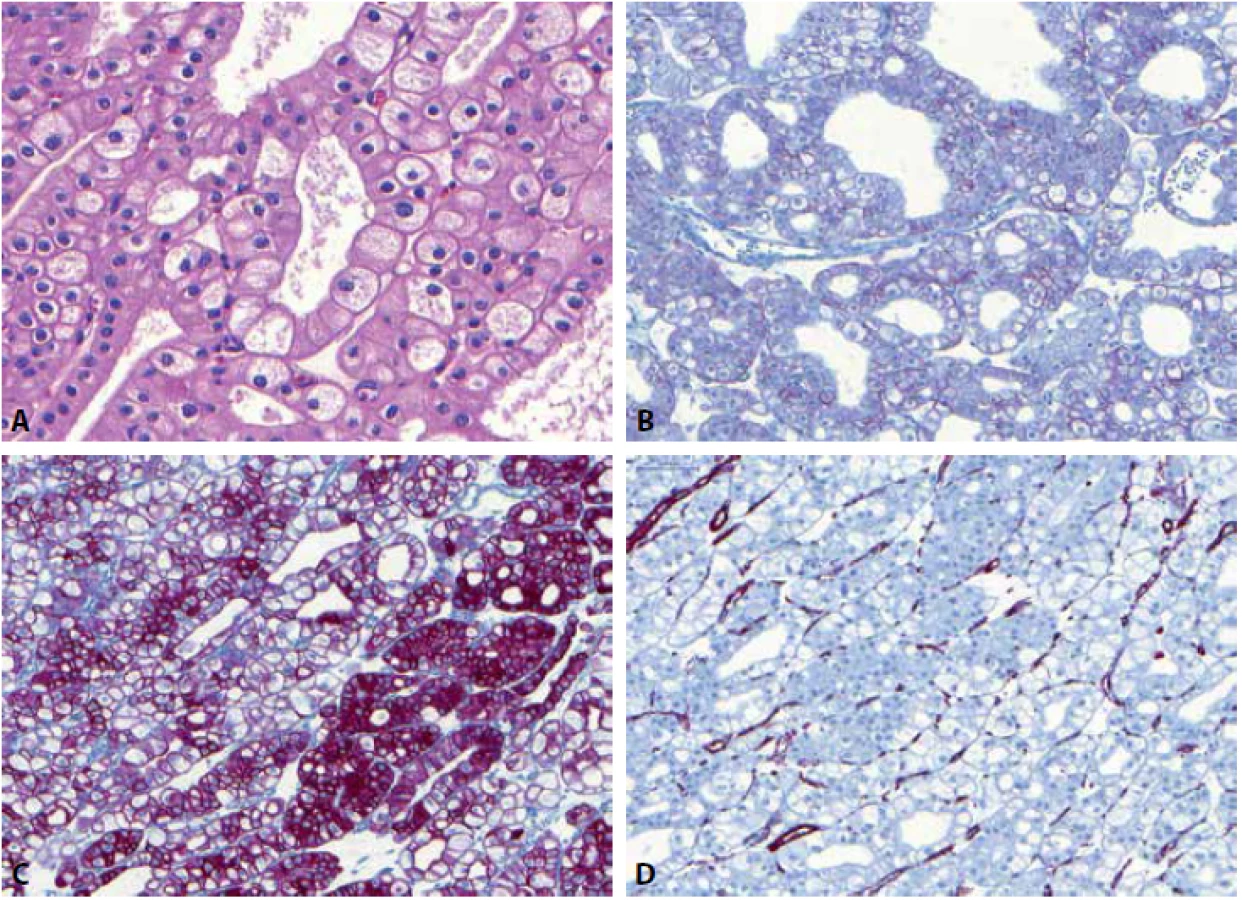
Eozinofilní solidní a cystický renální karcinom (ESC RCC) je recentně popsaná nádorová jednotka (89), která prozatím není zahrnuta ve WHO 2016, avšak je jednoznačně přijata komunitou urogenitálních patologů. Většina případů byla popsána u žen (90), jen ojedinělé případy byly dokumentovány u mužů (25,47). Případy ESC RCC se mohou vyskytovat u pacientů s tuberózní sklerózou, většina případů však vzniká sporadicky. Typicky je tato neoplázie spojována s indolentním klinickým průběhem, v literatuře jsou ale popsány i případy s metastatickým potenciálem (25,91). Jak je již z názvu této jednotky patrné, jedná se o neoplázii s okrsky solidního a cystického uspořádání (různě velké makro - a mikrocystické okrsky). Buňky mají objemnou eozinofilní cytoplazmu a hrubá až tečkovitá cytoplasmatická granula (granula jsou hrubá do té míry, že mohou připomínat až obraz při leishmaniové infekci – Obr. 5). Jádra jsou často iregulární s prominentními jadérky. Buňky jsou pozitivní v imunohistochemickém průkazu PAX8, AE1/3, CK8/18 a vimentinu. U ESC RCC je přítomna pro renální tumory relativně neobvyklá, avšak pro tento podtyp tumoru zcela charakteristická pozitivita cytokeratinu CK20 ve většině případů, zatímco CK7 je obvykle negativní či pozitivní jen fokálně (47,89,90). CD117 je negativní. Karyotyp těchto lézí vykazuje variabilní genomické alterace (90), četné studie prokázaly somatickou bialelickou ztráta TSC1/TSC2 genu u většiny ESC RCC (92-95).
Sukcynátdehydrogenáza deficientní renální karcinom (SDHRCC) je definován jako SDH deficientní neoplázie se ztrátou imunohistochemického barvení protilátkou proti SDHB (1) (protilátka SHDB detekuje ztrátu všech SDH podtypů). Mikroskopicky nádor většinou sestává z low-grade neoplastických buněk s eosinofilní jemně zrnitou cytoplazmou a cytoplazmatickými vakuolami (47,96) a kromě definující negativity SDHB imunohistochemicky je obvykle negativní v barvení CA-IX, CD117, většinou negativní CK7, CK20, AE1/3, CK8/18. Difúzně pozitivní je SDHRCC v PAX8, CD10 a EMA (96,97).
t(6;11)/TFEB translokační renální karcinom (t(6;11) TRCC) je v typické morfologii nádor diagnostikovatelný jednoduše i pouze z hematoxylinu a eosinu (bifazicky vypadající tumor s hnízdy větších epiteloidních buněk kombinovaných s klastry menších buněk obklopujících globulární eosinofilní materiál – tzv. pseudorosety (1)). Bohužel, existuje řada morfologických variant, kdy nejsou vyjádřené pseudorozety, epitelová komponenta je spíše světlobuněčná či lehce eosinofilní. Imunohistochemicky většina nádorů exprimuje HMB45, Melan A a kathepsin K. V případě atypické morfologie či imunofenotypu je nutné diagnózu podpořit molekulární genetikou (průkaz zlomu genu TFEB). U potvrzených případů pak dále nutno provést vyšetření amplifikace TFEB genu, neboť TFEB TRCC s touto amplifikací vykazují agresivní chování (98-100).
Hybridní onkocytom-chromofóbní tumory (HOCT) jsou ve WHO zmiňovány v kapitole chromofóbních renálních karcinomů jako onkocytom připomínající eosinofilní ChRCC (1). Jedná se o typ tumoru s překryvnými morfologickými charakteristikami RO a ChRCC – překryvným/smíšeným morfologickým vzhledem a imunoprofilem. Molekulárně geneticky a klinickým chováním se HOCT zdají být rozdílné od RO a od ChRCC (101). Hybridní onkocytom-chromofóbní tumor se může vzácně vyskytovat sporadicky (jako solitární neoplázie), častěji však vzniká jako multifokální tumor v souvislosti s renální onkocytózou či u pacientů s Birt-Hogg-Dubé syndromem. Tumory často připomínají RO ovšem s perinukleárním cytoplazmatickým projasněním, bez raisinoidních jader. U těchto tumorů byla prokázána pozitivita barvení v CK7, AE1/3, EMA, MIA a fokální pozitivita vimentinu; CD117 bylo slabě či alespoň fokálně exprimováno ve většině případů. Naopak negativní byly nádory v barvení CK20 (102). Molekukárně genetické znaky této jednotky byly prozatím popsány jen v ojedinělých studiích s částečně konfliktními výsledky (101-103).
Již takto nesnadná situace na poli diferenciální diagnostiky onkocytických tumorů je ještě dále komplikována recentně velmi diskutovanými jednotkami – low-grade onkocytický tumor (LOT) a high-grade onkocytický tumor (HOT). Low-grade onkocytický tumor (dříve nejspíše též popsán jako „ChRCC, onkocytární varianta“ (77)) je charakterizován jako CD117-/CK7+ tumor s indolentním chováním. Nádor je tvořen uniformní populací buněk s low-grade vzhledem, bez jaderných iregularit, fokálně s perinukleárním projasněním a relativně ostře oddělenými hypocelulární okrsky s řídce rozloženými nádorovými buňkami vytvářející trámce, řídkou síť či s individuálně roztroušenými buňkami (47) (tyto okrsky jsou morfologicky odlišné od okrsků typicky přítomných u RO) (53). Kromě typického imunoprofilu CD117-/CK7+ jsou nádory pozitivní v barvení AE1/3, PAX8, BerEP4 a MOC31 a negativní v CA-IX, CK20, vimentinu, HMB45 a MELAN A (53). U této jednotky nebyl prokázán konsistentní chromozomální aberační status (bez pevně prokázaných chromozomální ztrát a zisků) (53). High-grade onkocytický tumor sestává z buněk s objemnou eosinofilní či jasnou cytoplazmou s intracytoplazmatickými vakuolami. Jádra vykazují high-grade morfologii - jsou kulatá, s prominentními jadérky (WHO/ISUP grade 3) a většinou s hladkou konturou, bez raisinoidních změn (54). Nádor je ve většině pozitivní v průkazu AE1/3, CK18, PAX8, MIA, CD10, kathepsin K a negativní v TFE3, HMB45, MELAN A a vimentinu. Variabilní je reakce s CD117 a CK7 (většina HOT je CD117 pozitivní a CK7 negativní/jen fokálně pozitivní) (54). I přes high-grade morfologii nebylo u HOT dokumentováno agresivní chování. U tumoru nebyly prokázány kompletní chromozomální zisky ani ztráty, ojedinělé případy prokázaly ztrátu části chromozomů (data však velmi limitovaná malým počtem případů) (54). Recentní práce popisující tento tumor prokázala časté mutace v TSC1/TSC2 a mTOR (104).
Epiteloidní angiomyolipom (eAML) může být predominantně tvořen buňkami s hojnou eosinofilní cytoplazmou a výraznými cytologickými atypiemi. Nádor je na rozdíl od většiny RCC negativní v expresi PAX8 a pozitivní v průkazu HMB45, MELAN A (ale pozor, cytoplazmatická reaktivita může být jen fokální a slabá), či kathepsinu K (8).
UROTEL VERSUS RENÁLNÍ ORIGO?
Uroteliální karcinom (UC) postihující horní močový trakt dokáže velmi věrně napodobit primární renální karcinom a naopak. V ideálních případech je autory doporučován panel protilátek PAX8, GATA3 a p63 (u RCC PAX8+/GATA3-/p63-, naopak u UC PAX8-/GATA3+/p63+). Jedná se však o ideální případ, který v praxi samozřejmě úplně dobře nefunguje. Při indikaci a hodnocení imunohistohemických barvení je třeba myslet na to, že PAX8 vykazuje pozitivitu u 15 – 30 % UC horních močových cest (pozitivita může být silná a difúzní), včetně nenádorového urotelu (7, 105). Některé typy RCC jsou též GATA3 pozitivní a to ve velkém procentu případů (ChRCC 51 % (106), RO 17 % (106), CCPRCC 76 % případů (107)). Spíše než na imunohistochemický profil tak doporučujeme klást důraz na morfologii léze a především dostatečné zpracování materiálu/množství bloků, se zaměřením na oblast renální pánvičky (ve snaze najít uroteliální lézi – ať už uroteliální CIS či papilární uroteliální neoplázii). My, na základě našich zkušeností, v praxi využíváme kombinaci CK7, vimentin, GATA3. Pokud je vyšetřovaná léze CK7, GATA 3 pozitivní a vimentin negativní, spíše se kloníme k uroteliálnímu origu. Léze vimentin pozitivní a GATA3 negativní budou pak nejspíše origa renálního.
ZÁVĚR
Morfologie v diagnostice renálních lézí zůstává základním a hlavním diagnostickým kritériem. Klíčem pro správnou diagnózu je především nezapomínat na základní pravidlo při zpracování vzorku - dostatečně rozsáhlé zpracování materiálu (sampling), neboť některé typické morfologické znaky mohou být vyjádřeny pouze fokálně a lze je tedy zastihnout právě pouze při dostatečném až excesivním zablokování materiálu. V ideálních případech je molekulární genetika společně s imunohistochemií nemalou a velmi cennou pomocnou modalitou, tyto však patří spíše do rukou zkušeného uropatologa než do rutinní praxe.
Ačkoliv výše popsaná expanze morfologického a molekulárně genetického spektra renálních neoplázií zůstává prozatím bez přímého klinického dopadu (neboť léčebný algoritmus se u různých renálních nádorů t.č. zásadně neliší) v budoucnu a s rozvojem cílené léčby, by toto mělo nabýt na významu a mít klinické opodstatnění. Z pohledu patologa je pak znalost morfologického spektra a variabilit neodmyslitelnou součástí každodenní praxe a snad i zárukou správné diagnózy.
PODĚKOVÁNÍ
Podpořeno programem rozvoje vědních oborů Karlovy univerzity (Projekt Q39) a MZ ČR RVO (Fakultní nemocnice Plzeň – FNPl, 00669806).
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
∗ Adresa pro korespondenci:
MUDr. Kristýna Pivovarčíková, Ph.D.
Šiklův ústav patologie LF UK a FN Plzeň
Alej Svobody 80, 30460 Plzeň
tel.: +420377404633
e-mail: pivovarcikovak@fnplzen.cz
Sources
1. Moch H, Humphrey PA, Ulbright TM, Reuter VE. WHO classification of tumours of the urinary system and male genital organs. Lyon: IARC; 2016.
2. Wasco MJ, Pu RT. Comparison of PAX-2, RCC antigen, and antiphosphorylated H2AX antibody (gamma-H2AX) in diagnosing metastatic renal cell carcinoma by fine-needle aspiration. Diagn Cytopathol 2008; 36(8): 568-573.
3. Ozcan A, Liles N, Coffey D, Shen SS, Truong LD. PAX2 and PAX8 expression in primary and metastatic mullerian epithelial tumors: a comprehensive comparison. Am J Surg Pathol 2011; 35(12): 1837-1847.
4. Gokden N, Gokden M, Phan DC, McKenney JK. The utility of PAX-2 in distinguishing metastatic clear cell renal cell carcinoma from its morphologic mimics: an immunohistochemical study with comparison to renal cell carcinoma marker. Am J Surg Pathol 2008; 32(10): 1462-1467.
5. Reuter VE, Argani P, Zhou M, Delahunt B, Members of the IIiDUPG. Best practices recommendations in the application of immunohistochemistry in the kidney tumors: report from the International Society of Urologic Pathology consensus conference. Am J Surg Pathol 2014; 38(8): e35-49.
6. Sangoi AR, Karamchandani J, Kim J, Pai RK, McKenney JK. The use of immunohistochemistry in the diagnosis of metastatic clear cell renal cell carcinoma: a review of PAX-8, PAX-2, hKIM-1, RCCma, and CD10. Adv Anat Pathol 2010; 17(6): 377-393.
7. Laury AR, Perets R, Piao H, et al. A comprehensive analysis of PAX8 expression in human epithelial tumors. Am J Surg Pathol 2011; 35(6): 816-826.
8. Cox RM, Magi-Galluzzi C, McKenney JK. Immunohistochemical Pitfalls in Genitourinary Pathology: 2018 Update. Advances in anatomic pathology. 2018;25(6):387-99.
9. McGregor DK, Khurana KK, Cao C, et al. Diagnosing primary and metastatic renal cell carcinoma: the use of the monoclonal antibody ‘Renal Cell Carcinoma Marker’. Am J Surg Pathol 2001; 25(12): 1485-1492.
10. Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997; 10(6): 537-544.
11. Langner C, Wegscheider BJ, Ratschek M, Schips L, Zigeuner R. Keratin immunohistochemistry in renal cell carcinoma subtypes and renal oncocytomas: a systematic analysis of 233 tumors. Virchows Arch 2004; 444(2): 127-134.
12. Jiang F, Richter J, Schraml P, et al. Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol 1998; 153(5): 1467-1473.
13. Kovac M, Navas C, Horswell S, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat Commun 2015; 6 : 6336.
14. Yu W, Zhang W, Jiang Y, et al. Clinicopathological, genetic, ultrastructural characterizations and prognostic factors of papillary renal cell carcinoma: new diagnostic and prognostic information. Acta Histochem 2013; 115(5): 452-459.
15. Marsaud A, Dadone B, Ambrosetti D, et al. Dismantling papillary renal cell carcinoma classification: The heterogeneity of genetic profiles suggests several independent diseases. Genes Chromosomes Cancer 2015; 54(6): 369-382.
16. Gunawan B, von Heydebreck A, Fritsch T, et al. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of type 2 from type 1 tumors. Cancer Res 2003; 63(19): 6200-6205.
17. Antonelli A, Tardanico R, Balzarini P, et al. Cytogenetic features, clinical significance and prognostic impact of type 1 and type 2 papillary renal cell carcinoma. Cancer Genet Cytogenet 2010; 199(2): 128-133.
18. Saleeb RM, Brimo F, Farag M, et al. Toward Biological Subtyping of Papillary Renal Cell Carcinoma With Clinical Implications Through Histologic, Immunohistochemical, and Molecular Analysis. Am J Surg Pathol 2017; 41(12): 1618-1629.
19. Pitra T, Pivovarcikova K, Alaghehbandan R, Hes O. Chromosomal numerical aberration pattern in papillary renal cell carcinoma: Review article. Ann Diagn Pathol 2019; 40 : 189-199.
20. Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 2007; 31(10): 1578-1585.
21. Muller M, Guillaud-Bataille M, Salleron J, et al. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod Pathol 2018; 31(6): 974-983.
22. Smith SC, Trpkov K, Chen YB, et al. Tubulocystic Carcinoma of the Kidney With Poorly Differentiated Foci: A Frequent Morphologic Pattern of Fumarate Hydratase-deficient Renal Cell Carcinoma. Am J Surg Pathol 2016; 40(11): 1457-1472.
23. Ulamec M, Skenderi F, Zhou M, et al. Molecular Genetic Alterations in Renal Cell Carcinomas With Tubulocystic Pattern: Tubulocystic Renal Cell Carcinoma, Tubulocystic Renal Cell Carcinoma With Heterogenous Component and Familial Leiomyomatosis-associated Renal Cell Carcinoma. Clinicopathologic and Molecular Genetic Analysis of 15 Cases. Appl Immunohistochem Mol Morphol 2016; 24(7): 521-530.
24. Trpkov K, Hes O, Agaimy A, et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am J Surg Pathol 2016; 40(7): 865-875.
25. Li Y, Reuter VE, Matoso A, Netto GJ, Epstein JI, Argani P. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology 2018; 72(4): 588-600.
26. Pivovarčíková K, Martínek P, Trpkov K, et al. Fumarate hydratase deficient renal cell carcinoma and fumarate hydratase deficient-like renal cell carcinoma: Morphologic comparative study of 23 genetically tested cases. Cesk Patol 2019; 55(4): 244–249.
27. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 2001; 98(6): 3387-3392.
28. Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002; 30(4): 406-410.
29. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 2003; 73(1): 95-106.
30. Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2006; 43(1): 18-27.
31. Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 2011; 225(1): 4-11.
32. Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol 2014; 38(5): 627-637.
33. Kuroda N, Ohe C, Kawakami F, et al. Clear cell papillary renal cell carcinoma: a review. Int J Clin Exp Pathol 2014; 7(11): 7312-7318.
34. Aron M, Chang E, Herrera L, et al. Clear cell-papillary renal cell carcinoma of the kidney not associated with end-stage renal disease: clinicopathologic correlation with expanded immunophenotypic and molecular characterization of a large cohort with emphasis on relationship with renal angiomyoadenomatous tumor. Am J Surg Pathol 2015; 39(7): 873-888.
35. Deml KF, Schildhaus HU, Comperat E, et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am J Surg Pathol 2015; 39(7): 889-901.
36. Hes O, Comperat EM, Rioux-Leclercq N. Clear cell papillary renal cell carcinoma, renal angiomyoadenomatous tumor, and renal cell carcinoma with leiomyomatous stroma relationship of 3 types of renal tumors: a review. Ann Diagn Pathol 2016; 21 : 59-64.
37. Argani P, Zhong M, Reuter VE, et al. TFE3-Fusion Variant Analysis Defines Specific Clinicopathologic Associations Among Xp11 Translocation Cancers. Am J Surg Pathol 2016; 40(6): 723-737.
38. Argani P, Zhang L, Reuter VE, Tickoo SK, Antonescu CR. RBM10-TFE3 Renal Cell Carcinoma: A Potential Diagnostic Pitfall Due to Cryptic Intrachromosomal Xp11.2 Inversion Resulting in False-negative TFE3 FISH. Am J Surg Pathol 2017; 41(5): 655-662.
39. Xia QY, Wang XT, Zhan XM, et al. Xp11 Translocation Renal Cell Carcinomas (RCCs) With RBM10-TFE3 Gene Fusion Demonstrating Melanotic Features and Overlapping Morphology With t(6;11) RCC: Interest and Diagnostic Pitfall in Detecting a Paracentric Inversion of TFE3. Am J Surg Pathol 2017; 41(5): 663-676.
40. Wang XT, Xia QY, Ni H, et al. SFPQ/PSF-TFE3 renal cell carcinoma: a clinicopathologic study emphasizing extended morphology and reviewing the differences between SFPQ-TFE3 RCC and the corresponding mesenchymal neoplasm despite an identical gene fusion. Hum Pathol 2017; 63 : 190-200.
41. Hayes M, Peckova K, Martinek P, et al. Molecular-genetic analysis is essential for accurate classification of renal carcinoma resembling Xp11.2 translocation carcinoma. Virchows Arch 2015; 466(3): 313-322.
42. Udager AM, Pan J, Magers MJ, et al. Molecular and immunohistochemical characterization reveals novel BRAF mutations in metanephric adenoma. Am J Surg Pathol 2015; 39(4): 549-557.
43. Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. Mod Pathol 2003; 16(10): 1060-1063.
44. Gonzalez ML, Alaghehbandan R, Pivovarcikova K, et al. Reactivity of CK7 across the spectrum of renal cell carcinomas with clear cells. Histopathology 2019; 74(4): 608-617.
45. Williamson SR, Halat S, Eble JN, et al. Multilocular cystic renal cell carcinoma: similarities and differences in immunoprofile compared with clear cell renal cell carcinoma. Am J Surg Pathol 2012; 36(10): 1425-1433.
46. Parilla M, Alikhan M, Al-Kawaaz M, et al. Genetic Underpinnings of Renal Cell Carcinoma With Leiomyomatous Stroma. Am J Surg Pathol 2019; 43(8): 1135-1144.
47. Trpkov K, Hes O. New and emerging renal entities: a perspective post-WHO 2016 classification. Histopathology 2019; 74(1): 31-59.
48. Ross H, Martignoni G, Argani P. Renal cell carcinoma with clear cell and papillary features. Arch Pathol Lab Med 2012; 136(4): 391-399.
49. Fuzesi L, Gunawan B, Bergmann F, Tack S, Braun S, Jakse G. Papillary renal cell carcinoma with clear cell cytomorphology and chromosomal loss of 3p. Histopathology 1999; 35(2): 157-161.
50. Salama ME, Worsham MJ, DePeralta-Venturina M. Malignant papillary renal tumors with extensive clear cell change: a molecular analysis by microsatellite analysis and fluorescence in situ hybridization. Arch Pathol Lab Med 2003; 127(9): 1176-181.
51. Jia L, Jayakumaran G, Al-Ahmadie H, et al. USCAP 2018 Abstracts: Clear cell renal cell carcinoma with prominent papillary architecture: a rare morphologic variant supported by molecular evidence. Mod Pathol 2018; 31 : 323.
52. Alaghehbandan R, Ulamec M, Martinek P, et al. Papillary pattern in clear cell renal cell carcinoma: Clinicopathologic, morphologic, immunohistochemical and molecular genetic analysis of 23 cases. Ann Diagn Pathol 2019; 38 : 80-86.
53. Trpkov K, Williamson SR, Gao Y, et al. Low-grade Oncocytic Tumor of Kidney (CD117 Negative, Cytokeratin 7 Positive): A Distinct Entity? Histopathology 2019; 75(2): 174-184.
54. He H, Trpkov K, Martinek P, et al. “High-grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch 2018; 473(6): 725-738.
55. Trpkov K, Bonert M, Gao Y, et al. High-grade Oncocytic Tumor (HOT) of Kidney in a Patient with Tuberous Sclerosis Complex. Histopathology 2019; 75(3): 440-442.
56. Xia QY, Rao Q, Shen Q, et al. Oncocytic papillary renal cell carcinoma: a clinicopathological study emphasizing distinct morphology, extended immunohistochemical profile and cytogenetic features. Int J Clin Exp Pathol 2013; 6(7): 1392-1399.
57. Hes O, Brunelli M, Michal M, et al. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol 2006; 10(3): 133-139.
58. Park BH, Ro JY, Park WS, et al. Oncocytic papillary renal cell carcinoma with inverted nuclear pattern: distinct subtype with an indolent clinical course. Pathol Int 2009; 59(3): 137-146.
59. Lefevre M, Couturier J, Sibony M, et al. Adult papillary renal tumor with oncocytic cells: clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol 2005; 29(12): 1576-1581.
60. Han G, Yu W, Chu J, et al. Oncocytic papillary renal cell carcinoma: A clinicopathological and genetic analysis and indolent clinical course in 14 cases. Pathol Res Pract 2017; 213(1): 1-6.
61. Kunju LP, Wojno K, Wolf JS, Jr., Cheng L, Shah RB. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol 2008; 39(1): 96-101.
62. Michalova K, Steiner P, Montiel DP, et al. Chromosomal Aberration Pattern in Oncocytic Papillary Renal Cell Carcinoma: Analysis of 28 Cases. United States & Canadian Academy of Pathology 106th Annual Meeting; San Antonio: Mod Pathol 2017; p. 243 A.
63. Michalova K, Steiner P, Alaghehbandan R, et al. Papillary renal cell carcinoma with cytologic and molecular genetic features overlapping with renal oncocytoma: Analysis of 10 cases. Ann Diagn Pathol 2018; 35 : 1-6.
64. Tickoo SK, Amin MB, Linden MD, Lee MW, Zarbo RJ. Antimitochondrial antibody (113-1) in the differential diagnosis of granular renal cell tumors. Am J Surg Pathol 1997; 21(8): 922-930.
65. Hes O, Michal M, Boudova L, Mukensnabl P, Kinkor Z, Miculka P. Small cell variant of renal oncocytoma--a rare and misleading type of benign renal tumor. Int J Surg Pathol 2001; 9(3): 215-222.
66. Zhang W, Yu W, Wang Q, Jiang Y, Li Y. The clinicopathological, ultrastructural, genetic features and diagnosis of small cell variant renal oncocytoma. Acta Histochem 2015; 117(6): 505-511.
67. Rocca PC, Brunelli M, Gobbo S, et al. Diagnostic utility of S100A1 expression in renal cell neoplasms: an immunohistochemical and quantitative RT-PCR study. Mod Pathol 2007; 20(7): 722-728.
68. Skinnider BF, Folpe AL, Hennigar RA, et al. Distribution of cytokeratins and vimentin in adult renal neoplasms and normal renal tissue: potential utility of a cytokeratin antibody panel in the differential diagnosis of renal tumors. Am J Surg Pathol 2005; 29(6): 747-754.
69. Mathers ME, Pollock AM, Marsh C, O’Donnell M. Cytokeratin 7: a useful adjunct in the diagnosis of chromophobe renal cell carcinoma. Histopathology 2002; 40(6): 563-567.
70. Hes O, Michal M, Kuroda N, et al. Vimentin reactivity in renal oncocytoma: immunohistochemical study of 234 cases. Arch Pathol Lab Med 2007; 131(12): 1782-1788.
71. Wobker SE, Williamson SR. Modern Pathologic Diagnosis of Renal Oncocytoma. J Kidney Cancer VHL 2017; 4(4): 1-12.
72. Fuzesi L, Frank D, Nguyen C, Ringert RH, Bartels H, Gunawan B. Losses of 1p and chromosome 14 in renal oncocytomas. Cancer Genet Cytogenet 2005; 160(2): 120-125.
73. Joshi S, Tolkunov D, Aviv H, et al. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep 2015; 13(9): 1895-1908.
74. Anderson CB, Lipsky M, Nandula SV, et al. Cytogenetic analysis of 130 renal oncocytomas identify three distinct and mutually exclusive diagnostic classes of chromosome aberrations. Genes Chromosomes Cancer 2019; doi: 10.1002/gcc.22766. [Epub ahead of print]
75. Michal M, Hes O, Svec A, Ludvikova M. Pigmented microcystic chromophobe cell carcinoma: a unique variant of renal cell carcinoma. Ann Diagn Pathol 1998; 2(3): 149-153.
76. Dundr P, Pesl M, Povysil C, et al. Pigmented microcystic chromophobe renal cell carcinoma. Pathol Res Pract 2007; 203(8): 593-597.
77. Kuroda N, Tanaka A, Yamaguchi T, et al. Chromophobe renal cell carcinoma, oncocytic variant: a proposal of a new variant giving a critical diagnostic pitfall in diagnosing renal oncocytic tumors. Med Mol Morphol 2013; 46(1): 49-55.
78. Hes O, Vanecek T, Perez-Montiel DM, et al. Chromophobe renal cell carcinoma with microcystic and adenomatous arrangement and pigmentation--a diagnostic pitfall. Morphological, immunohistochemical, ultrastructural and molecular genetic report of 20 cases. Virchows Arch 2005; 446(4): 383-393.
79. Kuroda N, Iiyama T, Moriki T, Shuin T, Enzan H. Chromophobe renal cell carcinoma with focal papillary configuration, nuclear basaloid arrangement and stromal osseous metaplasia containing fatty bone marrow element. Histopathology 2005; 46(6): 712-713.
80. Parada DD, Pena KB. Chromophobe renal cell carcinoma with neuroendocrine differentiation. APMIS 2008; 116(9): 859-865.
81. Kuroda N, Tamura M, Hes O, Michal M, Gatalica Z. Chromophobe renal cell carcinoma with neuroendocrine differentiation and sarcomatoid change. Pathol Int 2011; 61(9): 552-554.
82. Foix MP, Dunatov A, Martinek P, et al. Morphological, immunohistochemical, and chromosomal analysis of multicystic chromophobe renal cell carcinoma, an architecturally unusual challenging variant. Virchows Arch 2016; 469(6): 669-678.
83. Gutierrez FJQ, Panizo A, Tienza A, et al. Cytogenetic and immunohistochemical study of 42 pigmented microcystic chromophobe renal cell carcinoma (PMChRCC). Virchows Arch 2018; 473(2): 209-217.
84. Williamson SR, Gadde R, Trpkov K, et al. Diagnostic criteria for oncocytic renal neoplasms: a survey of urologic pathologists. Hum Pathol 2017; 63 : 149-156.
85. Brunelli M, Eble JN, Zhang S, Martignoni G, Delahunt B, Cheng L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol 2005; 18(2): 161-169.
86. Kovacs A, Kovacs G. Low chromosome number in chromophobe renal cell carcinomas. Genes Chromosomes Cancer. 1992; 4(3): 267-268.
87. Sperga M, Martinek P, Vanecek T, et al. Chromophobe renal cell carcinoma--chromosomal aberration variability and its relation to Paner grading system: an array CGH and FISH analysis of 37 cases. Virchows Arch 2013; 463(4): 563-573.
88. Brunelli M, Gobbo S, Cossu-Rocca P, et al. Chromosomal gains in the sarcomatoid transformation of chromophobe renal cell carcinoma. Mod Pathol 2007; 20(3): 303-309.
89. Trpkov K, Hes O, Bonert M, et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol 2016; 40(1): 60-71.
90. Trpkov K, Abou-Ouf H, Hes O, et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol 2017; 41(10): 1299-1308.
91. McKenney JK, Przybycin CG, Trpkov K, Magi-Galluzzi C. Eosinophilic solid and cystic renal cell carcinomas have metastatic potential. Histopathology 2018; 72(6): 1066-1067.
92. Argani P. A Molecular Marker for Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol 2018; 74(4): 487-488.
93. Mehra R, Vats P, Cao X, et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol 2018; 74(4): 483-486.
94. Palsgrove DN, Li Y, Pratilas CA, et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas Harbor TSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am J Surg Pathol 2018; 42(9): 1166-1181.
95. Parilla M, Kadri S, Patil SA, et al. Are Sporadic Eosinophilic Solid and Cystic Renal Cell Carcinomas Characterized by Somatic Tuberous Sclerosis Gene Mutations? Am J Surg Pathol 2018; 42(7): 911-917.
96. Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 2014; 38(12): 1588-1602.
97. Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol 2015; 28(1): 80-94.
98. Peckova K, Vanecek T, Martinek P, et al. Aggressive and nonaggressive translocation t(6;11) renal cell carcinoma: comparative study of 6 cases and review of the literature. Ann Diagn Pathol 2014; 18(6): 351-357.
99. Gupta S, Johnson SH, Vasmatzis G, et al. TFEB-VEGFA (6p21.1) co-amplified renal cell carcinoma: a distinct entity with potential implications for clinical management. Mod Pathol 2017; 30(7): 998-1012.
100. Williamson SR, Grignon DJ, Cheng L, et al. Renal Cell Carcinoma With Chromosome 6p Amplification Including the TFEB Gene: A Novel Mechanism of Tumor Pathogenesis? Am J Surg Pathol 2017; 41(3): 287-298.
101. Ruiz-Cordero R, Rao P, Li L, et al. Hybrid oncocytic/chromophobe renal tumors are molecularly distinct from oncocytoma and chromophobe renal cell carcinoma. Mod Pathol 2019; doi: 10.1038/s41379-019-0304-y. [Epub ahead of print].
102. Petersson F, Gatalica Z, Grossmann P, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch 2010; 456(4): 355-365.
103. Pote N, Vieillefond A, Couturier J, et al. Hybrid oncocytic/chromophobe renal cell tumours do not display genomic features of chromophobe renal cell carcinomas. Virchows Arch 2013; 462(6): 633-638.
104. Chen YB, Mirsadraei L, Jayakumaran G, et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol 2019; 43(1): 121-131.
105. Tong GX, Yu WM, Beaubier NT, et al. Expression of PAX8 in normal and neoplastic renal tissues: an immunohistochemical study. Mod Pathol 2009; 22(9): 1218-1227.
106. Miettinen M, McCue PA, Sarlomo-Rikala M, et al. GATA3: a multispecific but potentially useful marker in surgical pathology: a systematic analysis of 2500 epithelial and nonepithelial tumors. Am J Surg Pathol 2014; 38(1): 13-22.
107. Mantilla JG, Antic T, Tretiakova M. GATA3 as a valuable marker to distinguish clear cell papillary renal cell carcinomas from morphologic mimics. Hum Pathol 2017; 66 : 152-158.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2020 Issue 3
Most read in this issue
- Lymph node metastasis of parotid gland high-grade adenoid-cystic carcinoma
- Immunohistochemistry in hollow urinary tract
- Immunohistochemistry and renal neoplasias
- Immunohistochemistry in prostate pathology