
Nádory CNS – klinické a radiologické aspekty
CNS Tumors – clinical and radiological aspects
Tumors of the central nervous system (CNS) include primary tumors - itraaxial, growing from brain and spinal cord cells (neuroepithelial tumors) or extraaxial, growing from surrounding structures (brain and spinal cord, nerve sheaths, vascular structures, lymphatic tissue, germ cells, malformations, pituitary glands). Much more often they are located in the intracranial space a solitary or multiple metastatic spread of malignancy originating from another organ (eg lung, breast, malignant melanoma, Grawitz’s tumor). The occurrence of metastases of solid tumors is then in the intraaxial or extraaxial region, leptomeningeal or dural. Even morphologically benign tumors with their occurrence in a closed CNS compartment can have malignant behaviour and cause severe slowly developing to acute neurological symptoms, including intracranial hypertension. Primary tumors of the central nervous system present 1-2% of all cancers, with a higher incidence in adults after the age of 60, with a slight predominance in men, with higher mortality in men than in women. About 5% of CNS tumors are hereditary (e.g., Li-Fraumeni syndrome, neurofibromatosis type I, II). The causes of most brain and spinal cord tumors are unclear, the effect of radiation has been definitely demonstrated, there is an increased risk in transplant patients and AIDS (Acquired Immune Deficiency Syndrome) patients, and the potentiating effects of some chemicals and viruses on the development of CNS neoplasms are uncertain.
The effectiveness of treatment of brain and spinal cord tumors is influenced by the existence of the so-called hematoencephalic barrier, which protects the brain from the penetration of toxic substances, but at the same time prevents the penetration of most cytostatics to the tumor target. Another obstacle may be the localization of the tumor in areas difficult to access for histological verification (brain stem, optical chiasma) due to the high risk of complications even after stereotactic biopsy. In some cases, in an effort not to cause an irreversible neurological deficit by inconsiderate tissue collection, the sample of histological material can then become inconclusive to tumor cells, i.e., tumor cells are not captured. Last but not least, the radiosensitivity of some brain structures is also limiting, which makes it impossible to apply a higher dose of ionizing radiation to a tumor affecting sensitive tissues or located near of these sensitive tissues.
The rapid development of immunohistochemical (IHC) and molecular genetic analysis methods has significantly refined diagnostics and thus theoretically facilitates the choice of the optimal treatment procedure for the individual patient. While advances in modern conformal photon and particle (currently the most frequently proton) radiotherapy, stereotactic radiosurgery has enabled accurately targeted irradiation of the CNS tumor site and at the same time spare the high-risk brain structures, thereby significantly reduce the risk of acute and late neurotoxicity, pharmacotherapy options are still limited. Just molecular-genetic knowledge already provides us with predictive and prognostic information. They should increasingly stratify patients for targeted therapy.
Keywords:
Tumors of the central nervous system – diagnosis and treatment of CNS tumors – immunohistochemical methods – molecular analysis
Authors:
Renata Emmerová 1; Jana Engelová 2,3; Stěpan Vinakurau 3,4; Barbora Ondrová 3,4
Authors‘ workplace:
Odděleni klinické a radiační onkologie, Krajská nemocnice Liberec, a. s.
1; Radiodiagnostické oddělení Nemocnice Jablonec nad Nisou
2; Centrum protonové léčby v Praze
3; Onkologická klinika 2. LF UK a FN Motol Praha
4
Published in:
Čes.-slov. Patol., 58, 2022, No. 3, p. 150-160
Category:
Reviews Article
Overview
K nádorům centrální nervové soustavy (CNS) patří nádory primární, intraaxiální – vyrůstající z buněk mozkové a míšní tkáně (neuroepitelové nádory) nebo extraaxiální, které rostou z okolních struktur (mozkových a míšních plen, nervových obalů, cévních struktur, lymfatické tkáně, zárodečných buněk, malformací, hypofýzy). Mnohem častěji se v nitrolebním prostoru vyskytuje solitární nebo vícečetný metastatický rozsev malignity původem z jiného orgánu, např. maligního tumoru plíce, prsu, metastáza maligního melanomu, Grawitzova tumoru. Výskyt metastáz solidních nádorů je pak v oblasti intraaxiální či extraaxiální, leptomeningeální nebo durální. I nádory morfologicky benigní se svým výskytem v uzavřeném kompartmentu CNS mohou projevovat maligně a způsobovat závažné pomalu se rozvíjející, až akutně vzniklé neurologické symptomy, včetně nitrolební hypertenze.
Primární nádory centrálního nervového systému tvoří 1-2 % všech zhoubných nádorů, s častějším výskytem u dospělých po 60. roce věku, s mírnou převahou výskytu u mužů, s vyšší mortalitou u mužů než u žen. Kolem 5 % CNS nádorů je podmíněno dědičně (např. Li-Fraumeni syndrom, neurofibromatóza typ I, II). Příčiny vzniku většiny nádorů mozku a míchy jsou nejasné, jednoznačně prokázán byl vliv radioaktivního záření, zvýšené riziko je u pacientů po transplantaci a nemocných AIDS (Acquired Immune Deficiency Syndrome), nejisté jsou potenciační účinky některých chemikálií a virů na vznik CNS neoplasií.
Účinnost léčby nádorů mozku a míchy je ovlivněna existencí tzv. hematoencephalické bariéry, která chrání mozek před průnikem toxických látek, ale zároveň znemožňuje průnik většiny cytostatik k nádorovému cíli. Další překážkou může být lokalizace nádoru v oblastech obtížně přístupných histologické verifikaci (mozkový kmen, optické chiasma) pro vysoké riziko komplikací i po stereotaktické biopsii. V některých případech, ve snaze nezpůsobit ireverzibilní neurologický deficit nešetrným odběrem tkáně, se pak může stát vzorek histologického materiálu nevýtěžný, to znamená, že nejsou zachyceny nádorové buňky. Limitující je v neposlední řadě i radiosenzitivita některých mozkových struktur, která znemožňuje aplikovat vyšší dávku ionizujícího záření do nádoru postihujícího senzitivní tkáně CNS nebo nacházejícího se v blízkosti těchto senzitivních tkání.
Prudký rozvoj metod imunohistochemické (IHC) a molekulárně-genetické analýzy výrazně zpřesnil diagnostiku a tím teoreticky usnadňuje volbu optimálního léčebného postupu pro individuálního pacienta. Zatímco pokrok v moderní technice konformní fotonové, částicové (v současné době nejčastěji protonové) radioterapie, stereotaktické radiochirurgie umožnil přesné cílené ozáření nádorového ložiska CNS se šetřením rizikových mozkových struktur a tím výrazně snížil riziko akutní a pozdní neurotoxicity, možnosti farmakoterapie jsou stále limitované. Právě molekulárně-genetické poznatky nám již nyní poskytují prediktivní a prognostické informace. Do budoucna by měly stále více stratifikovat pacienty k cílené terapii.
Klíčová slova:
Nádory centrální nervové soustavy – diagnostika a léčba nádorů CNS – metody imunohistochemické – molekulární analýza
Nová WHO klasifikace CNS malignit dává možnost přesnějšího stanovení prognózy, v řadě případů terapeutického postupu a v neposlední řadě může usnadnit stratifikaci pacientů do klinických studií s využitím nových molekulárních znaků. Zvláště u histologicky podobných a u morfologicky heterogenních CNS tumorů, kdy chirurgický „sampling“ nezachytí vždy relevantní diagnostické znaky, může molekulárně genetické testování přesněji oddiferencovat jednotlivé typy nádorů a přinést informaci o prognóze, v některých případech usnadňuje léčebnou rozvahu. Přestože tradiční histopatologické vyšetření zůstává fundamentální pro klasifikaci CNS novotvarů, díky molekulárním znalostem nastal další pokrok v možnosti precizní klinické diagnostiky CNS tumorů. Konvenční histopatologické mikroskopické metody jsou stále častěji kombinovány s možností vyšetření mnohočetných mutací pomocí NGS (next generation sequencing) panelů a s vyšetřením metylačního profilu genů. Budoucnost snad přinese díky tomu i efektivnější cílené terapeutické postupy.
Histologické a molekulární vyšetření umožňuje mnohem přesnější diagnózu především difuzních gliomů a embryonálních nádorů CNS (1-5). Velkým význam má separace glioblastomu, jako high grade gliového nádoru, který se dříve verifikoval podle přítomnosti nekróz, mikrovaskularní proliferace a zahrnoval 10 % IDH – pozitivních tumorů a cca 90 % IDH – negativních tumorů, které mají ve skutečnosti odlišný průběh onemocnění a prognózu. V nové klasifikaci jsou jako glioblastom označovány již jen IDH – negativní nádory. Ke glioblastomu se řadí též tumory, které mají jeden nebo více z následujících parametrů – mutaci TERT promotoru, amplifikaci EGFR genu nebo kombinaci přítomnosti chromozomu 7 a absenci chromozomu 10 [+7/−10] (6,7).
Role neurochirurga je klíčová pro získání tkáně k imunohistochemické a molekulárně genetické analýze. Rozsah neurochirurgického výkonu, tj. makroskopicky radikální resekce (gross total resection – GTR) versus subtotální resekce (subtotal resection – STR), pozitivně koreluje s celkovým přežitím (overall survival – OS) a dobou do progrese (progression free survival – PFS). Radikalita chirurgického výkonu je zásadní předpoklad úspěšnosti následné onkologické léčby, zvláště u gliomů grade 3 a 4 (high grade glioma – HGG) a embryonálních nádorů CNS. Zatímco u gliomů grade 1 a 2 (low grade glioma – LGG) může po GTR následovat tzv.„watch and wait“ strategie, verifikace HGG opravňuje k podání agresivní onkologické léčby. I při elokventně uložené tumorózní infiltraci, či při suspekci na lymfom CNS, je vždy cílem získat tkáň otevřenou biopsií, potvrdit (vyloučit) malignitu CNS a rozhodnout o další léčbě.
Pro evaluaci a interpretaci nových poznatků z klinických studií byla vytvořena skupina vedoucích neuropatologů – the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy – Not Official WHO (cIMPACT-NOW). Tato skupina publikuje výsledky své práce pravidelně v četných up-date článcích (8,9).
DIFUZNÍ GLIOMY
Stěžejní molekulární marker pro difuzní gliomy jsou IDH1/ IDH2 mutace, 1p19q kodelece a mutace genů v histonu H3 (15). IHC je vyšetřována IDH1 mutace (R132H), což je standard pro všechny pacienty s gliomem (16). V případě negativity R132H, zvláště u pacientů mladších 55 let, je doporučeno sekvenování IDH1 a IDH2 pro záchyt méně častých mutací. IDH mutace definuje WHO grade 2 a 3 astrocytomy, oligodendrogliomy a sekundární grade 4 astrocytomy (17). Přítomnost IDH mutace (IDH1 nebo IDH2) odlišuje low grade gliomy od primárních glioblastomů, které jsou vždy IDH nemutované (IDH wildtype – wt). IDH mutaci nenajdeme u neinfiltrativních grade 1 gliomů (pilocytický astrocytom, gangliogliom). Pokud je IDH mutace zachycena, jedná se přinejmenším o difusní infiltrativní gliom grade 2 (18). U gliomů nízkého stupně malignity s nemutovanou variantou IDH, zvláště u těch, které mají izolovanou TERT promotor mutaci, je vysoké riziko agresivního chování gliomu a tyto nádory by měly být léčeny jako high grade gliomy (19).
Pokud má difuzní infiltrativní astrocytom IDH mutovaný typické znaky pro grade 4 (nekrózu, mikrovaskulární proliferaci) a/ nebo obsahuje homozygotní deleci CDKN2A/B, bude mít nejspíše tendenci chovat se podobně agresivně jako glioblastom, bude tak i léčen a bude patologem popsán jako astrocytom IDH mutovaný WHO grade 4 (20-22).
Některé difuzně infiltrativní astrocytomy IDH-wt, bez typických histologických znaků pro glioblastom (nekróza a/nebo mikrovaskulární proliferace), ale s průkazem pozitivity alespoň jednoho z následujících vyšetření – amplifikace EGFR, mutace promotoru TERT, zisk chromosomu 7 a ztráta chromosomu 10 (7+/10-) by měly být diagnostikovány a léčeny na základě molekulárního gradingu jako glioblastom, WHO grade 4 (4,17).
MRI charakteristika IDH mutovaných gliomů: predominantní lokalizace ve frontálním laloku, postižení kortexu, méně agresivní vzhled – dobré ohraničení, absence edému, málo výrazné sycení, DWI (difuzně vážené obrazy): vyšší hodnoty ADC mapy, PWI (perfuzně vážené obrazy): nižší hodnoty rCBV (regional cerebral blood volume), MRS: nižší hodnoty cholin (Cho)/ kreatin (Cr), přítomnost 2-hydroxyglutarátu, častější MGMT methylace (obr. 1, 2).


OLIGODENDROGLIOM
Průkaz kodelece 1p19q je zásadní pro molekulární diagnostiku oligodendrogliomu (ODG), zvláště u tumorů s neurčitými histologickými znaky. Zároveň je pro ODG typický průkaz IDH mutace. Takto klasifikovaný tumor CNS bude s velkou pravděpodobností dobře léčebně ovlivnitelný alkylační cytostatickou léčbou a radioterapií a bude mít poměrně příznivou prognózu (23).
Zatímco přítomnost mutace promotoru TERT v kombinaci s IDH mutací a 1p/19q kodelecí je charakteristická pro ODG, nádory s poměrně příznivou prognózou (24), je naopak záchyt mutace promotoru TERT u difusně infiltrativních gliomů IDH-wt asociován s agresivním průběhem onemocnění refrakterním na radioterapii a chemoterapii a kratším celkovým přežitím (25,26).
MRI charakteristika ODG: predominantní frontální lokalizace, rozsáhlejší tumor, SWI (susceptibility weighted imaging): vícečetné ITSS (intratumoral susceptibilty signal), T2W/FLAIR – absence tzv. “mismatch„ obrazů v T2W a ve FLAIR čase – homogenní vzhled, PWI: vyšší rCBV než astrocytomy, DWI: vyšší ADC, MRS: vyšší Cho (obr. 3, 4).


DIFUZNÍ „MIDLINE“ GLIOM A DIFUZNÍ HEMISFERICKÝ GLIOM
S histonovými mutacemi se nejčastěji setkáváme u difuzních „midline“ (středočárových) gliomů a difuzních hemisferických gliomů u dětí a mladých dospělých.
Difuzní středočárový gliom (DMG) má typicky mutaci v histonu H3K27M (27). Vzhledem k biologické povaze nádoru a s ohledem na anatomickou lokalizaci, ve které gliom vyrůstá, se jedná o devastující onemocnění, dosud bez možnosti efektivní léčby. V rámci klinických studií se zkouší různé metody cílené prolongované kontinuální aplikace léčiva do obtížně přístupné mozkové krajiny pomocí katetru, kterými je překonána hematoencefalická bariéra-CED (conventional – enhanced delivery) (28). Cílená terapie by v budoucnu mohla být nadějí pro dětské pacienty, ale i pro dospělé nemocné, u kterých se sporadicky „midline“ gliomy s histonovými mutacemi také vyskytují (29).
MRI charakteristika DMG: heterogenní vzhled nádoru, lokalizace kmen, thalamus, mícha, heterogenní, méně výrazné sycení, expanzivní vzhled, kortikální invaze, leptomeningeální postižení, nízké hodnoty ADC, vyšší rCBV (obr. 5-8).


Rozsáhlá intramedulární infiltrace nízkého signálu v T2W čase i na ADC
mapě při agresivním růstu tumoru, s cystickou porcí uvnitř.


U difuzního hemisferického gliomu je cytogeneticky potvrzena přítomnost mutace v genu histonu H3.3G34. Difuzní hemisferický gliom se vyskytuje nejčastěji v mozkových hemisférách adeloscentů a mladých dospělých a může se histologicky jevit jako glioblastom nebo mít mikroskopický fenotyp embryonálního nádoru (30). Vzhledem k lokalizaci, rozsahu postižení nejsou tyto nádory neurochirurgicky řešitelné, verifikace je někdy obtížná i za pomoci stereotaktické biopsie. Tyto nádory jsou IDH nemutované, ATRX a TP53 mutované. Mají tendenci chovat se jako high grade gliomy (31).
MRI charakteristika hemisferického gliomu: rozsáhlá infiltrativní, supratentoriální, neostře ohraničená léze, expanzivní charakter, nevýrazné sycení, vyšší hodnoty ADC a nižší hodnoty rCBV, absence ITSS, MRS: vyšší hodnoty Cho/Cr (obr. 9-10).


GLIOBLASTOM
Vyšetření metylace promotoru MGMT by mělo být standardem pro molekulární diagnostiku všech high grade gliomů. Pacienti s multiformním glioblastomem (GBM) bez průkazu metylace promotoru MGMT mají z léčby alkylačním temozolomidem menší benefit než ti, jejichž tumor je MGMT promotor metylovaný. Metylace promotoru MGMT dává nemocným s glioblastomem šanci na delší přežití (32). U starších pacientů s glioblastomem, při průkazu metylace promotoru MGMT, vzhledem k předpokládané vyšší chemosenzitivitě, může onkologická léčba individuálně spočívat v samotné chemoterapii temozolomidem. Dle nových EANO doporučení má být podáno u anaplastických gliomů 12 cyklů udržovací terapie temozolomidem po ukončené chemoradioterapii (33). U glioblastomu však délka aplikace udržovacího temozolomidu dle EANO doporučení spočívá v podání 6 cyklů. A to s ohledem na skutečnost, že u glioblastomu, dle randomizované multicentrické studie faze 2 GEINO 14-01(n=159), nebyl potvrzen benefit pro celkové přežití (OS) a dobu do progrese (PFS) při prodloužení aplikace udržovacího temozolomidu na více než 6 cyklů (34).
Je doporučeno testovat glioblastomy na „driver“ mutace (BRAF V600E aktivační mutace, NTRK fúze) pro event. možnost cílené terapie, zatím spíše v rámci klinických studií (35).
MRI charakteristika GBM (který je vždy IDH wild type): agresivní vzhled, větší, nehomogenně se sytící léze, s cystami, nekrózami, hemoragiemi, a s peritumorálním edémem, SWI: vysoký grade ITSS, DWI: nižší hodnoty ADC v sytící se porci, vysoké v nekróze, PWI: vyšší rCBV, MRS: peak Cho a Lac, vyšší hodnoty Cho/Cr a rCBV i v peritumorálním infiltrativním edému (obr. 11-12).


IDH WILD-TYPE HIGH-GRADE ASTROCYTÁRNÍ GLIOM S PILOIDNÍMI RYSY
IDH wild-type high-grade astrocytární nádory s pilocytárními rysy se vyskytují relativně často v zadní jámě lební dospělých pacientů. Při molekulární diagnostice mají tyto nádory často prokázanou deleci cyklin – dependentního kinázového inhibitoru genu 2A/B (CDKN2A/B), méně často ATRX mutaci. Jedná se o agresivní typy nádoru většinou se špatnou prognózou, navzdory prováděné neurochirurgické a onkologické léčbě (36).
EPENDYMOM
Ependymom patří v dospělém věku k vzácným nádorům. Vliv radikality resekčního výkonu se odráží v doporučeních EANO, kdy adjuvantní radioterapie u dospělých pacientů je indikována v případě neradikálního výkonu u grade 2 tumoru a vždy u grade 3 tumoru. V terapii ependymomu hraje hlavní roli maximální chirurgická resekce a adjuvantní radioterapie (37). V současné době se do celkové terapeutické strategie u ependymomů promítají také nové poznatky molekulární diagnostiky (38).
Supratentoriální ependymomy mají aktivační fúzi ZFTA/ C11orf95, která je považována za reprezentativnější pro tento typ nádoru než dříve uváděná RELA. Tato fúze se nevyskytuje u ependymomů zadní jámy lební a u míšních ependymomů, znamená agresivnější průběh onemocnění než u supratentoriálních ependymomů bez ZFTA/ C11orf95 fúze. V klinické praxi by tento molekulární znak měl upozornit na vyšší riziko recidivy i po chirurgické a onkologické léčbě. Naopak supratentoriální ependymomy s fúzí YAP1 jsou typické pro dětský věk, mají dobrou prognózu (39,40).
Infratentoriální ependymomy, neboli PF (posterior fossa) ependymomy, se dělí na PFA, typické pro kojence, (tzv. dětský typ) se špatnou prognózou a PFB, typické pro starší děti a dospělé, (tzv. dospělý typ) prognosticky příznivější (obr. 13-14).


Míšní ependymomy (spinal cord ependymoma – SCE) - myxopapilární typ – nově WHO grade 2 - má horší prognózu než klasický typ míšního ependymomu – WHO grade 2/3. SCE s MYC amplifikací je anaplastický, WHO grade 3, často diseminovaný ependymom se špatnou prognózou (41).
MEDULOBLASTOM
Meduloblastom je nádor mozečku typicky diagnostikovaný u dětí, ale raritně i u dospělých. Cytogenetické vyšetření umožní zařadit meduloblastom do jedné ze 4 skupin a zahájit onkologickou léčbu dle standardu, WNT tumory mají výrazně lepší prognózu u dětí (15,42).
Klinické a MRI charakteristiky meduloblastomu podle skupin
WNT subgroup: představuje 10 % meduloblastomů, má nejlepší prognózu, je lokalizován v cerebropontinním (CP) úhlu či CP cisterně, s homogenním sycením, typicky bez hydrocephalu, raritně se vyskytují makrometastázy, střední edém, mikrocysty intratumorózně (obr. 15-16).


SHH subgroup: tvoří 30 % všech meduloblastomů, se střední prognózou, nejčastěji se jedná o heterogenní větší tumor, se středním sycením, lokalizovaný v laterální část mozečkové hemisféry. U této skupiny meduloblastomu dominuje edém, vyskytují se laminární metastázy, nejsou přítomny hemoragie, ani mikro a makrocysty, při magnetická rezonanční spektroskopii (MRS) dominuje peak Cho/lipid, nízký je peak Cr.
Group 4: má u meduloblastomů 35% zastoupení, se silnou predominancí u mužů, se střední prognózou, se středočárovou lokalizací, především v oblasti IV. mozkové komory, v její dolní části. Pro tuto skupinu je charakteristické žádné či minimální sycení, absence hemoragií, edému. Lokalizace ložisek bývá ependymální, u metastáz infundibulární a supraselární. Metastázy mívají restrikci difuze a absenci sycení.
Group 3: se verifikuje u 30 % pacientů s diagnózou meduloblastomu, predominantně u mužů, tento meduloblastom má nejhorší prognózu. Je lokalizován středočárově v oblasti vermis mozečku, IV.komory. Typickými znaky jsou heterogenní obláčkovité sycení a přítomnost leptomeningeálních či spinálních metastáz (obr. 17-18).


PINEALOBLASTOM
Blastomy v oblasti epifýzy (pinealoblastomy) jsou extrémně vzácné embryonální nádory s malým výskytem cytogenetických alterací, např. DICER1 mutace (43). Tato mutace se vyskytuje také u dětských intrakraniálních sarkomů (44). Dosud nebyly nalezeny jednoznačné molekulární znaky potvrzující diagnózu pineoblastomu (obr. 19-20).
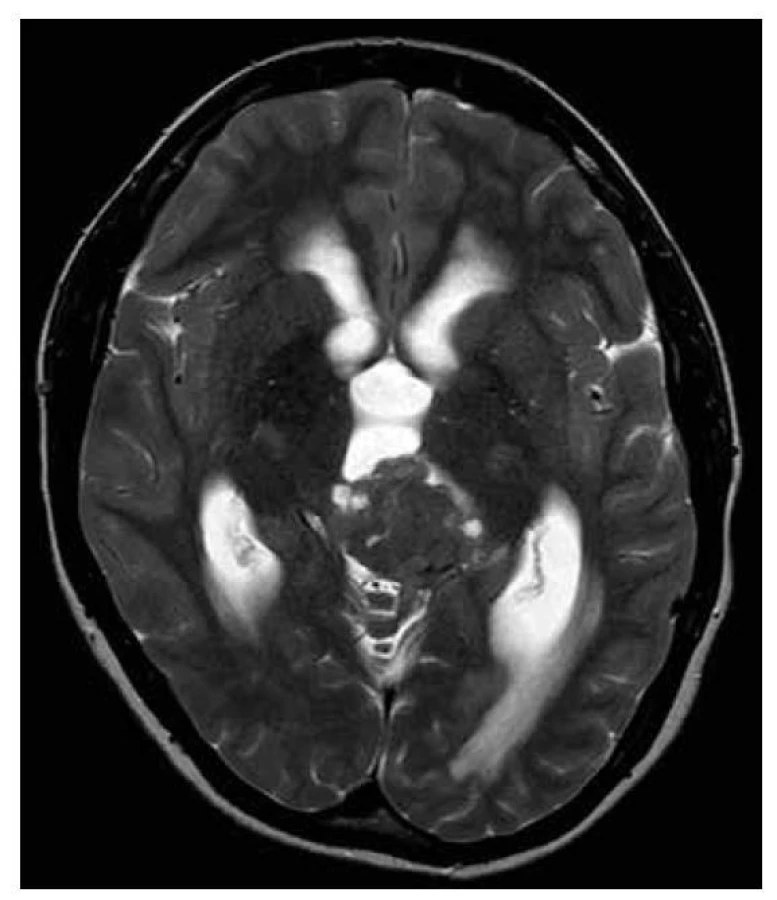

SOLITÁRNÍ FIBRÓZNÍ TUMOR
Solitární fibrózní tumory CNS/hemangiopericytomy (SFTs/ HPCs) obsahují typicky fúzi NAB2-STAT6, a i přes rozdílné histologické znaky patří do jedné skupiny nádorů (45) (obr. 21-22).


MENINGIOM
Často se vyskytující nádor vyrůstající z mozkových a míšních obalů je meningeom. Objevuje se převážně u starších pacientů (v 6. a 7. dekádě). Ženy onemocní meningeomem častěji než muži, což zřejmě souvisí s vlivem steroidních hormonů na patogenezi meningeomů (46,47). K dalším významným rizikovým faktorům patří terapeutické ozáření, zvláště v dětském věku. Radiací podmíněné meningeomy bývají vícečetné a mívají agresivní klinicko-patologické znaky (48,49). Podle WHO klasifikace je většina meningeomů grade 1 (80 %), tj. benigní histologie a indolentního chování (50). Zbylých 20 % tumorů jsou grade 2 a 3, atypické až maligní histologie a chovají se agresivněji (51,52). 70-80 % meningeomů může vyléčit chirurgická resekce (52). Nejspolehlivější prognostický faktor je u těchto tumorů WHO grade a rozsah chirurgické resekce (Simpson grade) (52 - 54).
Poslední dekáda přinesla nové poznatky v oblasti molekulárního profilu meningeomů. Identifikace genetických a epigenetických alterací u meningeomů umožňuje objasnit jejich biologické chování a usnadňuje prognostickou stratifikaci. Obecně platí, že cytogenetické změny se hromadí s rostoucím stupněm dediferenciace nádoru, proto atypické a anaplastické meningeomy vykazují komplexní cytogenetické profily (55,56). Ztráta chromozomu 22 byla zjištěna na počátku cytogenetické éry (57) a vedla k identifikaci NF2 (umístěného na chromozomu 22q) jako silného řídícího genu (58). Ztráta chromozomu 1p je druhou nejčastější změnou a souvisí s vyšším rizikem recidivy (59).
Somatické mutace se obvykle nacházejí u meningeomů grade 1. Pro NF2 – nemutované meningeomy jsou charakteristické mutace KLF4 genu umístěného na 9q31 (60), AKT1 genu umístěného na 14q32.33 (61,62), mutace V600E v genu BRAF kódujícího B-Raf proto-onkogen (63) a další. Přítomnost TERT mutace je u meningiomů, dle literárních zdrojů, znakem agresivnějšího klinického chování (64).
Ačkoliv je výskyt většiny meningeomů sporadický, několik zřídka se vyskytujících familiárních syndromů zvyšuje riziko vzniku meningeomů. Mezi jinými je to germinální mutace u neurofibromatózy typu 2 - mutace NF2 genu umístěného na 22q12.2 (65). U Gorlinova syndromu (nevoid basal cell carcinoma syndrome) vede mutace PTCH1 genu (Drosophila patched gene) k aberantní aktivaci SHH (sonic hedgehog) signální dráhy a zvyšuje tak riziko vzniku meningeomu (66,67). Jedinci se syndromem BAP1 (BRCA1-associated protein 1) mají zvýšené riziko vzniku četných tumorů (např. uveálních a kožních melanomů, pleurálních a peritoneálních mezoteliomů, renálních karcinomů) včetně meningeomů s agresivním chováním (68).
Objevují se důkazy, že profily metylace DNA mohou přesněji předpovídat recidivu a chování nádoru a jsou užitečné pro molekulární klasifikaci meningiomů (50). V roce 2017 publikovali Olar et al. první studii. V této studii profilovali 140 případů meningiomů a rozpoznali dvě odlišné metylační podskupiny meningiomů, které pojmenovali jako prognosticky příznivé (MM - -FAV) a nepříznivé (MM-UNFAV) podskupiny (69).
Stupeň diferenciace dle WHO a rozsah resekce jsou stále nejdůležitějšími prognostickými faktory pro meningiom. Uplatnění kritérií WHO klasifikace však ne vždy spolehlivě předpoví recidivu nádoru nebo riziko transformace meningiomu na vyšší stupeň. Molekulární charakteristiky meningiomů umožňují jeho subtypizaci, která se stává součástí patologické klasifikace a může napomoci rozvoji individualizované medicíny (obr. 23-24).


Zhodnocení všech nálezů a diskuse o patologické diagnóze u každé CNS malignity je záležitostí multidisciplinárního týmu tvořeného (neuro)radiodiagnostikem, neurochirurgem, onkologem, (neuro)patologem. To je klíčové pro transformaci získaných výsledků vyšetření v optimální terapeutický postup u každého jednotlivého pacienta.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
MUDr. Renata Emmerová
Oddělení klinické a radiační onkologie
Husova 357/10, 460 63, Liberec
tel: 485 312 226
Sources
1. Hegi ME, Stupp R. Withholding TMZ in glioblastoma patients with unmethylated MGMT promoter-still a dilemma? Neuro Oncol 2015; 17 : 1425-1427.
2. Weller M, Pfister SM, Wick W, et al. Molecular neuro-oncology in clinical practice : a new horizont. Lancet Oncol 2013; 14 : 370-379.
3. Dubbink HJ, Atmodimedjo PN, Kros JM, et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol 2016; 18 : 388-400.
4. Brat DJ, Verhaak RG, et al. Cancer Genome Atlas Research N. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 2015; 372 : 2481-2498.
5. Capper D, Jones DTW, Sill M, et al. DNA methylation based classification of central nervous system tumours. Nature 2018; 555 : 469-474.
6. Wen PY, Packer RJ. The 2021 WHO Classification of Tumors of the Central Nervous System: clinical implications, Neuro-Oncology 2021; 23(8): 1215-1217.
7. Gonzalez Castro LN, Wesseling P. The cIMPACT - NOW updates and their significance to current neuro-oncology practice. Neurooncol Pract 2020; 29(1): 4-10.
8. Louis DN, Aldape K, Brat DJ, et al. Announcing cIMPACT-NOW: the Consortium to inform molecular and practical approaches to CNS tumor taxonomy. Acta Neuropathol 2017; 133(1): 1-3.
9. Louis DN, Wesseling P, Paulus W, et al. cIMPACT - NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol 2018; 135(3): 481-484.
10. Louis DN, Giannini C, Capper D, et al. cIMPACT - NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol 2018; 135(4): 639–642.
11. Brat DJ, Aldape K, Colman H, et al. cIMPACT - NOW update 3: recom mended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV.”Acta Neuropathol 2018; 136(5): 805–810.
12. Ellison DW, Hawkins C, Jones DTW, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAFV600E mutation. Acta Neuropathol 2019; 137(4):683–687.9.
13. Louis DN, Ellison DW, Brat DJ, et al. cIMPACT - NOW: a practical summary of diagnostic points from Round 1 updates. Brain Pathol 2019; 29(4): 469–472.
14. Yeaney GA, Brat DJ. What every neuropathologist needs to know: update on cIMPACT - NOW. J Neuropathol Exp Neurol 2019; 78(4): 294–296.
15. Louis DN, Wesseling P, Aldape K, et al. cIMPACT - NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 2020; 30(4): 844–856.
16. Kristensen BW, Priesterbach-Ackley LP, Petersen JK, et al. Molecular pathology of tumors of the central nervous system. Ann Oncol 2019; 30(8): 1265-1278.
17. Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial defferentiation and age:a study of 1,010 diffuse gliomas. Acta Neuropathol 2009; 118 : 469-474.
18. Horbinsky C. What do we know about IDH1/2 mutation so far, and how do we use it? Acta Neuropathol 2013; 125 : 621-636.
19. Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to TMZ in low-grade gliomas. Neurology 2010; 75 : 1560-1566.
20. Berzero G, Di Stefano AL, Ronchi S, et al. IDH-wildtype lower-grade diffuse gliomas: the importance of histological grade and molecular assessment for prognostic stratification. Neuro Oncol 2021; 23(6): 955-966.
21. Shirahata M, Ono T, Stichel D, et al. Novel, impoved grading system(s) for IDH mutant astrocytic gliomas. Acta Neuropathol 2018; 136 : 153-166.
22. Reis GF, Pekmezci M, Hansen HM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol 2015; 74 : 442-452.
23. Yang RR, Shi ZF, Zhang ZY, et al. IDH mutant lower grade (WHO grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol 2020; 30 : 541-553.
24. Caincross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J Clin Oncol 2013; 31 : 344-350.
25. Arita H, Matsushita Y, Machida R, et al. TERT promoter mutation confers favorable prognosis regardless of 1p/19q status in adult diffuse gliomas with IDH1/2 mutations. Acta Neuropathol Commun 2020; 8 : 201.
26. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promotor mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 2013; 110 : 6021-6026.
27. Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma groups based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 2015; 372 : 2499-2508.
28. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482 : 226-231.
29. BT, Zhang L, Daniels DJ. Treatment Strategies in Diffuse Midline Gliomas With the H3K27M Mutation: The Role of Convection - Enhanced Delivery in Overcoming Anatomic Challenges. Front Oncol 2019; 9 : 31.
30. Meyronet D, Esteban-Mader M, Bonnet C, et al. Characteristics of H3 K27M-mutant gliomas in adult. Neuro Oncol 2017; 19 : 1127 - 1134.
31. Yoshimoto K, Hatae R, Sangatsuda Y, et al. Prevalence and clinicopathological features od H3.3 G34-mutant high-grade gliomas: A retrospective study of 411 consecutive glioma cases in a single institution. Brain Tumor Pathol 2017; 34 : 103-112.
32. Korshunov A, Capper D, Reuss D, et al. Histological distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 2016; 131 : 137-146.
33. Wick W, Platten M, Meisner C, et al. TMZ chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 2012; 13 : 707-715.
34. Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021; 18(3): 170-186.
35. Balana C. Vaz MA, Sepúlveda JM, et al. A phase II randomized, multicenter, open-label trial of continuing adjuvant temozolomide beyond six cycles in patients with glioblastoma (GEINO 14-01). Neuro Oncol 2020; 22(12): 1851-1861.
36. Natsumeda M, Chang M, Gabdulkhaev R, et al. Predicting BRAF V600E mutation in glioblastoma: utility of radiographic features. Brain Tumor Pathol 2021; 38(3): 228-233.
37. Bender K, Perez E, Chirica M, et al. High grade astrocytoma with piloid features (HGAP): the Charité experience wit a new central nervous system tumor entity. J Neurooncol 2021; 153(1): 109-120.
38. Rudà R, Reifenberger G, Frappaz D, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol 2018; 20(4): 445-456.
39. Delgado-López PD, Corrales-García EM, Alonso-García E, et al. Central nervous system ependymoma: clinical implications of the new molecular classification, treatment guidelines and controversial issues. Clin Transl Oncol 2019; 21(11): 1450-1463.
40. Pajtler KW, Mack SC, Ramaswamy V, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 2017; 133 : 5-12.
41. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 2015; 27 : 728-743.
42. Fukuoka K, Kanemura Y, Shofuda T, et al. on behalf of the Japan Pediatric Molecular Neuro-Oncology Group (JPMNG). Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 2018; 6 : 134; 2018.
43. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012; 123 : 473-484.
44. Kock L, Sabbaghian N, Druker H, et al. Germ-line and somatic DICER1 mutations in pinealoblastoma. Acta Neuropathol 2014; 128(4): 583-595.
45. Lee JC, Villanueva-Meyer JE, Ferris SP, et al. Primary intracranial sarcomas with DICER1 mutation often contain prominent eosinophilic cytoplasmic globules and can occur in the setting of neurofibromatosis type 1. Acta Neuropathol 2019; 137(3): 521-525.
46. Fritchie KJ, Jin L, Rubin BP, et al. NAB2 - STAT6 Gene Fusion in Meningeal Hemangiopericytoma and Solitary Fibrous Tumor. J Neuropathol Exp Neurol 2016; 75(3): 263-271.
47. Huisman TW, Tanghe HJ, Koper JW, et al. Progesterone, oestradiol, somatostatin and epidermal growth factor receptors on human meningiomas and their CT characteristics. E J Cancer Clin Oncol 1991; 27 : 1453-1457.
48. Sioka C, Kyritsis AP. Chemotherapy, hormonal therapy, and immunotherapy for recurrent meningiomas. J Neurooncol 2009; 92 : 1-6.
49. Sadetzki S, Flint-Richter P, Ben-Tal T, et al. Radiation-induced meningioma: a descriptive study of 253 cases. J Neurosurg 2002; 97 : 1078-1082.
50. Phillips LE, Frankenfeld CL, Drangsholt M, et al. Intracranial meningioma and ionizing radiation in medical and occupational settings. Neurology 2005; 64 : 350-352.
51. Suppiah S, Nassiri F, Bi WL, et al. Molecular and translational advances in meningiomas. Neuro Oncol 2019; 21: i4-17.
52. Ostrom QT, Gittleman H, Truitt G, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol 2018; 20 : 1-86.
53. Preusser M, Brastianos PK, Mawrin C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol 2018; 14 : 106-115.
54. Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry 1957; 20 : 22-39.
55. Gousias K, Schramm J, Simon M. The Simpson grading revisited: aggressive surgery and its place in modern meningioma management. J Neurosurg 2016; 125 : 551-560.
56. Weber RG, Boström J, Wolter M, et al. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci U S A 1997; 94 : 14719-14724.
57. Aizer AA, Abedalthagafi M, Bi WL, et al. A prognostic cytogenetic scoring system to guide the adjuvant management of patients with atypical meningioma. Neuro Oncol 2016; 18 : 269-274.
58. Zankl H, Zang KD. Cytological and cytogenetical studies on brain tumors. 4. Identification of the missing G chromosome in human meningiomas as no. 22 by fluorescence technique. Humangenetik 1972; 14 : 167-169.
59. Seizinger BR, de la Monte S, Atkins L, et al. Molecular genetic approach to human meningioma: loss of genes on chromosome 22. Proc Natl Acad Sci U S A 1987; 84 : 5419-5423.
60. Lamszus K. Meningioma pathology, genetics, and biology. J Neuropathol Exp Neurol 2004; 63 : 275-286.
61. Tetreault MP, Yang Y, Katz JP. Kruppel-like factors in cancer. Nat Rev Cancer 2013; 13 : 701-713.
62. Brastianos PK, Horowitz PM, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 2013; 45 : 285-289.
63. Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007; 448 : 439-444.
64. Loo E, Khalili P, Beuhler K, Vasef MA. BRAF V600E mutation across multiple tumor types: correlation between DNA-based sequencing and mutation-specific immunohistochemistry. Appl Immunohistochem Mol Morphol 2018; 26 : 709-713.
65. Spiegl-Kreinecker S, Lötsch D, Neumayer K, et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro Oncol 2018; 20(12): 1584-1593.
66. Kerr K, Qualmann K, Esquenazi Y, Hagan J, et al. Familial syndromes involving meningiomas provide mechanistic insight Into sporadic disease. Neurosurgery 2018; 83 : 1107-1118.
67. Smith MJ, Beetz C, Williams SG, et al. Germline mutations in SUFU cause Gorlin syndrome - associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 2014; 32 : 4155 - 4161.
68. Ng JM, Curran T. The Hedgehog‘s tale: developing strategies for targeting cancer. Nat Rev Cancer 2011; 11 : 493-501.
69. Haugh AM, Njauw CN, Bubley JA, et al. Genotypic and phenotypic features of BAP1 cancer syndrome: a report of 8 new families and review of cases in the literature. JAMA Dermatol 2017; 153 : 99-1006.
70. Olar A, Wani KM, Wilson CD, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 2017; 133 : 431-444.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2022 Issue 3
Most read in this issue
- News in WHO 2021 classification of tumours of the central nervous system
- CNS Tumors – clinical and radiological aspects
- Mucormycosis: Case report
- Giant cell fibroblastoma: a case report