
Crohnova choroba a ulcerózna kolitída – súčasný pohľad na genetickú determináciu, imunopatogenézu a biologickú liečbu
Crohn’s disease and ulcerative colitis – current view on genetic determination, immunopathogenesis and biologic therapy
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory disorders of the intestine, also called inflammatory bowel diseases (IBD), which are not caused by pathogenic microorganisms but result from non-specific inflammatory processes in the bowel. IBD are polygenic diseases, as evidenced by the genome-wide association studies (GWAS), which have discovered more than 200 genes or genetic regions to be associated with IBD. Some of them are specific for CD or UC; however, there are 110 overlapping genes. In the pathogenesis of CD, activation of adaptive immunity mediated by TH1, TH17, or TH1/TH17 cells is induced because of disturbances in the mechanisms of innate immunity and autophagocytosis. By comparison, the major events that trigger autoimmune processes in UC are an increased translocation of commensal bacteria into the submucosa because of loose inter-epithelial connections with subsequent activation of ILC2, TH9, TH2, and NKT cells. Knowledge of the pathogenesis of a disease enables an effective therapy, which is especially true for biological therapy. It is noteworthy that monoclonal antibodies directed against the major protagonists underlying both CD and UC have failed. It points to the complexity of immunopathologic processes that run in both diseases. One can suppose that a blockade of one inflammatory pathway is circumvented by an alternative pathway. TNF is the principal pro-inflammatory cytokine that plays a major role in CD and UC as well. It was therefore decided to treat IBD patients with anti-TNF monoclonal antibodies, infliximab or adalimumab. Approximately one half of the CD patients and one third of the UC patients respond to this treatment.
Keywords:
Crohn – ulcerative colitis – autophagocytosis – autoantibodies – T cells subsets – innate lymphoid cells – anti-TNF treatment
Authors:
M. Buc
Authors‘ workplace:
Imunologický ústav, Lekárska fakulta UK, Bratislava, Slovenská republika
Published in:
Epidemiol. Mikrobiol. Imunol. 66, 2017, č. 4, s. 189-197
Category:
Review Article
Overview
Crohnova choroba (CD) a ulcerózna kolitída (UC) patria k chronickým zápalovým chorobám (IBD) čreva, ktoré nevyvolávajú patogénne mikroorganizmy, ale ide o nešpecifický zápalový proces čreva. CD aj UC sú polygénovo podmienené. Poznáme už viacero génov, resp. genetických oblastí, o ktorých sa dokázalo, že majú určitú spojitosť s ich vývojom. GWAS-štúdie doteraz odhalili viac ako 200 génov, niektoré z nich sú špecifické len pre CD, iné iba pre UC, ale až 110 je prekrývajúcich sa. V patogenéze CD dochádza k aktivácii adaptívnej imunity mediovanej TH1 a TH17 - a TH1/TH17 lymfocytmi, a to v dôsledku porúch v mechanizmoch prirodzenej imunity a autofagocytózy. Naproti tomu pri UC sa ako hlavná porucha spustenia autoimunitných procesov považuje zvýšený prestup komenzálnych baktérií do submukózy pre nepevnosť interepitelových spojení s následnou aktiváciou ILC2-buniek, TH9 a TH2-lymfocytov a NTK-buniek.
Poznanie patogenézy choroby umožňuje aj racionálnu terapiu, najmä pokiaľ ide o biologickú liečbu. Zaujímavé je, že liečba monoklonovými protilátkami proti hlavným protagonistom, ktorí rozvíjajú zápalové procesy či už pri CD, alebo pri UC neuspela, čo poukazuje na zložitosť imunopatologických procesov, ktoré pri oboch chorobách prebiehajú. Dá sa predpokladať, že pri zablokovaní určitej zápalovej kaskády sa táto dá obísť zapojením inej, alternatívnej. Pri zápalovom procese pri oboch chorobách významnú úlohu zohráva TNF, preto sa začalo liečbou monoklonovými protilátkami proti nemu (infliximab, adalimumab). Na túto liečbu odpovedá asi polovica pacientov s CD a tretina s UC.
Kĺúčová slová:
morbus Crohn – ulcerózna kolitída – autofagocytóza – subpopulácie T lymfocytov – prirodzené lymfoidné bunky – autoprotilátky – anti-TNF liečba
ÚVOD
Crohnova choroba (CD − Crohn disease) a ulcerózna kolitída (UC − ulcerative colitis) patria k chronickým zápalovým chorobám čreva, ktoré nevyvolávajú špecifické patogénne mikroorganizmy, ale ide o nešpecifický zápalový proces čreva (inflammatory bowel diseases – IBD). Crohnova choroba a ulcerózna kolitída sa považujú za relatívne nové choroby, hoci o ich existencii máme záznamy už 150 rokov. Dôvodom tohto názoru je, že IBD sú choroby civilizácie, vyskytujú sa častejšie v industrializovaných (prevalencia v západnej Európe je 1 : 1 000) ako v menej vyvinutých krajinách a že ich výskyt sa značne zvýšil najmä v ostatných 50 rokoch. Typicky to ukazuje situácia v Hong Kongu, kde incidencia v r. 1985 bola 1 pacient na 1 milión obyvateľov, kým v r. 2014 už viac ako 30. Odhaduje sa, že Ázia dohoní Európu, USA a Kanadu za 20–30 rokov. Predpokladá sa, že príčina toho nárastu spočíva na zmene črevnej mikroflóry v dôsledku zmeny v stravovacích návykov, keď ľudia jedia málo vláknitej potravy a veľa komerčne upravených potravín [1, 2].
Príčinu CD a UC zatiaľ nepoznáme; súčasné poznatky však naznačujú, že obe choroby majú autoimunitný základ, ktorý sa rozvíja sa na genetickom pozadí v interakcii s faktormi prostredia, najmä s dysbalanciou črevnej flóry. Riziko získania choroby je väčšie u detí, ktorých obaja rodičia trpia buď na UC, alebo CD. Pri CD sa prevalencia choroby medzi prvostupňovými príbuznými pohybuje medzi 13,4–36%, pri UC tieto čísla varírujú medzi 4,2 –17,9 %. Silnú rodinnú predispozíciu najlepšie odráža rizikový index súrodencov λs, ktorý pri CD dosahuje hodnotu 15–42 a pri UC 7 až 17.(Index λs sa vypočíta, keď riziko choroby súrodencov sa vydelí rizikom choroby v populácii; číslo 1,0 vyjadruje hodnotu pre choroby, ktoré rodinnú záťaž nemajú).
Koexistencia iných autoimunitných chorôb je pri CD chorobe zriedkavá (2%), pozoruje sa najmä autoimunitná hemolytická anémia, celiakia a autoimunitné zápaly štítnej žľazy. Pomer ženského a mužského pohlavia je 1,5 : 1 [3, 4].
GENETICKÁ DETERMINÁCIA CROHNOVEJ CHOROBY A ULCERÓZNEJ KOLITÍDY
Crohnova choroba aj ulcerózna kolitída, podobne ako iné autoimunitné choroby, sú polygénovo podmienené. Poznáme už viacero génov, resp. genetických oblastí, o ktorých sa dokázalo, že majú určitú spojitosť s ich vývojom. GWAS-štúdie (genome wide associated studies) doteraz odhalili viac ako 200 génov či genetických oblastí, niektoré z nich sú špecifické len pre CD, iné iba pre UC, ale až 110 je prekrývajúcich sa [5, 6]. Asociované gény sú buď tie, ktoré podporujú TH1 (STAT1, IL-12B, IFNG a IL-18RAP) alebo TH17 (IL-23R, STAT3, RORC a CCR6) imunitnú odpoveď. Niektoré z nich (IL-12B, IFNG, IL-23R, RORC) sa podieľajú aj na diferenciácii a funkcii ILC-buniek (innate lymphoid cells) [7]. Pre CD sú charakteristické predovšetkým gény NOD2 (kóduje intracelulárny vzorkový receptor), IRGM, ATG16L1 (regulujú procesy autofagocytózy) a XBP1 (determinuje transkripčný faktor regulujúci skupinu génov zodpovedných za správnu funkciu endoplazmového retikula); pre UC gény HNF4A (transkripčný faktor, ktorý reguluje expresiu apikálnych interepitelových proteínov), LAMB1 (kóduje laminín v bazálnej membráne črevného epitelu), CDH1 (kóduje E-kadherín) a GNA12 (kóduje GTP-ázu, ktorá sa podieľa na vzniku pevných interepitelových spojení), ktoré zodpovedajú za funkčnosť intestinálnej bariéry, ktorá je pri UC porušená [5, 6].
Ako najvýznamnejšie sa ukazujú gény pre HLA-molekuly, NOD2, HDAC3 a FOXO3. Asociácia HLA-molekúl s Crohnovou chorobou sa prejavuje mierne zvýšenou frekvenciou alely HLA-DRB1*07, ulcerózna kolitída zase s HLA-DRB1*01 : 03; pacienti, ktorí sú nositeľmi tejto sú viac náchylnejší na vývoj pankolitídy [8, 9]. Uvedené gény kódujú molekuly, ktoré viažu cudzorodé antigény, preto možno usudzovať, že iniciačný faktor bude nejaký mikrobiálny alebo environmentálny antigén, ktorý tieto molekuly preferenčne prezentujú T-lymfocytom [2, 4].
Ako jeden z najdôležitejších génov pre vznik morbus Crohn sa však ukazuje NOD2. Homozygoti s mutáciou v NOD2-géne majú väčšiu pravdepodobnosť vývoja choroby ako heterozygoti, riziko je približne 20 - až 40-násobne vyššie oproti zdravým osobám; pri heterozygotnej mutácii riziko klesá na hodnotu 2–4 (údaje platia pre kaukazoidnú populáciu, v aziatickej a africkej populácii sa asociácia NOD2 s m. Crohn nepozoruje). Mutácie sa spájajú so zníženou funkciu tohto vzorkového receptora, ktorá vyúsťuje k nedostatočnej tvorbe transkripčného faktora NFκB, ktorý je potrebný pre aktiváciu početných génov imunitnej odpovede a v nedostatočnej inhibícii ďalšieho vzorkového receptora − TLR2 (pozri ďalej) [10].
Ďalšou poruchou, ktorá sa pri mutácii NOD2 pozoruje, je porucha tvorby autofagozómu v dôsledku nedostatočnej interakcie NOD2 s iniciačným autofagozómovým proteínom ATGL1 [11]. Pri autofagozóme ide o tvorbu dvojmembránových vezikúl okolo ivadujúcich intracelulárne parazitujúcich baktérií alebo cytoplazmového materiálu obsahujúceho poškodené organely alebo proteíny a pod. Autofagozóm splýva s lyzozómami na definitívnu degradáciu svojho obsahu, ide teda o hrubý degradačný proces.
Crohnova choroba sa spája aj s mutáciami v samotných génoch, ktoré kódujú zložky autofagozómu, konkrétne ATGL1 a IRGM, čo opäť podčiarkuje význam autofagocytózy na iniciácii zápalových procesov [11, 12].
U pacientov s CD aj s UC sa pozorovala znížená expresia deacetylázy histónov 3 [HDAC3], enzýmu, ktorá sa podieľa na epigenetickej modifikácii génov. (Deacetylácia histónov spôsobuje represiu transkripcie génov). Táto skutočnosť naznačuje, že HDAC3 sa bude spolupodieľať na rozvoji oboch chorôb. Pri myšiach, ktorým sa tento gén znefunkčnil, sa pozoroval úbytok Panethových buniek, zvýšila sa proliferácia intestinálnych epitelových buniek (IEC) a znížila ich antimikrobiálna aktivita, čo spôsobilo dysmikróbiu. Súčasne sa zistila poškodená bariérová funkcia čreva a jeho zvýšená vnímavosť na poškodenie a zápal. Zaujímavou skutočnosťou však bolo pozorovanie, že fyziologická funkcia HDCA3 závisela od prítomnosti komenzálnych baktérií, ktoré zatiaľ ešte neznámym spôsobom ovplyvňovali jeho aktivitu [13]. HDCA3 v prítomnosti komenzálnych baktérií svojou aktivitou teda zodpovedá za koordinovanú expresiu génov, ktoré ovplyvňujú funkciu IEC a intestinálnu homeostázu; porucha v tejto vzájomnej interakcii prispieva k rozvoju choroby [13, 14].
Doteraz sa vždy hľadali gény, ktoré podmieňujú vznik autoimunitnej choroby, menej sa venovala pozornosť génom, ktoré majú na jej priebeh pozitívny účinok, ktoré zmierňujú jej priebeh. Nedávno sa objavila správa o existencii takého génu práve pri m. Crohn; ide o gén pre transkripčný faktor FOXOP3. V jeho intrónovej oblasti sa identifikoval polymorfizmus jednotlivého nukleotidu (SNP; rs12212067; T → G); T-alela je častejšia, kým G-alela naopak. Ak sa G-alela nachádza v homozygotnom stave, podmieňuje mierny priebeh choroby. Vysvetlenie asociácie sa zakladá na existencii nukleovej obnovy (nuclear recovery), ktorá je u jedincov s G-alelou v homozygotnom stave rýchlejšia. Pri zápalovom procese sa FOXOP3 premiestňuje z cytoplazmy do jadra bunky a následne sa zase dostáva von do cytoplazmy. Translokácia FOXOP3 z jadra von sa v monocytoch a makrofágoch spája so zvýšenou syntézou prozápalových cytokínov (TNF, IL-1β, IL-6, IL-8). Pri návrate späť do jadra indukuje však zvýšenie syntézy IL-10, TGF-β a útlm tvorby prozápalových cytokínov. Autori práce dokázali, že FOXOP3 sa priamo viaže na promótorovú oblasť génu kódujúceho TGF-β, a navodí tak jeho zvýšenú syntézu [15, 16] a tým aj potlačenie zápalových procesov.
Patogenéza Crohnovej choroby
Súčasné poznatky naznačujú, že Crohnova choroba sa vyvíja trojstupňovo. Prvý stupeň charakterizuje penetrácia luminálneho obsahu čreva do steny čreva pre zvýšenú intestinálnu permeabilitu [17, 18]. Túto predstavu podporuje asociácia CD a génmi MUC19 (mucín 19) [19] a PTGE4R (prostaglandin E4 receptor) [20], ktoré zodpovedajú za permeabilitu čreva. Penetrácia fekálneho obsahu za fyziologických okolností navodí adekvátnu akútnu zápalovú odpoveď. Pri CD je však táto porušená, v dôsledku zníženého influxu neutrofilov pre nižšiu produkciu prozápalových cytokínov ako sú IL-8 a IL-1β; neutrofily sú plne funkčné [21]. Výsledkom je prechod zápalového procesu do tretej fázy, kde primárnu úlohu už zohrávajú bunky adaptívnej imunity, ktoré v konečnom dôsledku vedú k vytvoreniu typických zápalových zmien v črevách a vývoju klinického obrazu. Tretia fáza imunopatologického procesu začína akumuláciou lymfocytov a plazmocytov v blízkosti slizničných krýpt; nasleduje influx makrofágov, z ktorých niektoré sa neskôr menia na veľké epiteloidné bunky, a tak prispievajú k vzniku granulómu. Nahromadenie buniek na spodine krýpt sa napokon prejaví pri tvorbe abscesov, zatiaľ čo podobný proces na povrchu krýpt spôsobí skôr vznik vredov [22, 23].
Zrelé lézie v čreve charakterizuje zmiešaný celulárny infiltrát – nachádzajú sa tu lymfocyty T, B a makrofágy. B-lymfocyty, resp. plazmocyty, produkujú protilátky triedy IgG (IgG2), IgM, menej IgA, aj to skôr IgA1 ako IgA2. Tvorba J-reťazca je znížená, takže IgA je skôr monomérového ako dimérového charakteru [24]. T-lymfocyty v čreve sú aktivované, čomu naznačuje skutočnosť, že exprimujú molekuly HLA-DR, IL-2R (receptor pre IL-2; možno ho vo zvýšených koncentráciách dokázať aj v plazme pacientov) a CD45RO. Pomer lymfocytov CD4+ a CD8+ ostáva nezmenený. O význame T-lymfocytov v patogenéze choroby svedčí aj pozorovanie pacientov, ktorí mali súčasne Crohnovu chorobu a AIDS; Crohnova choroba sa dostala do úplnej remisie [25]. Pre Crohnovu chorobu je typická predovšetkým zvýšená aktivita TH1-subpopulácie, ako to dokazuje aj vysoká expresia transkripčného faktora T-BET (Transkripčný faktor T-BET polarizuje naivné T-lymfocyty do diferenciácie v smere TH1-subpopulácie, naopak transkripčný faktor GATA3 polarizuje vývoj T-lymfocytov do TH2-subpopulácie [12]).
V T-lymfocytoch lamina propria čreva, kým pri ulceróznej kolitíde rovnako lokalizované T-lymfocyty tento transkripčný faktor nemajú. Lymfocyty izolované z lamina propria produkujú in vitro veľa IL-12, IFN-γ a TNF [26].
Zvýšenú aktivitu TH1-lymfocytov podmieňuje dysregulácia ich aktivácie spôsobenej nefunkčnosťou proteínu NOD-2 v dôsledku mutácie jeho génu (príbuzný receptor, NOD-1, zostáva nepoškodený). NOD-2 patrí k intracelulárnym vzorkovým receptorom; po nadviazaní ligandu aktivuje IκB-aktivačný komplex, výsledkom ktorého je presun transkripčného faktora NFκBκ do jadra bunky. Tento sa podieľa na transkripcii viacerých génov, o. i. aj pre cytokíny a adhezívne molekuly [27].
Peptidoglykány (PGN) bez ohľadu na to či obsahujú muramyl-dipetid (MDP, ligand receptora NOD-2) alebo g-D-glutamyl-meso-diaminopimelovú kyselinu (iE-DAP; ligand receptora NOD-1), rozpoznávajú aj receptory TLR2. PGN-indukovaná signalizácia cez TLR2 prostredníctvom podpory tvorby NFκB vedie k produkcii IL-12 a tým polarizácii imunitnej odpovede do TH1-smeru. Keďže TLR2 rozpoznáva PGN a receptory NOD zase jeho zložky, usudzovalo sa, že obe signalizačné dráhy by mohli byť prepojené a navzájom sa ovplyvňovať. Tento predpoklad sa potvrdil; zistilo sa, že signalizácia cez NOD-2 inhibuje signalizáciu cez TLR2 [28, 29]. Pri m. Crohn dochádza v dôsledku mutácie génu NOD-2 k výpadku negatívnej kontroly aktivity TLR2, a tým premrštenej aktivácii TH1-imunitnej odpovede tak typickej pre imunopatogenézu tejto choroby (obr. 1).
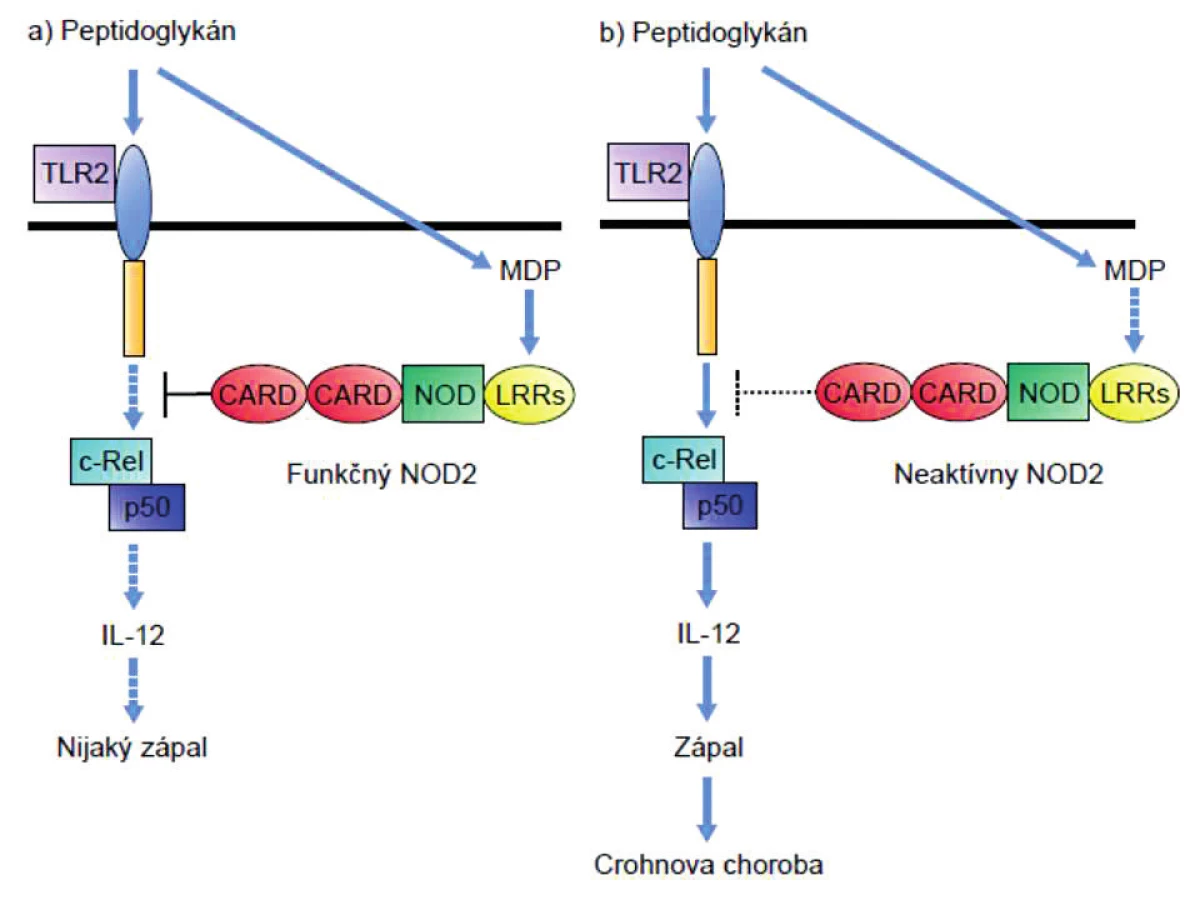
Panethove bunky exprimujú NOD2 tiež a jeho znížená aktivita sa prejaví aj zníženou syntézou antibaktériových látok (beta-defenzínov HBD2, HBD3 a HBD4), ktoré produkujú, čo opäť naznačuje na nedostatočnú likvidáciu baktérií a ich zvýšenú translokáciu z lúmenu do submukózy [30].
Za fyziologických okolností zápalové a imunitné procesy v čreve udržuje pod svojou kontrolou imunosupresívny cytokín TGF-β. Zistilo sa, že pri m. Crohn je aktivita tohto cytokínu znížená. Pri skúmaní, prečo nedochádza k presadeniu jeho fyziologickej funkcie sa došlo k poznaniu, že za ňu zodpovedá zvýšená aktivita SMAD-7, čo je negatívny regulátor signalizačných procesov indukovaných TGF-β (inhibícia SMAD7 je perspektívnym terapeutickým zásahom pri liečbe tejto choroby) [31]. Receptor pre TGF-β sa skladá z dvoch reťazcov – I a II. TGF-β sa viaže na receptor II, ktorý sa autofosforyluje a následne fosforyluje receptor I. Aktivovaný TGF-βRI v ďalšom fosforyluje, a tým aktivuje, proteíny SMAD-2 a SMAD-3. Oba sa potom spájajú a vytvárajú heterodimér, ktorý viaže SMAD-4. Takto vytvorený oligomér SMAD-2, -3, -4 sa napokon presúva do jadra bunky na promótorovú oblasť pre inhibítor transkripčného faktora NFκB (IκB) a indukuje jeho transkripciu. IκB sa v cytoplazme viaže na NFκB a blokuje jeho aktivitu (obr. 2). Aktiváciu SMAD-2 a SMAD-3 kontroluje SMAD-7, ktorý sa viaže na TGFβRI a zabraňuje fosforylácii SMAD-1 či SMAD-2. Takto sa zabráni vytvoreniu heterotriméru SMAD-2, -3, -4 a následne transkripcii IκB-génu, výsledkom čoho je nekontrolovaný zápalový proces. Navyše opäť je tu aj nedostatočná kontrola aktivity TH1-lymfocytov, pretože TGF-β za fyziologických okolností inhibuje syntézu dvoch principiálnych molekúl, transkripčného faktora T-BET a receptora pre IL-12 a keď túto svoju aktivitu nemôže uplatniť, polarizácia T-lymfocytov do TH1-subpopulácie pokračuje ďalej. Polarizácii napomáha aj zvýšená tvorba substancie P, ktorá podporuje aj ich syntézu IFN-γ [32, 33]
![Transkripčné faktory SMAD pri aktivácii a represii génov – fyziologický stav
Receptor pre TGF-β sa skladá z dvoch reťazcov I a II. TGF-β sa viaže na receptor II, ktorý sa autofosforyluje a následne fosforyluje receptor I. Aktivovaný TGF-βRI v ďalšom fosforyluje a tým aktivuje proteíny SMAD-2 a SMAD-3. Tieto sa potom spolu spájajú a vytvárajú heterodimér, ktorý sa spája so SMAD-4. Takto vytvorený oligomér sa napokon presúva do jadra bunky na promótorovú oblasť pre inhibítor transkripčného faktora NFκB [IκB] a indukuje jeho transkripciu. IκB sa v cytoplazme viaže na NFκB a blokuje jeho aktivitu. Aktiváciu SMAD-2 a SMAD-3 kontroluje SMAD-7, ktorý sa viaže na TGFβRI a zabraňuje fosforylácii SMAD-1 či SMAD-2. Takto sa zabráni vytvorenie heterotriméru SMAD-2, -3, -4 a následne transkripcia IκB-génu, výsledkom čoho je nekontrolovaný zápalový proces.
(podľa Monteleone et al. Trends Immunol 2004; 25(10): 513-7, modifikované).
Figure 2. SMAD transcription factors in gene activation and repression – physiological state
The TGF-β receptor consists of two chains designated I and II. TGF-β binds to receptor II, which autophosphorylates and then phosphorylates receptor I. Activated TGF-βRI phosphorylates, thus activating SMAD-2 and SMAD-3 proteins. They bind to each other to form a heterodimer, which binds to SMAD-4. The resultant oligomer translocates to the cell nucleus, to the promotor region for the NFκB transcription factor inhibitor [IκB], inducing its transcription. In the cytoplasm, IκB binds to NFκB and blocks its activity. SMAD-2 and SMAD-3 activation is controlled by SMAD-7, which binds to TGFβRI and inhibits SMAD-1 or SMAD-2 phosphorylation. Consequently, heterotrimer formation by SMAD-2, SMAD-3, and SMAD-4 and subsequent transcription of the IκB gene are inhibited, whichresults in an uncontrolled inflammatory process.
(adapted from Monteleone et al. Trends Immunol 2004; 25(10): 513–517)](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/2000bbd07ebb52032f27289e15d88d87.jpg)
Predstava o rozvoji imunopatologického procesu je taká, že T-lymfocyty začnú reagovať proti nejakým antigénom črevných baktérií, aktivujú sa a začnú syntetizovať cytokíny, najmä IL-12, ktoré vedú k polarizácii imunitnej odpovede v smere TH1-typu. O akumuláciu T-lymfocytov do zápalového ložiska sa stará chemokín CCL25 (TECK), ktorý produkujú epitelové bunky čreva. Cytokíny TH1-typu indukujú expresiu HLA-molekúl na epitelových bunkách, ktoré následne začnú plniť funkciu buniek prezentujúcich antigén a prezentovať početné autoantigény a tak rozvíjať autoimunitný proces v celom jeho rozsahu. Cytokíny, najmä IL-2, IL-6 a IL-15 zase indukujú rezistenciu T-lymfocytov na apoptotické podnety, čo spôsobuje, neprimeranú akumuláciu T-lymfocytov v ložisku a rozvoj chronického zápalu (obr. 3). Novšie sa však zistilo, že v lamina propria sa vyskytujú vo zvýšenej miere aj TH17-lymfocyty (produkujú prozápalový IL-7) a TH1/TH17-lymfocyty (syntetizujú aj IL-17 aj IFN-γ), čo naznačuje, že tieto bunky by tiež mohli podstatne prispievať k zápalovému procesu [7, 34–36]. S týmito nálezmi sú však v protiklade experimentálne údaje o pozitívnej úlohe IL-17, ktorý signalizáciou cez svoj receptor bráni zvýšeniu permeability a prestupu baktérií do submukózy [37]. Potvrdzovali by to aj neúspešné výsledky liečby pacientov s m. Crohn monoklonovými protilátkami anti-IL-17 (secukinumab) [38], resp. proti jeho receptoru (brodalumab) [37].

IL-23, ktorý sa spolu s IL-6 a IL-1β podieľa na polarizácii naivných TH-lymfocytov do TH17-subopopulácie, súčasne potláča pozitívnu aktivitu IL-33. Tento cytokín pôsobí ako alarmín a produkujú ho epitelové bunky čreva, kde podporuje proliferáciu a akumuláciu nTreg-lymfocytov. Ich pozitívne, t. j. imunosupresívne pôsobenie však ruší nadmerná syntéza IL-23 makrofágmi a mDC, ktorý v nTreg-lymfocytoch znižuje expresiu receptora pre IL-33 (ST2) [39, 40].
Nedávne štúdie ukázali, že na rozvoji zápalového procesu sa podieľajú aj ILC-bunky, konkrétne NCR+ ILC3 a ILC1. Množstvo a aktivita NCR+ ILC3 je znížená, kým pri ILC1-bunkách je to zase naopak, čo poukazuje na celkovú dysreguláciu produkcie cytokínov a následnú dysreguláciu imunitných procesov [41, 42].
NCR+ ILC3 syntetizujú IL-22, ktorý stimuláciou epitelových buniek podporuje syntézu antimikrobiálnych látok, ktoré inhibujú aj prílišný rast komenzálnych baktérií, najmä SFB-baktérií (segmented filamentous bacteria), a tým bránia ich prechodu do submukózy a indukcii zápalových procesov. IL-22 podporuje aj syntézu amfiregulínu a LIF (leukemia inhibitory factor), ktoré podporujú rast enterocytov [43]. NCR+ ILC3-bunky produkujú aj BAFF (B-cell-activating factor), faktor, ktorý podporuje syntézu IgA, čo sú významné slizničné protilátky. Zníženie aktivity uvedenej skupiny buniek teda prispieva k rozvoju zápalového procesu pri m. Crohn [41, 42].
Aktivita ILC1-buniek je pri m. Crohn zvýšená. Tieto bunky sa vyznačujú cytotoxickou rekciou a produkciou IFN-γ, ktorý ďalej podporuje aktivitu TH1-lymfocytov, čím podporujú ich imunopatologické pôsobenie, alebo prispievajú k ich polarizácii [41].
Patogenéza ulceróznej kolitídy
Ulcerózna kolitída (UC) sa dlhodobo považovala za chorobu, na ktorej patogenéze sa podieľajú TH2-lymfocyty. Novšie údaje ukazujú, že v patogenetických procesoch budú hrať významnejšiu úlohu nedávno identifikované prirodzené lymfocyty, a to druhej skupiny – ILC2 [44–47], resp. ukazuje sa, že ILC-2-bunky by mali byť na samom začiatku imuno-patologických procesov a až následne by sa do nich zapájali TH2-lymfocyty. Nedávnym výskumom sa totiž zistilo, že pri UC sa vo veľkej miere tvorí IL-33, ktorý aktivuje početné ďalšie bunky do rozvoja zápalového procesu.
IL-33 produkujú početné bunky nášho organizmu, najmä fibroblasty, epitelové a endotelové bunky. V neprítomnosti stimulu sa po svojej syntéze presúva do jadra bunky, kde pôsobí ako dôležitý transkripčný faktor mnohých génov. Za fyziologických okolností IL-33 zabezpečuje pevnosť interepitelových spojení, podporuje proliferáciu epitelových buniek a produkciu hlienu, čím dopĺňa funkciu IL-22. Do vonkajšieho prostredia sa môže dostať pri poškodení buniek a pôsobí teda ako alarmín, ale aj po stimulácii ich vzorkových receptorov, cytokínmi, LPS a inými látkami [48, 49].
Pri UC poškodené epitelové bunky vo zvýšenej miere tvoria IL-33. V submukóze aktivuje ILC2-bunky a aj početné ďalšie bunky, najmä mastocyty. Aktivované mastocyty začnú produkovať chymázu, tryptázu a najmä prostaglandín E2, ktoré ďalej zvyšujú aktiváciu ILC-2 buniek. Tieto sa vyznačujú syntézou IL-5,IL-6, IL-9 a IL-13. IL-5 je diferenciačným a aktivačným faktorom pre eozinofily, ktoré svojim hlavným bázickým proteínom poškodzujú epitel čreva, čo spôsobí vznik ulcerácií. IL-9 podmieňuje aktiváciu mastocytov, ktoré sú značným zdrojom TNF. Tento indukuje zápalové procesy, najmä aktiváciou neutrofilov, ktoré svojimi cídnymi produktmi prispievajú k poškodeniu epitelu čreva. IL-13 aktivuje NKT-bunky, ktoré sa pri UC nachádzajú v submukóze tiež vo zvýšenej miere. Tieto svojou cytotoxickou aktivitou spôsobujú poškodenie epitelu tiež [45, 50, 51]. IL-13 navyše podporuje apoptózu epitelových buniek a uvoľnenie interepitelových spojení [52]. Aktivované ILC-2 bunky exprimujú HLA-molekuly druhej triedy a kostimulačné molekuly, čím vytvárajú podmienky na vznik TH2-lymfocytov, ktoré svojimi cytokínmi ďalej prehlbujú zápalový proces; podobne môžu aktivovať B-lymfocyty, aby produkovali pre UC charakteristické autoprotilátky [53].
IL-33 podporuje aj diferenciáciu naivných T-lymfocytov do TH9-subpopulácie, ktorá v porovnaní s pacientmi s CD a aj zdravými jedincami zvýšená, podobne ako expresia receptorov pre IL-9 na epitelových bunkách čreva. Samotný IL-9 sa podieľa na uvoľnení interepitelových spojení. O význame TH9 v rozvoji imunopatologických stavov svedčí aj korelácia ich aktivity s vážnosťou choroby [54, 55]. Na tvorbe ulcerácií sa podieľa aj znížená aktivita TH22-lymfocytov [56], ktorých IL-22 za fyziologických okolností prispieva k antibaktériovej obranyschopnosti podporou syntézy mikrobicídnych látok a proliferácie epitelových buniek. Napokon treba uviesť, že na zápalových procesoch, podobne ako pri CD, sa podieľa aj aktivita TH17-lymfocytov [57].
O význame IL-33 v imunopatogenéze UC svedčí aj pokles jeho hladín v sérach od pacientov liečených infliximabom [58]. Otázka zostáva, čo indukuje jeho syntézu. Genetické štúdie v rámci GWAS odhalili, že s UC asociu-jú aj gény, ktoré sa podieľajú na determinácii molekúl zodpovedných za interepitelové spojenia (HNF4A, LAMB1, CDH1 a GNA12) [5, 59]. Sú údaje, ktoré dokazujú, že pri UC je ozaj zvýšená priepustnosť týchto spojení a tým zvýšený prestup komenzálnych baktérií do submukózy; tu by aktivovali bunky, ktoré dokážu produkovať IL-33 (pozri vyššie).
Z uvedenej predstavy o imunopatogenéze UC aj vyplýva, prečo asi len tretina pacientov odpovedá na liečbu anti-TNF protilátkami. Vidíme, že TNF je významným hráčom na poli v rozvoji zápalových procesov, ale nie rozhodujúcim, na patogenéze sa zúčastňujú aj iné cytokíny, najmä IL-33 a IL-9.
Biomarkery pre stanovenie diagnózy IBD
Prirodzenou snahou lekárov je urobiť správnu a včasnú diagnózu choroby. Za týmto účelom sa využíva okrem klinického zhodnotenia stavu pacienta, endoskopického, histologického, či CT - alebo MRI-vyšetrenia aj stanovovanie biomarkerov. Pri IBD ide predovšetkým o fekálne a sérologické biomarkery. Z fekálnych biomarkerov sa využíva najmä stanovovanie hladín kalprotektínu a laktoferínu. Kalprotektín je dimér proteínov S100A8 a S10019. Predstavuje až 60 % solubilných cytozolových proteínov neutrofilov. Má bakteriostatické a fungicídne vlastnosti, v prítomnosti vápnika dokáže sekvestrovať zinok a mangán. Odoláva enzýmovému štiepeniu, takže v stolici sa ľahko dokáže [60]. Laktoferín je glykoproteín prítomný v sekrečných tekutinách (materské mlieko, sliny, slzy, nazálny sekrét) a tiež v granulách neutrofilov. Viaže voľné železo, čo spôsobuje, že má antibaktériové vlastnosti. Oba biomarkery nepriamo odrážajú prítomnosť neutrofilov v sliznici čreva. Senzitívnosť a špecifickosť pre identifikáciu pacientov s IBD oproti zdravým jedincom sa pohybuje na úrovni 82–89 % pre senzitívnosť a 81 % pre špecifickosť. Fekálne biomarkery korelujú lepšie s ulceróznou kolitídou ako s m. Crohn [61].
Zo sérologických biomarkerov sa treba zmieniť o C-reaktívnom proteíne (CRP), ktorého hladina je zvýšená u 100 % pacientov s morbus Crohn, ale len u 50% pacientov s ulceróznou kolitídou. Pri úspešnej liečbe hladiny CRP klesajú [62].
Špecifickejšie ako stanovovanie vyššie uvedených biomarkerov je určovanie prítomnosti protilátok asociovaných s jednou, alebo druhou chorobou.
Až 79 % chorých trpiacich na Crohnovu chorobu má v plazme protilátky proti manózovým oligosacharidom v stene Saccharomyces cerevisiae (ASCA); vyššie titre sa pozorovali najmä u pacientov so skorším začiatkom choroby a vážnejším klinickým priebehom (fibrotizácia čreva, tvorba fistúl). Päť až dvadsaťpäť percent chorých s CD tvorí protilátky proti perinukleovým proteínom (pANCA − perinuclear anti-neutrophil cytoplasmic antibodies); tieto sú však charakteristické viac pre UC. Takíto pacienti majú charakter choroby viac sa približujúci k UC s ľavostranným postihnutím hrubého čreva, krvácaním, tvorbou hlienu. Nasvedčuje to, že Crohnova choroba a ulcerózna kolitída sú opačné konce celého spektra klinických príznakov IBD; napokon asi u 10 % chorých sa nedá jasne odlíšiť, či ide o UC alebo CD [63]. Špecifickými protilátkami pri CD sa ukazujú aj tie, ktoré rozpoznávajú GP2-antigén (granule membrane glycoprotein 2). Vyskytuje sa v membránach enterocytov ako aj M-buniek Peyerových plakov, hoci pôvodne sa identifikoval na bunkách pankreasu [64].
V ostatných rokoch sa pri m. Crohn opísali aj ďalšie protilátky proti niektorým baktériám – protilátka anti-OmpC (rozpoznáva vonkajší antigén bunkovej steny Escherichia coli − Outer membrane porin C) sa nachádza u 55 % chorých [65]. Ďalšie protilátky, anti-I2 a anti-CBir1 sú namierené proti antigénom Pseudomonas fluorescens; tiež sa dajú dokázať asi u polovice chorých [66] – viď tabuľka 1.
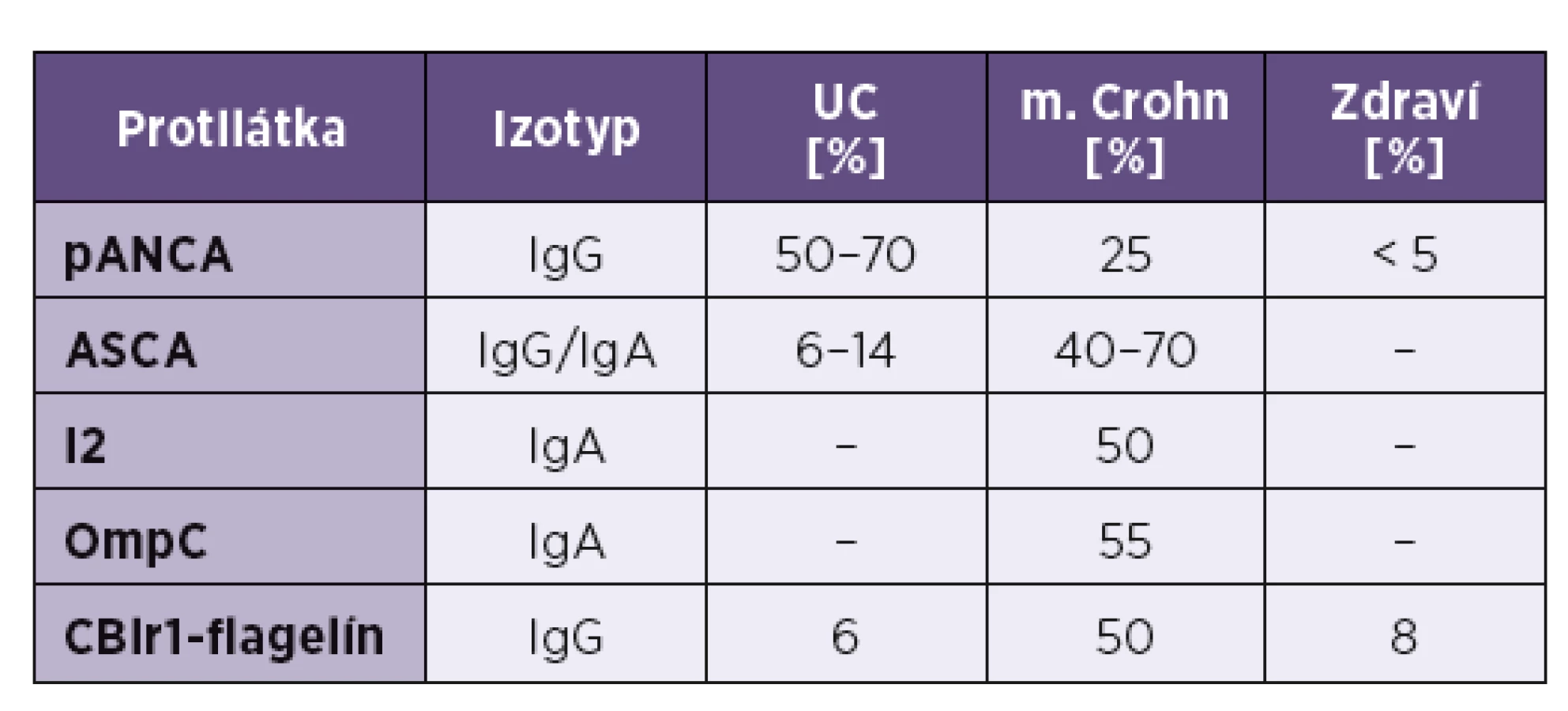
Pacienti trpiaci na ulceróznu kolitídu charakteristicky vyvíjajú pANCA-protilátky, ktorých terčom je laktoferín a katepsín G. V svojej plazme ich má 50–70% pacientov; spájajú sa tiež s vývojom pankolitídy; terčovým antigénom pANCA-protilátok býva laktoferín a katepsín G.Prítomnosť pANCA a chýbanie ASCA naznačuje, že pacient trpí skôr na UC ako na CD [63].
Klasická a biologická liečba pri CD a UC
Poznanie patogenézy choroby umožňuje aj racionálnu terapiu. Klasicky, pri oboch chorobách sa lieči stupňovito, najprv sa podáva sulfasalazín, neskôr glukokortikoidy. Pri vyvinutí rezistencie na glukokortikoidy, ako sa to často stáva pri m. Crohn, alebo pri ťažších formách oboch chorôb, sa aplikuje azatioprín, pri UC cyklosporín A alebo takrolimus [7].
V ostatnej dobe sa začalo aj s biologickou liečbou. Zaujímavé je, že liečba monoklonovými protilátkami (mAb) proti hlavným protagonistom, ktorí rozvíjajú zápalové procesy či už pri CD alebo pri UC neuspela. Neosvedčila sa ani anti-IFN-γ (fontolimumab) [67], resp. anti-IL-17A (secukinumab) [38, 68] liečba pri m. Crohn a ani anti-IL-13 (tralokinumab ) liečba pri UC [69], čo poukazuje na zložitosť imunopatologických procesov, ktoré pri týchto chorobách prebiehajú. Dá sa predpokladať, že pri zablokovaní určitej zápalovej kaskády sa táto dá obísť zapojením inej, alternatívnej. Pretože pri zápalovom procese pri oboch chorobách významnú úlohu zohráva TNF, začalo sa liečbou monoklonovými protilátkami proti nemu − infliximab je chimérová a adalimumab zase plne humanizovaná mAb. Na túto liečbu odpovedá asi polovica pacientov s CD a tretina s UC, ale aj u tých často neúplne a dočasne [70–72]. Otázka je, ako predvídať či liečba bude úspešná. Určité náznaky tu sú. Zistilo sa, že u tých, ktorí majú v svojej plazme vysoké hladiny CRP, liečba zaberá lepšie [73]. Odporúča sa preto začať s anti-TNF liečbou ak choroba trvá kratšie ako dva roky, hladiny CRP sú vyššie ako 10 mg.l-1 a pacienti nemajú abscesy, fistuly či striktúry. Podobne sa zistilo, že ak bunky imunitného systému exprimujú membránovo viazaný TNF, tak na anti-TNF liečbu zaberajú tiež lepšie (problémom je, že táto metóda nepatrí medzi rutinné). CD-pacientom, ktorí na anti-TNF liečbu neodpovedajú, sa odporúča podávať ustekinumab. Sú to monoklonové protilátky namierené proti spoločnému reťazcu IL-12 a IL-23, t. j. p40, takže zasahujú do aktivity aj TH1 - and TH17-lymfocytov [74]. Účinnejšie sa však ukazujú mAb proti samotnému IL-23, jeho p19 reťazcu (tildrakizumab a guselkumab) [37], takže IL-17 neneutralizujú.
V terapii m. Crohn sa osvedčili aj monoklonové protilátky proti molekule α4 (natalizumab), ktorá je súčasťou integrínu α4β1 (VLA4), resp. α4β7 (α4-integrín sa nachádza na aktivovaných lymfocytoch a monocytoch. Jeho partnerská molekula je VCAM-1 [pre VLA4], resp. MAdCAM-1 [pre α4β7], ktoré sa nachádzajú na endotelových bunkách. Vzájomná interakcia uvedených molekúl uľahčuje prechod lymfocytov a monocytov do zápalového ložiska. Pri liečbe m. Crohn sa uplatňuje blokáda integrínu α4β7.) Sú rovnako účinné ako infliximab, ale pre možnosť indukcie multifokálnej leukoencefalopatie ich používanie v Európe EMA (European Medicines Agency) nepovolila [75].Lepšie je na tom novšia mAb, vedolizumab, ktorá je namierená len proti α4β7-integrínu (tzn., že neblokuje prechod lymfocytov do CNS ako natalizumab) a pozitívne pôsobí aj pri CD aj pri UC [7, 76]. Sľubným liekom sa zdá byť tofacitinib, ktorý selektívne inhibuje JAK-kinázy (JAK1 a JAK3). Tieto tyrozínkinázy sprostredkúvajú prenos signálu z receptorov pre tie cytokíny, ktoré využívajú spoločný gama-reťazec; ide o cytokíny IL-2, -4, -7, -9, -15 a -21. Sú to interleukíny, ktoré zabezpečujú aktiváciu lymfocytov, ich proliferáciu a funkciu. Navyše inhibícia JAK1-kinázy oslabuje signalizáciu prozápalových cytokínov IL-6 a IFN-γ. Tofacitinib je účinný pri zvládaní strednej a ťažkej formy UC, pri liečbe Crohnovej choroby je neúčinný [77].
Napokon pri m. Crohn sa treba zmieniť aj o pomerne nezvyčajnej terapii – podávajú sa pri nej vajíčka červa Trichuris suis, z ktorých sa síce vyvinú dospelé červy, sú však pre človeka nepatogénne. Indukujú TH2-imunitnú odpoveď, ktorá má vyvážiť patogenetickú TH1, ktorá u chorých riadi imunopatologické procesy. Prvé skúsenosti sú pozitívne, i keď zatiaľ ešte chýbajú výsledky veľkých kontrolných štúdií. Racionálne jadro tejto liečby vychádza z hygienickej hypotézy. Mikroorganizmy po tisícročia „cvičili“ imunitný systém človeka tak, aby sa im čo najlepšie vedel brániť. Moderný svet zrazu tento „vzájomný boj o prežitie“ zvrátil; ak si napr. zoberieme infestáciu obyvateľstva helmintmi, tak táto vo vyspelých krajinách buď neexistuje, alebo je značne redukovaná. Vieme, že helminty indukujú imunitnú odpoveď TH2-typu a pôvodne sa myslelo, že umelé infikovanie chorých s m. Crohn „prehodí“ ich TH1-imunitnú odpoveď na TH2 [78, 79]. Doterajšie štúdie však jednoznačne účinnosť a bezpečnosť tejto terapie nepotvrdili, vyžadujú sa ďalšie sledovania [80].
V experimentálnom štádiu sú terapeutické postupy využívajúce transplantácie autológnej kostnej drene pre pacientov, ktorí sú už refraktérni na predchádzajúcu liečbu chemoterapeutikami či biologikami a transplantácie mezenchýmových kmeňových buniek, pre liečbu análnych fistúl [81]. V neposlednom rade sa treba ešte zmieniť o fekálnej terapii (FMT – faecal microbiota therapy; ide o prenos mikroorganizmov čreva od zdravých dobrovoľníkov chorému), ktorá sa úspešne ujala pri liečbe kolitídy spôsobovanej Clostridium difficile [82].
Do redakce došlo dne 28. 12. 2016.
Adresa pro korespondenci:
prof. MUDr. Milan Buc, DrSc.
Imunologický ústav LFUK
Odborárske námestie 14
813 72 Bratislava Slovenská republika
e-mail: milan.buc@fmed.uniba.sk
Sources
1. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology, 2004;126(6): 1504–1517.
2. Chi KR. Epidemiology: Rising in the East. Nature, 2016;540(7634): S100–S102.
3. Oostenbrug LE, van Dullemen HM, te Meerman GJ, Jansen PL. IBD and genetics: new developments. Scand J Gastroenterol Suppl, 2003(239): 63–68.
4. Halme L, Paavola-Sakki P, Turunen U, et al. Family and twin studies in inflammatory bowel disease. World J Gastroenterol, 2006;12(23): 3668–3672.
5. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut, 2011;60(12): 1739–1753.
6. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 2012;491(7422): 119–124.
7. Biancheri P, Powell N, Monteleone G, et al. The challenges of stratifying patients for trials in inflammatory bowel disease. Trends Immunol, 2013;34(11): 564–571.
8. Ahmad T, Marshall SE, Jewell D. Genetics of inflammatory bowel disease: the role of the HLA complex. World J Gastroenterol, 2006;12(23): 3628–3635.
9. Silverberg MS, Mirea L, Bull SB, Murphy JE, Steinhart AH, Greenberg GR, et al. A population - and family-based study of Canadian families reveals association of HLA DRB1*0103 with colonic involvement in inflammatory bowel disease. Inflam Bowel Dis, 2003;9(1): 1–9.
10. Abreu MT, Taylor KD, Lin YC, Hang T, Gaiennie J, Landers CJ, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn‘s disease. Gastroenterology, 2002;123(3): 679–688.
11. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol, 2010;11(1): 55–62.
12. Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet, 2007;39(2): 207–211.
13. Alenghat T, Osborne LC, Saenz SA, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature, 2013;504(7478): 153–157.
14. Kugelberg E. Immune homeostasis: balancing the gut. Nat Rev Immunol, 2013;13(12): 848–849.
15. Lee JC, Espeli M, Anderson CA, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell, 2013;155(1): 57–69.
16. Leavy O. Immunogenetics: SNPing at FOXO3 to limit inflammation. Nat Rev Immunol, 2013;13(11): 771.
17. Olaison G, Leandersson P, Sjodahl R, Tagesson C. Intestinal permeability to polyethyleneglycol 600 in Crohn‘s disease. Peroperative determination in a defined segment of the small intestine. Gut, 1988;29(2): 196–199.
18. Ukabam SO, Clamp JR, Cooper BT. Abnormal small intestinal permeability to sugars in patients with Crohn‘s disease of the terminal ileum and colon. Digestion, 1983;27(2): 70–74.
19. Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn‘s disease. Nat Genet, 2008;40(8): 955–962.
20. Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet, 2007;3(4): e58.
21. Marks DJ, Harbord MW, MacAllister R, et al. Defective acute inflammation in Crohn‘s disease: a clinical investigation. Lancet, 2006;367(9511): 668–678.
22. Sewell GW, Marks DJ, Segal AW. The immunopathogenesis of Crohn‘s disease: a three-stage model. Curr Opin Immunol, 2009;21(5): 506–513.
23. Segal AW. Making sense of the cause of Crohn‘s − a new look at an old disease. F1000Res, 2016;5 : 2510–2545.
24. MacDermott RP, Nash GS, Bertovich MJ, et al. Altered patterns of secretion of monomeric IgA and IgA subclass 1 by intestinal mononuclear cells in inflammatory bowel disease. Gastroenterology, 1986;91(2): 379–385.
25. Yoshida EM, Chan NH, Herrick RA, et al. Human immunodeficiency virus infection, the acquired immunodeficiency syndrome, and inflammatory bowel disease. J Clin Gastroenterol, 1996;23(1): 24–28.
26. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol, 2003;3(7): 521–533.
27. Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol, 2009;9(11): 778–788.
28. Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol, 2004;5(8): 800–808.
29. Netea MG, Kullberg BJ, de Jong DJ, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn‘s disease. Eur J Immunol, 2004;34(7): 2052–2059.
30. Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn‘s disease and ulcerative colitis. Inflam Bowel Dis, 2003;9(4): 215–223.
31. Lukas D, Yogev N, Kel JM, et al. TGF-beta inhibitor Smad7 regulates dendritic cell-induced autoimmunity. Proc Ntl Acad Sci USA, 2017, v tlači.
32. Monteleone G, Pallone F, MacDonald TT. Smad7 in TGF-beta-mediated negative regulation of gut inflammation. Trends Immunol, 2004;25(10): 513–517.
33. Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature, 1997;389(6651): 631–635.
34. Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut, 2003;52(1): 65–70.
35. Neurath MF, Finotto S, Fuss I, et al. Regulation of T-cell apoptosis in inflammatory bowel disease: to die or not to die, that is the mucosal question. Trends Immunol, 2001;22(1): 21–26.
36. Verdier J, Begue B, Cerf-Bensussan N, Ruemmele FM. Compartmentalized expression of Th1 and Th17 cytokines in pediatric inflammatory bowel diseases. Inflamm Bowel Dis, 2012;18(7): 1260–1266.
37. Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-Independent IL-17 production regulates intestinal epithelial permeability. Immunity, 2015;43(4): 727–738.
38. Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn‘s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut, 2012;61(12): 1693–1700.
39. Schiering C, Krausgruber T, Chomka A, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature, 2014;513(7519): 564–568.
40. Kugelberg E. Regulatory T cells: alarmin(g) control. Nat Rev Immunol, 2014;14(9): 579.
41. Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol, 2013;14(3): 221–229.
42. Magri G, Miyajima M, Bascones S, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol, 2014;15(4): 354–364.
43. Hepworth MR, Sonnenberg GF. Regulation of the adaptive immune system by innate lymphoid cells. Curr Opin Immunol, 2014;27 : 75–82.
44. Cortez VS, Robinette ML, Colonna M. Innate lymphoid cells: new insights into function and development. Curr Opin Immunol, 2015;32C: 71–77.
45. Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Cin Invest, 2004;113(10): 1490–1497.
46. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells − how did we miss them? Nat Rev Immunol, 2013;13(2): 75–87.
47. Di Sabatino A, Biancheri P, Rovedatti L, et al. New pathogenic paradigms in inflammatory bowel disease. Inflamm Bowel Dis, 2012;18(2): 368–371.
48. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol, 2016;17(2): 122–131.
49. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity, 2015;42(6): 1005–1019.
50. Sponheim J, Pollheimer J, Olsen T et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol, 2010;177(6): 2804–2815.
51. Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol, 2011;7(6): 321–329.
52. Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology, 2005;129(2): 550–564.
53. Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol, 2013;14(6): 536–542.
54. Hufford MM, Kaplan MH. A gut reaction to IL-9. Nat Immunol, 2014;15(7): 599–600.
55. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol, 2014;15(7): 676–686.
56. Leung JM, Davenport M, Wolff MJ, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol, 2014;7(1): 124–133.
57. Geremia A, Biancheri P, Allan P, et al. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev, 2014;13(1): 3–10.
58. Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Ntl Acad Sci USA, 2010;107(17): 8017–8022.
59. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 2012;491(7422): 119–124.
60. Joshi S, Lewis SJ, Creanor S, Ayling RM. Age-related faecal calprotectin, lactoferrin and tumour M2-PK concentrations in healthy volunteers. Ann Clin Biochem, 2009;47(3): 259–263.
61. Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology, 2011;140(6): 1817–1826.
62. Poullis AP, Zar S, Sundaram KK, Moodie SJ, Risley P, Theodossi A, et al. A new, highly sensitive assay for C-reactive protein can aid the differentiation of inflammatory bowel disorders from constipation - and diarrhoea-predominant functional bowel disorders. Eur J Gastroenterol Hepatol, 2002;14(4): 409–412.
63. Reese GE, Constantinides VA, Simillis C, et al. Diagnostic precision of anti-Saccharomyces cerevisiae antibodies and perinuclear antineutrophil cytoplasmic antibodies in inflammatory bowel disease. Am J Gastroenterol, 2006;101(10): 2410–2422.
64. Roggenbuck D, Hausdorf G, Martinez-Gamboa L, et al. Identification of GP2, the major zymogen granule membrane glycoprotein, as the autoantigen of pancreatic antibodies in Crohn‘s disease. Gut, 2009;58(12): 1620–1628.
65. Papp M, Altorjay I, Norman GL, et al. Seroreactivity to microbial components in Crohn‘s disease is associated with ileal involvement, noninflammatory disease behavior and NOD2/CARD15 genotype, but not with risk for surgery in a Hungarian cohort of IBD patients. Inflamm Bowel Dis, 2007;13(8): 984–992.
66. Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn‘s disease-associated I2 sequence and T-cell superantigen. Infect Immun, 2002;70(12): 6567–6575.
67. Reinisch W, Hommes DW, Van Assche G, et al. A dose escalating, placebo controlled, double blind, single dose and multidose, safety and tolerability study of fontolizumab, a humanised anti-interferon gamma antibody, in patients with moderate to severe Crohn‘s disease. Gut, 2006;55(8): 1138–1144.
68. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol, 2014;14(9): 585–600.
69. Biancheri P, Di Sabatino A, Ammoscato F, et al. Absence of a role for interleukin-13 in inflammatory bowel disease. Eur J Immunol, 2014;44(2): 370–385.
70. Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn‘s disease. New Engl J Med, 2010;362(15): 1383–1395.
71. Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. New Engl J Med, 2005;353(23): 2462–2476.
72. Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol, 2014;7(1): 6–19.
73. Jurgens M, Mahachie John JM, Cleynen I, et al. Levels of C-reactive protein are associated with response to infliximab therapy in patients with Crohn‘s disease. Clin Gastroenterol Hepatol, 2011;9(5): 421–427.
74. Sandborn WJ, Gasink C, Gao LL, et al. Ustekinumab induction and maintenance therapy in refractory Crohn‘s disease. N Engl J Med, 2012;367(16): 1519–1528.
75. Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm, 2013;2013 : 963748.
76. Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. New Engl J Med, 2013;369(8): 699–710.
77. Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med, 2012;367(7): 616–624.
78. Wickelgren I. Immunotherapy. Can worms tame the immune system? Science, 2004;305(5681): 170–171.
79. Weinstock JV, Elliott DE. Translatability of helminth therapy in inflammatory bowel diseases. Int J Parasitol, 2013;43(3–4): 245–251.
80. Garg SK, Croft AM, Bager P. Helminth therapy (worms) for induction of remission in inflammatory bowel disease. Cochrane Database Syst Rev, 2014;1: CD009400.
81. Bender E. Cell-based therapy: Cells on trial. Nature, 2016;540(7634): S106–S108.
82. Drew L. Microbiota: Reseeding the gut. Nature, 2016;540(7634): S109–S112.
Labels
Hygiene and epidemiology Medical virology Clinical microbiologyArticle was published in
Epidemiology, Microbiology, Immunology

2017 Issue 4
Most read in this issue
- Humánní alveolární echinokokóza a přehled výskytu tasemnic Echinococcus multilocularis u zvířat v České republice
- Crohnova choroba a ulcerózna kolitída – súčasný pohľad na genetickú determináciu, imunopatogenézu a biologickú liečbu
- Průtoková cytometrie v mikrobiologii
- Mycological diagnosis of pulmonary Aspergillus infections with a focus on serological methods