
Arytmogenní ventrikulární kardiomyopatie
Arytmogenic ventricular cardiomyopathy
Arrhythmogenic ventricular cardiomyopathy is considered to be a primary cardiomyopathy. Over the last few decades, although being a relatively rare disease with its prevalence 1 : 2000 – 1 : 5000, there were numerous studies performed with the aim to elucidate the underlaying causes, pathogenesis, diagnostical aspects and possible treatment options of the disease. Arrhythmogenic ventricular cardiomyopathy is genetically conditioned disease where proteins of the cell-cell junctions are involved. Mutations of the myocardial intercalated dics proteins, mainly desmosomal proteins (e.g.plakoglobin), are held to be responsible for electromechanical instability of the myocardium which causes regressive changes in cardiomyocytes in most cases of arrhythmogenic ventricular cardiomyopathy. Subsequent morphological changes include fibrofatty replacement and inflammation of the myocardium. The condition results in structural changes of the heart hence arrhytmias and other signs of heart disease. There are 3 variants of this cardiomyopathy: ‚classical‘ variant with predominant right ventricular involvement, biventricular and variant with left ventricular predominance. Clinical findings in patients with arrhythmogenic ventricular cardiomyopathy suggested the most appropriate means of the diagnostics and helped to create Task Force Criteria for in vivo diagnosis of the disease. The major pitfall and significance of arrhythmogenic ventricular cardiomyopathy lies in its common presentation as sudden cardiac death affecting mostly young adults.
Keywords:
arrhythmogenic cardiomyopathy – intercalated disc – plakoglobin – desmosome – sudden death
Authors:
Petr Tomášek 1,2; Petra Dohnalová 1; Tereza Kubíková 2; Milena Králíčková 2; Michal Beran 1; Zbyněk Tonar 2
Authors‘ workplace:
Ústav soudního lékařství 2. LF UK a Nemocnice Na Bulovce, Praha
1; Ústav histologie a embryologie LF UK v Plzni
2
Published in:
Soud Lék., 60, 2015, No. 4, p. 51-56
Category:
Review
Overview
Arytmogenní ventrikulární kardiomyopatie je primární strukturální chorobou s fibrolipomatózní náhradou myokardu. Postihuje zpravidla pravou komoru, existují ale i formy biventrikulární a levostranné. Vzniká na podkladě mutací strukturálních proteinů mezibuněčných spojů v interkalárních discích, především v desmozomech a vede k poruše mechanické i elektrické stability myokardu. Manifestuje se u mladších dospělých, primomanifestací je často náhlá srdeční smrt. Prevalence je asi 1 : 2000 až 1 : 5000.
Klíčová slova:
arytmogenní kardiomyopatie – interkalární disk – plakoglobin – desmozom – náhlé úmrtí
Arytmogenní ventrikulární kardiomyopatie (AVC) je geneticky podmíněná choroba, postihující převážně pravou komoru srdeční (PK) a projevující se rozmanitými morfologickými i klinickými příznaky, z nichž nezávažnějším je náhlá srdeční smrt (NSS), která je často i primomanifestací choroby. Prevalence je odhadována asi 1 : 2000 až 1 : 5000, vyšší výskyt je prokázán v oblasti Benátek, a též na řeckém ostrově Naxos, kde se vyskytuje v kombinaci s palmoplantární keratózou a „vlněnými“ vlasy (Naxos Disease). Muži jsou AVC postiženi častěji než ženy, v poměru asi 3 : 1 (1-3). Onemocnění se manifestuje zpravidla v mladším dospělém věku, při fyzické námaze (často u atletů). Objev AVC lze s nadsázkou přisuzovat Itálii. V roce 1736 Giovanni Maria Lancisi (profesor anatomie na univerzitě La Sapienza v Římě a osobní lékař papeže) popsal v knize „De Motu Cordis et Aneurysmatibus“ historii rodiny, kde se po 4 generace vyskytovala choroba, projevující se příznaky dnes popisovanými u AVC (palpitace, dilatace a aneurysmata PK, srdeční selhání, náhlá smrt) (4). Původně byla AVC považována za vrozenou vývojovou vadu a označována za dysplazii (5). Pozdější výzkumy potvrdily, že jde o geneticky podmíněnou chorobu, nikoli o vrozenou fokální absenci myokardu PK a pojem „dysplazie“ v názvu choroby ztratil opodstatnění. V roce 1995 byla tato chorobná jednotka Světovou zdravotnickou organizací zařazena mezi primární kardiomyopatie (6). Vzhledem k tomu, že fenotyp choroby je značně heterogenní a zahrnuje i formy postihující převážně levou komoru (LK), či obě komory, bylo doporučeno užívat širší název arytmogenní kardiomyopatie (7,8). Nejnověji pak byla arytmogenní kardiomyopatie/dysplazie pravé komory (ARVC/D) společnostmi Heart Rhythm Society (HRS) a European Heart Rhythm Association (EHRA) přejmenována na arytmogenní ventrikulární kardiomyopatii (AVC) (9).
ETIOPATOGENEZE
AVC je geneticky podmíněnou chorobou mezibuněčných spojení (10,11). Její příčinou jsou mutace genů kódujících proteiny interkalárního disku (IKD), především desmozomů (12). Změněné proteiny, vzniklé na podkladě genových mutací, podmiňují ultrastrukturální abnormity desmozomů, vedou k jejich remodelaci, zkrácení, fragmentaci a k rupturám IKD (5,13). Ztráta integrity desmozomů může podstatně ovlivnit i další mezibuněčné spoje, jak gap junctions (nexy, které metabolicky i elektricky propojují cytoplazmu sousedních buněk), tak sodíkové membránové kanály. To vše pak vede ke snížení mechanické i elektrické stability myokardu a ke vzniku komorových arytmií ještě před rozvojem zjevného strukturálního poškození (14-16). Rozpojení poškozených IKD napomáhá těžká fyzická námaha. Faktické rozpojení kardiomyocytů (KMC) vede k buněčné smrti cestou apoptózy i nekrózy, doprovázené chronickým zánětem s lymfocytární infiltrací v intersticiu (17,18). Zánik KMC je provázen také jejich progresivní fibrolipomatózní náhradou (19). Tyto histologické změny vytvářejí podmínky pro fenomen reentry (krouživý vzruch) a opožděnou aktivaci komor, spouštějící komorové arytmie (8). Matthes et al. (20) prokázali studií buněčných kultur epikardiálních buněk ze srdcí novorozených potkanů postrádajících gen pro plakophilin-2 (PKP2) zvýšenou rychlost migrace a proliferace těchto buněk a vyslovili hypotézu, že desmozomální mutace mohou vést k myokardiální infiltraci fibroblasty a adipocyty z epikardu. To by bylo v souladu s již prokázaným poznatkem, že fibrolipomatózní infiltrace u AVC postupuje směrem od epikardu k endokardu (8,20). Některé studie naznačovaly též možnou souvislost mezi AVC a virovými, či bakteriálními myokarditidami. Prevalence virového genomu v endomyokardiálních biopsiích (EMB) u pacientů s AVC se udává 0 - 75 %, ale kauzální vliv virů na rozvoj AVC je obtížné prokázat (8,21,22).
MORFOLOGICKÉ PROJEVY, PODTYPY CHOROBY
Dříve byla rozlišována lipomatózní a fibrolipomatózní varianta choroby (23). V recentní literatuře se toto rozdělení neobjevuje a mezi patognomonickými znaky choroby je uváděna fibrolipomatózní náhrada zaniklých KMC. V současné době se uvádějí 3 podtypy (9): 1) klasická AVC s pravostrannou dominancí. PK může být dilatovaná, její stěna ztenčená i zesílená, s drobnými aneurysmaty. Fibrolipomatózní infiltrace progreduje směrem od epikardu k endokardu, někdy je až transmurální, trámčina a papilární svaly však nebývají postiženy (24,25). Časnější rozvoj a maximum změn je v PK obvykle popisováno v tzv. trojúhelníku dysplazie, tedy ve vtokovém traktu subtrikuspidálně, ve výtokovém traktu (right ventricular outflow tract, RVOT) a v hrotu (26). Tento trojúhelník je v současnosti zpochybňován s tím, že k výraznému rozvoji histopatologických změn v hrotu dochází až v pozdních stadiích pravostranné formy (27). 2) biventrikulární AVC je charakterizována časným rozvojem patologických změn v obou komorách současně a v pokročilém stadiu je spojena se systolickou dysfunkcí a dilatací obou komor, s obrazem městnavého srdečního selhávání. 3) AVC s levostrannou dominancí se projevuje časnými histopatologickými změnami v LK, zatímco globální funkce PK není narušená (28-30).
KLINICKÉ PROJEVY
Obvyklou primomanifestací AVC je buď NSS, nebo komorová tachyarytmie, která má u klasické pravostranné AVC původ v PK a morfologii bloku levého raménka Tawarova (LBBB) (31,32). Levostranná AVC se může projevovat komorovou tachykardií (KT) s původem v LK, s morfologií bloku pravého raménka Tawarova (RBBB). Biventrikulární AVC pak může mít morfologii bloku obou Tawarových ramének (30).
Polymorfní KT, či fibrilace komor s letálními následky se zpravidla objevují v latentní fázi choroby, setrvalá monomorfní KT spíše v pokročilejších stádiích, kdy jsou vytvořeny podmínky pro makroreentry (8). AVC může progredovat v těchto fázích: 1) latentní forma, 2) zjevné arytmie, 3) selhání PK, 4) selhání obou komor s progresivní dilatací komor, která může imitovat dilatovanou kardiomyopatii (33,34).
DIAGNOSTIKA
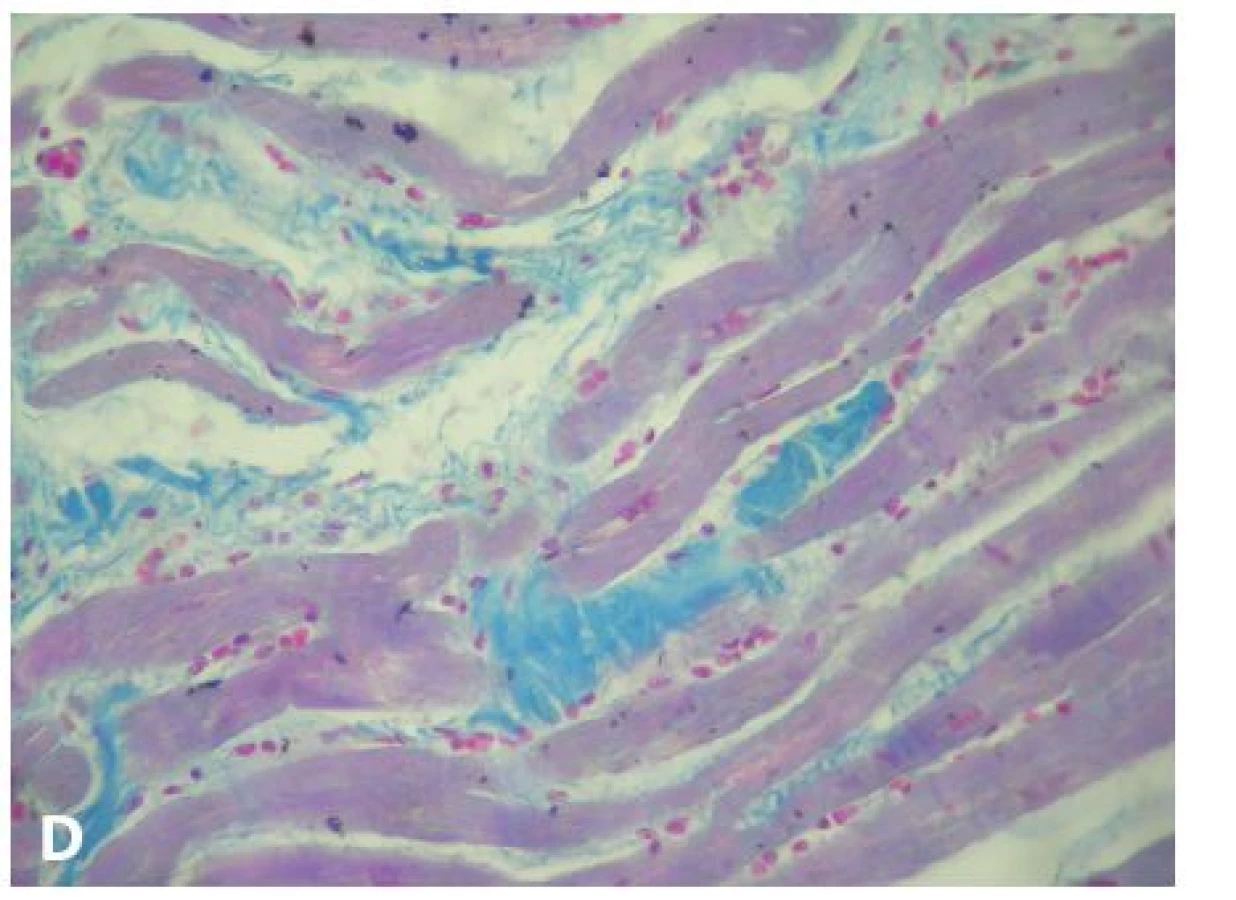
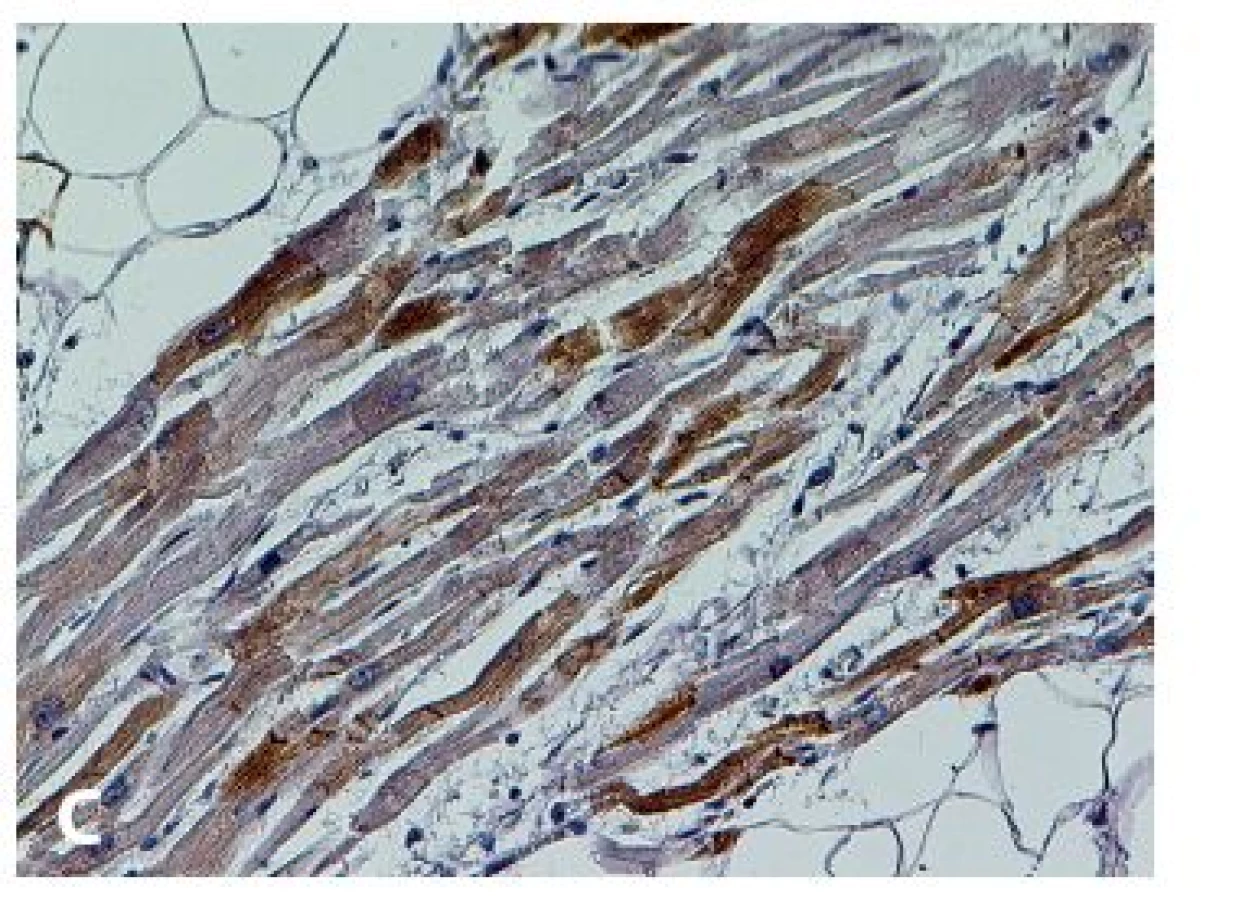
V roce 1994 byla zavedena diagnostická kritéria pro in vivo diagnostiku, vyžadující kvalitativní posouzení těchto parametrů: 1) globální nebo regionální dysfunkce a strukturální poškození, 2) tkáňová charakterizace stěny, 3) repolarizační abnormity, 4) depolarizační abnormity a poruchy vedení, 5) arytmie a 6) rodinná anamnéza. Modifikace kritérií v roce 2010 zvýšila jejich diagnostickou senzitivitu při zachování specificity zavedením kvantitativních parametrů pro zobrazovací metody (magnetická rezonance), EMB (tab. 1) a EKG, a také začleněním genetické analýzy (35,36). Histopatologické změny byly už zmíněny v úvodu. Diagnosticky nejdůležitější je průkaz signifikantní „replacement“ fibrózy, resp. fibrolipomatózní náhrady myokardu (obr. 1). Diagnózu nelze opřít o pouhou lipomatózní infiltraci svaloviny! V některých případech jsou přítomny i degenerativní změny KMC a lymfocytární a histiocytární infiltráty. V oblastech s fibrolipomatózní náhradou lze na reziduálních KMC pozorovat také buněčnou hypertrofii a pyknózu jader (7,23). Imunohistochemická (IHC) analýza není v současné době rutinní a diagnózu spolehlivě prokazující metodou. Proto nelze dát ani doporučení, jaká škála vyšetření by měla být rutinně prováděna při podezření na AVC. Byla provedena řada studií s odlišnými výsledky. Např. Tavora et al. prokázali jen mírný nebo dokonce žádný pokles exprese proteinů mezibuněčných spojů (plakoglobin, plakophilin, desmoplakin, connexin-43, N-cadherin) ve vzorcích s AVC oproti negativním kontrolám a nezjistili ani signifikantní rozdíly v imunoreaktivitě mezi pravou a levou komorou. Současně připouštějí, že na výsledcích studie se mohla do jisté míry podílet použitá metodika. Uvádějí ale zajímavý poznatek, když využili průkaz a rovnoměrnost pozitivity desminu jako kontrolu fixace tkáně (37). Naopak Kwon et al. prokázali, že IHC analýza plakoglobinu má relativně vysokou senzitivitu i specificitu, jako jedinou metodu k průkazu diagnózy AVC ji ale použít nelze (38).




DOPORUČENÍ K POST MORTEM DIAGNOSTICE
S ohledem na výše uvedené rozdíly ve výsledcích IHC metod, které mohou mít podklad nikoli ve fenotypu, ale i v metodologii analýzy, lze pro postmortální diagnostiku doporučit fixaci celého, standardními postupy odpitvaného srdce v pufrovaném formolu, který na rozdíl od formolu kyselého umožňuje delší uchování tkáňových antigenních struktur pro eventuální pozdější opakování, či rozšíření IHC analýzy a je výhodnější i pro analýzu genetickou. V každém případě je nejvhodnější odebrat vzorky z fixovaného srdce již po několikadenní fixaci a převést materiál do parafinových bločků, což umožní zachovat antigenicitu tkáně po dobu podstatně delší než při uložení ve fixačním mediu.
DIFERENCIÁLNÍ DIAGNOSTIKA
Škála chorobných jednotek, které je třeba vyloučit in vivo, je poměrně široká a zahrnuje i změny, které post mortem buď nejsou postižitelné, protože jde o záležitosti funkční (zejména poruchy srdečního rytmu), nebo je lze naopak pitvou dobře ozřejmit a přestávají pak být diferenciálně diagnostickým problémem (např. patologie trikuspidální chlopně, pravokomorový infarkt myokardu či defekt septa síní aj. (39). Okruh chorobných jednotek, které mohou činit potíže v nekroptické diagnostice je tedy o něco užší než in vivo, přesto je ale diferenciální diagnóza AVC obtížná a je nutné postupovat velmi opatrně, aby nedošlo k mylné interpretaci, nadhodnocení či podhodnocení jednotlivých histopatologických nálezů. Nepochybně je nutné odlišit myokarditidu, je-li to možné. Obrovskobuněčná myokarditida a myokarditida při sarkoidóze může být imunohistochemicky neodlišitelná od AVC, naopak virová myokarditida nezpůsobuje redistribuci plakoglobinu v interkalárním disku (40). Uhlova anomálie je někdy považována za jednotku příbuznou AVC. Jde o kongenitální fokální absenci parietálního myokardu PK, zejména v infundibulu, takže stěnu v postiženém místě tvoří pouze endokard s epikardem. Oproti AVC se projevuje už v dětském věku, a to městnavým srdečním selháním (7,41). Levostrannou formu AVC je třeba odlišit od idiopatické myokardiální fibrózy. tj. difuzní intersticiální i „replacement“ fibrózy, postihující zejména zadní stěnu LK za současné nepřítomnosti koronární aterosklerózy a jiných morfologických abnormit (8). U adipositas cordis (cor adiposum) je lipomatóza subepikardiální, s maximem anterolaterálně podél ostré hrany a prakticky s nepřítomností tuku na spodní stěně komor. Množství tuku stoupá s tělesnou hmotností; větší množství tukové tkáně je u žen než u mužů (42). Lipomatózní infiltrace myokardu PK: určité množství tukové tkáně v myokardu vmezeřené mezi svazky KMC je ve volné stěně PK považováno za normální nález, zejména anterolaterálně a apikálně. Hranice mezi tukovou a svalovou tkání je obvykle ostrá, nebo jen lehce setřená (23).
GENETIKA
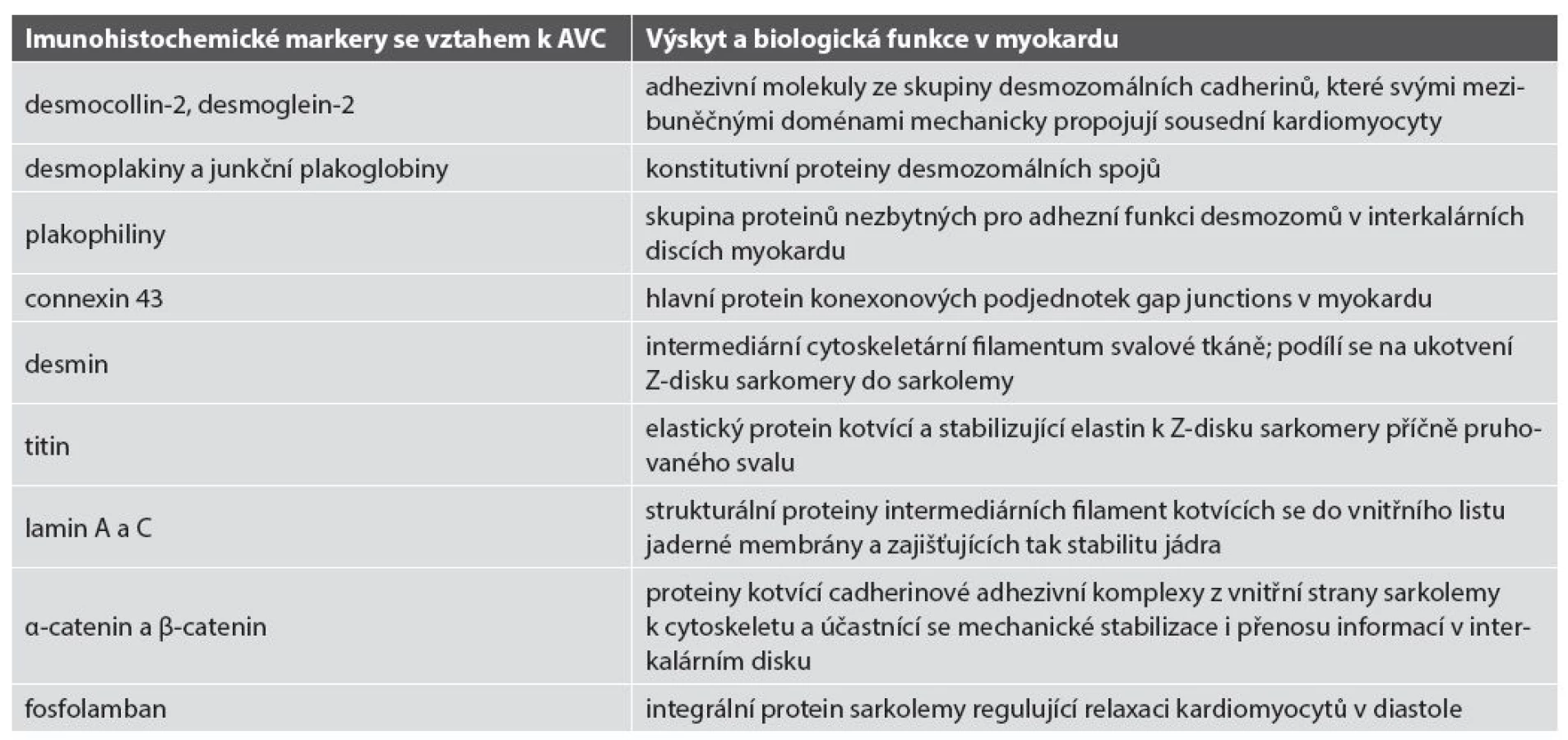
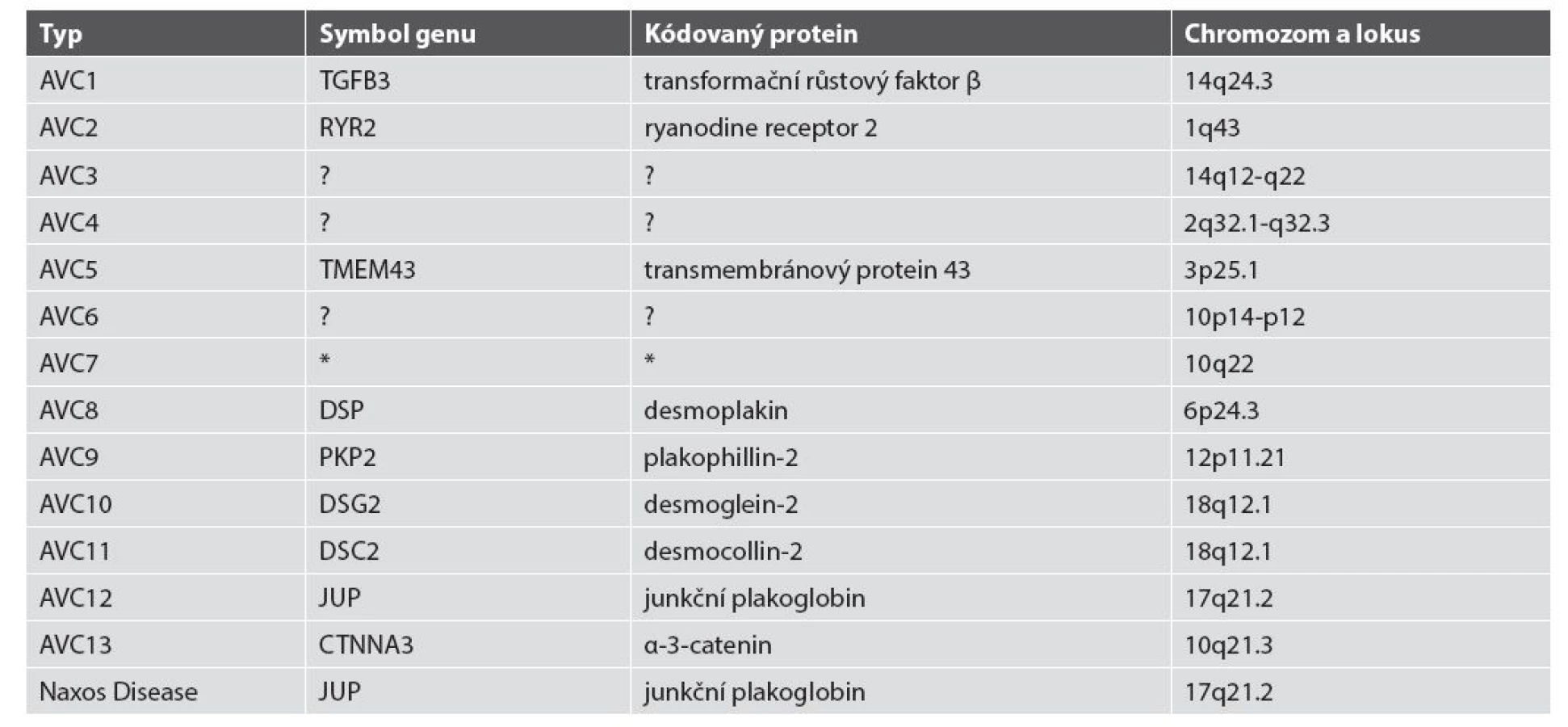
Ve více než 60 % případů AVC byly prokázány mutace genů pro desmozomální proteiny (desmocollin-2, desmoglein-2, desmoplakin, junkční plakoglobin, plakophilin-2) (36). U menší části pacientů s fenotypem AVC byly prokázány i genové mutace pro non-desmozomální proteiny: transformační růstový faktor β, transmembránový protein 43, desmin, titin, lamin A/C, α-3-catenin a fosfolamban (40,43-49) (tab. 2) Doposud bylo uváděno 12 typů AVC dle genových mutací (3,50). Databáze Online Mendelian Inheritance in Man (OMIM, www.omim.org) v roce 2015 uvádí i typ 13. Souhrn dosud známých typů AVC - viz tab. 3.
TERAPEUTICKÉ MOŽNOSTI
Kauzální léčba AVC neexistuje. U všech pacientů je nutná restrikce fyzické aktivity. Léčba se po potvrzení diagnózy odvíjí od klinické stratifikace rizika. Základními terapeutickými strategiemi jsou prevence náhlé smrti a léčba srdečního selhání, k čemuž lze využít farmakoterapii antiarytmiky, katetrovou ablaci, implantabilní kardioverter - defibrilátor (ICD) a v krajních případech transplantaci srdce (51). Experimentálně byla ověřena účast komplementu na progresi choroby. U myší s fenotypem AVC, které měly buď nulové alely pro desmin a receptor pro C5a složku komplementu, nebo nulovou alelu pro desmin a C5a složka komplementu u nich byla inhibována farmakologicky, bylo prokázáno zřetelné zlepšení srdečních funkcí, arytmií, histopatologických nálezů i přežití po vytrvalostní námaze (52).
ZÁVĚR
Navzdory dobře propracované diagnostice AVC in vivo zůstává mnoho otazníků, co je nepodkročitelným minimem pro stanovení této diagnózy post mortem, zejména u pacientů, kde je po této stránce neznámá či zcela němá anamnéza. Za základní kámen k postavení diagnózy lze považovat histopatologické změny, postižitelné ve světelné mikroskopii klasickými barvicími metodami a imunohistochemií (obr. 1,2), popřípadě i genetické vyšetření zajištěného materiálu. Taktéž pečlivý diferenciálně diagnostický rozbor je zde značně důležitý, neboť po potvrzení diagnózy AVC by měl následovat klinický i genetický screening příbuzných.


PODĚKOVÁNÍ
Práce byla podpořena projekty Prvouk P36 a SVV 260 047 Univerzity Karlovy v Praze.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Zkratky
ARVC/D - arytmogenní kardiomyopatie/dysplazie pravé komory
AVC - arytmogenní ventrikulární kardiomyopatie
EMB - endomyokardiální biopsie
IHC - imunohistochemie
IKD - interkalární disk
KMC - kardiomyocyty
KT - komorová tachykardie
LK – levá komora srdeční
NSS - náhlá srdeční smrt
PK – pravá komora srdeční
PKP2 - plakophilin-2
Adresa pro korespondenci:
MUDr. Petr Tomášek
Ústav soudního lékařství 2. LF UK a Nemocnice Na Bulovce,
Budínova 2, 180 81 Praha 8
tel.: +420266083437
fax: +420266083450
e-mail: petr.tom@seznam.cz
Sources
1. Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998; 97 : 2049–2058.
2. Herren T, Gerber PA, Duru F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a not so rare “disease of the desmosome” with multiple clinical presentations. Clin Res Cardiol 2009; 98 : 141–158.
3. Azaouagh A, Churzidse S, Konorza T, Erbel R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. Clin Res Cardiol 2011; 100 : 383–394.
4. Thiene G. Arrhythmogenic cardiomyopathy: from autopsy to genes and transgenic mice (SCVP Achievement Award Lecture, San Antonio, TX, February 27, 2011), Cardiovasc Pathol 2012; 21(4): 229-239.
5. Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J 1996; 132 : 203–206.
6. Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93 : 841–842.
7. Šteiner I. Kardiopatologie. Praha: Galén; 2010 : 51-52.
8. Saguner AM, Brunckhorst C, Duru F. Arrhythmogenic ventricular cardiomyopathy: A paradigm shift from right to biventricular disease. World J Cardiol 2014; 6(4): 154–174.
9. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011; 13 : 1077–1109.
10. Thiene G, Corrado D, Basso C. Cardiomyopathies: is it time for a molecular classification? Eur Heart J 2004; 25 : 1772–1775.
11. Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113 : 1807–1816.
12. Fressart V, Duthoit G, Donal E, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 2010; 12 : 861–868.
13. Basso C, Czarnowska E, Della Barbera M, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J 2006; 27 : 1847-1854.
14. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 2010; 107 : 700–714.
15. Sato PY, Coombs W, Lin X, et al. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res 2011; 109 : 193–201.
16. Gomes J, Finlay M, Ahmed AK, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J 2012; 33 : 1942–1953.
17. Bauce B, Basso C, Rampazzo A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J 2005; 26 : 1666–1675.
18. Khan A, Mittal S, Sherrid MV. Arrhythmogenic right ventricular dysplasia: from genetics to treatment. Anadolu Kardiyol Derg 2009; 9(Suppl 2): 24–31.
19. McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994; 71 : 215-218.
20. Matthes SA, Taffet S, Delmar M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun Adhes 2011; 18 : 73–84.
21. H Fischer A, van der Loo B, M Shär G, et al. Serological evidence for the association of Bartonella henselae infection with arrhythmogenic right ventricular cardiomyopathy. Clin Cardiol 2008; 31 : 469–471.
22. Bowles NE, Ni J, Marcus F, Towbin JA. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2002; 39 : 892–895.
23. Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc Pathol 2005; 14 : 37-41
24. Corrado D, Basso C, Nava A, Rossi L, Thiene G. Sudden death in young people with apparently isolated mitral valve prolapse. G Ital Cardiol 1997; 27 : 1097–1105.
25. Tabib A, Loire R, Chalabreysse L, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003; 108 : 3000–3005.
26. Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982; 65 : 384–398.
27. Te Riele AS, James CA, Philips B, et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol 2013; 24 : 1311–1320.
28. Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008; 52 : 2175–2187.
29. Kjaergaard J, Hastrup Svendsen J, Sogaard P, et al. Advanced quantitative echocardiography in arrhythmogenic right ventricular cardiomyopathy. J Am Soc Echocardiogr 2007; 20 : 27–35.
30. Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med 2010; 61 : 233-253.
31. Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A. Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 2000; 31 : 988–998.
32. Marcus FI, Fontaine G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: a review. Pacing Clin Electrophysiol 1995; 18 : 1298–1314.
33. Corrado D, Fontaine G, Marcus FI, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000; 101(11): E101-106.
34. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996; 94(5): 983-991.
35. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31 : 806-814.
36. Riele A, Tandri H, Bluemke D. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update, J Cardiovasc Magn Reson 2014; 16(1): 50.
37. Tavora F, Zhang M, Cresswell M, Li L, Fowler D, Franco M, Burke A. Quantitative Immunohistochemistry of Desmosomal Proteins (Plakoglobin, Desmoplakin and Plakophilin), Connexin-43, and N-cadherin in Arrhythmogenic Cardiomyopathy: An Autopsy Study. Open Cardiovascular Medicine Journal 2013; 7 : 28–35.
38. Kwon YS, Park TI, Cho Y, Bae MH, Kim S. Clinical usefulness of immunohistochemistry for plakoglobin, N-cadherin, and connexin-43 in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Int J Clin Exp Pathol 2013; 6(12): 2928-2935.
39. Romero J, Mejia-Lopez E, Manrique C, Lucariello R. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC/D): A Systematic Literature Review. Clin Med Insights Cardiol 2013; 7 : 97–114.
40. Groeneweg JA, van der Heijden JF, Dooijes van Veen TAB, van Tintelen JP, Hauer RN. Arrhythmogenic cardiomyopathy: diagnosis, genetic background, and risk management. Neth Heart J 2014; 22(7-8): 316–325.
41. Gerlis LM, Schmidt-Ott SC, Ho SY, Anderson RH. Dysplastic conditions of the right ventricular myocardium: Uhl’s anomaly vs arrhythmogenic right ventricular dysplasia. British Heart Journal 1993; 69(2): 142-150.
42. Schejbal V. Epicardial fatty tissue of the right ventricle – morphology, morphometry and functional significance. Pneumologie 1989; 43 : 490-499.
43. Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 2005; 65(2): 366–373.
44. Merner ND, Hodgkinson KA, Haywood AF, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008; 82(4): 809–821.
45. Van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009; 6(11): 1574–1583.
46. Taylor M, Graw S, Sinagra G, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011; 124(8): 876–885.
47. Quarta G, Syrris P, Ashworth M, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2012; 33(9): 1128–1136.
48. Van Hengel J, Calore M, Bauce B, et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2013; 34(3): 201–210.
49. Van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail 2012; 14(11): 1199–1207.
50. McNally E, MacLeod H, Dellefave-Castillo L. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. 2005 Apr 18 [Updated 2014 Jan 9]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1131/
51. Rigato I, Corrado D, Basso C. Pharmacotherapy and other therapeutic modalities for managing arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Drugs Ther 2015; 29 : 171–177.
52. Mavroidis M, Davos C H, Psarras S. Complement system modulation as a target for treatment of arrhythmogenic cardiomyopathy. Basic Res Cardiol 2015; 110(3): 27.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Forensic Medicine

2015 Issue 4
Most read in this issue
- Traumatická asfyxie: Autoptická kazuistika
- Arytmogenní ventrikulární kardiomyopatie
- Analýza krevních stříkanců na příkladech z praxe: Jsou výpočty po aplikaci parabolické trajektorie využitelné?
- Traumatická pseudoaneuryzma descendentnej torakálnej aorty riešená aortálnym stentgraftom s následnou fatálnou aortoezofageálnou fistulou