
Karcinom pankreatu: Molekulární biologie a časná diagnostika
Pancreatic cancer: Molecular biology and early detection
Pancreatic cancer is a disease with high malignant potential and is the fourth leading cause of cancer-related death for both men and women. Most patients die within 1 year of diagnosis. It is possible that one out of six cancers occurs in a setting of inherited risk. Like all epithelial malignancies, pancreatic cancer arises by a stepwise progression from non-invasive precursors called PanIN (Pancreatic intraepithelial Neoplasia1, 2, 3), which together with IPMN (Intraductal papillary mucinous Neoplasms) and MCN (Mucinous Cystic Neoplasms) represent lesions with malignant potential. Their histological progression follows genetic progression and some of them can be detected by imaging methods. Identification of patients with hereditary predisposition, early detection of pancreatic cancer precursors and identification of specific molecular alterations to enable rational therapy provide hope for early diagnosis and effective treatment.
Key words:
pancreatic cancer, pathogenesis, screening.
Authors:
A. Ságlová; J. Špičák; T. Hucl
Authors‘ workplace:
Přednosta: prof. MUDr. Julius Špičák, CSc.
; Klinika hepatogastroenterologie IKEM
Published in:
Prakt. Lék. 2010; 90(8): 489-493
Category:
Diagnostis
Overview
Karcinom pankreatu se řadí mezi onemocnění s vysokým maligním potenciálem a je čtvrtou nejčastější příčinou úmrtí na zhoubné onemocnění u obou pohlaví. Většina pacientů umírá méně než do 1 roku od stanovení diagnózy. Přibližně až jeden ze šesti karcinomů se rozvíjí na podkladě vrozené predispozice. Podobně jako je tomu u jiných epitelových nádorů, vzniká karcinom pankreatu postupnou progresí z neinvazivních prekurzorů, jimiž jsou tzv. PanIN (Pancreatic Intraepithelial Neoplasia1, 2, 3 ), které společně s IPMN (Intraductal Papillary Mucinous Neoplasms) a MCN (Mucinous Cystic Neoplasms) tvoří skupinu lézí s maligním potenciálem. Jejich histologická progrese sleduje progresi genetickou a některé z nich lze detekovat zobrazovacími metodami. Identifikace pacientů s vrozenou predispozicí, včasné rozpoznání prekurzorů karcinomu pankreatu či nalezení specifických molekulárních alterací umožňujících racionální terapii tak představují nadějné způsoby včasné diagnostiky a účinné léčby.
Klíčová slova:
karcinom pankreatu, patogeneze, screening.
Úvod
Incidence karcinomu pankreatu v České republice se pohybuje kolem 9 / 100 tisíc obyvatel (dle dat z Národního onkologického registru) a lineárně stoupá po 50 letech věku (1). V naprosté většině případů se jedná o adenokarcinom z duktálního epitelu. Iniciálními příznaky onemocnění jsou:
- dyspepsie,
- váhový úbytek,
- anorexie,
- únava,
- bolest,
- změna defekačních stereotypů, nebo
- vznik ikteru či diabetu.
Vyšetřením je odhaleno ložisko lokalizované většinou v hlavě pankreatu. Hlavním důvodem špatné prognózy je fakt, že v čase diagnózy se většina pacientů nachází již v pokročilém stádiu onemocnění (2). Jedinou léčbou pro tyto nemocné je paliace (např. implantace stentu do obturovaných žlučových cest) či nekurativní chemoterapie nebo chemoradioterapie. Jen nevelké procento nemocných jsou kandidáty chirurgické resekce, která jako jediná léčebná metoda prokazatelně prodlužuje medián přežití (3). I tito však nakonec většinou podlehnou generalizaci.
Identifikace vysoce rizikových pacientů a jejich screening může vést k detekci a léčbě asymptomatických časných stádií. Narůstající poznatky o molekulární patogenezi karcinomu pankreatu představují naději na pochopení, a tím i účinnou diagnostiku a léčbu této choroby.
Genetika
V průběhu onkogeneze duktálního adenokarcinomu dochází postupně k akumulaci genetických a epigenetických alterací v onkogenech, tumor supresorových genech či genech udržujících neporušený genom.
Onkogeny jsou výsledkem mutace genů zvaných protoonkogeny. Somatické mutace v protoonkogenu KRAS2 se vyskytují u více než 95 % duktálních adenokarcinomů pankreatu (4,5). Je lokalizován na 12. chromozomu a jeho mutace jsou časné v patogenezi adenokarcinomu pankreatu.
Karcinom pankreatu je dále asociován s inaktivací tří tumor supresorových genů:
- p53,
- CDKN2A (p16), a
- MADH4 (DPC4, SMAD4).
Ztráta jejich funkce vede k selhání regulace buněčného cyklu.
Gen p53 je lokalizován na 17. chromozomu, kóduje transkripční faktory regulující expresi genů důležitých pro buněčný cyklus, apoptózu a opravu DNA. Jeho inaktivace se vyskytuje u 50 až 75 % pankreatického duktálního adenokarcinomu (6, 7).
Gen CDKN2Alokalizovaný na 16. chromozomu je inaktivován téměř u všech pankreatických karcinomů, jeho produktem jsou proteiny inhibující vstup do S fáze buněčného cyklu inhibicí cyklin-dependentní kinázy. Jeho inaktivace je zapříčiněna delecí, mutací se ztátou heterozygosity nebo hypermethylací promotoru.
Gen MADH4 (DPC4, SMAD4) je inaktivovaný mutací či homozygotní delecí u asi 50 % nádorů (8). Gen je lokalizován na 18. chromozomu, kóduje mediátory TGF-beta dráhy, která inhibuje buněčný růst.
Genová rodina Fanconiho anémie (FA) se skládá z nejméně 13 genů. Defekty v genech FA se kromě Fanconiho anémie vyskytují také u solidních nádorů v běžné populaci. FANCC , FANCG a BRCA2 geny jsou mutované u karcinomů pankreatu.
Existuje skupina genů vykazujících somatické aberace s nižší frekvencí. Většina nádorů pak nese mutaci alespoň jednoho z těchto nízkofrekvenčních genů:
- TGF-beta receptory I a II (TGFBR1 a TGFBR2) (9),
- aktivinové receptory IB a II (ACVR1B a ACVR2) (10),
- LKB1/STK11 kináza (11),
- RB1 substrát p16 regulovaných cyklin dependentních kináz (12),
- MKK4 mediátor stresem aktivované dráhy protein kináz (13),
- EP 300,
- BAX,
- BRAF,
- BUB1 (14–24).
Mutace mitochondriálního genomu jsou přítomny u téměř všech dobře studovaných případů (25).
Vedle mutací bylo popsáno několik míst genové amplifikace AKT2, MYB, MDM2, KRAS2, AIB1 (19-21).
Jedním z významů znalostí molekulárních alterací je jejich využití v diagnostice. Nejvíce prozkoumanou genetickou alterací u adenokarcinomu pankreatu je K-ras mutace. Tuto mutaci lze detekovat v pankreatické šťávě, v aspirátu tenkou jehlou, v duodenálním obsahu, ve žluči a ve stolici u pacientů s karcinomem pankreatu (26). Stejně je tomu tak i u mutací p53, CDKN24 a MADH4. K-ras mutace může být ale identifikována i u pacientů s chronickou pankreatitidou, což limituje její specificitu.
Detekce p53 mutace se zdá být více slibná pro svou větší specificitu, objevuje se však v patogenezi karcinomu později. Spíše než přítomnost mutace CDKN24 je přínosnější detekce hypermethylace CDKN24. Užití těchto technik v běžné klinické praxi prozatím chybí.
Familiární predispozice
Patrně až 1 ze 6 karcinomů pankreatu vzniká na pozadí vrozené predispozice (27, 28). Toto číslo je mezi karcinomy neobvykle vysoké. Pacienty s vrozenou dispozicí můžeme rozdělit do 3 skupin:
- geneticky definované syndromy, jejichž součástí je karcinom pankreatu,
- nakupení karcinomu pankreatu bez zjevného genetického syndromu,
- karcinom pankreatu u přímých příbuzných pacientů s jiným tumorem (nikoli pankreatu).
V rodinách se 2 postiženými členy má každý další člen rodiny 18-ti násobné riziko vzniku karcinomu pankreatu. Rodiny se 3 členy mají riziko zvýšené 60 krát (29).
BRCA2 mutace
proteinový produkt BRCA2 genu hraje roli v interakci s proteiny zapojenými do regulace buněčného cyklu, regulace transkripce a opravy DNA. Mutace v BRCA2 genu typicky způsobují zdánlivě sporadické formy karcinomu díky jejich nízké penetranci. Relativní riziko rozvoje pankreatického karcinomu u nositele této mutace je 5,9 (30).
Familial Atypical Multiple Mole and Melanoma syndrom (FAMMM)
je autozomálně dominantní syndrom s inkompletní penetrancí způsobený vrozenou mutací genu CDKN2A. Je charakterizován mnohočetnými atypickými névy a spojený se zvýšeným rizikem výskytu melanomu a karcinomu pankreatu (31), který je druhým nejčastějším karcinomem v rámci tohoto syndromu (32) a riziko jeho vzniku je 16 % (33).
Peutz-Jeghers syndrom
zodpovědnou genetickou abnormalitou je mutace genu LKB1/STK11, který kóduje serin/threonin kinázu. Postižení trpí gastrointestinálními polypy typu hamartomů a asi 30 % karcinomem pankreatu (11, 34).
Syndrom hereditárního nepolypózního kolorektálního karcinomu (HNPCC)
způsobený vrozenými defekty v genech HMSH2, HMLH1 a HPMS2 opravujících záměny v DNA také predisponuje ke karcinomu pankreatu. Tyto karcinomy jsou charakterizovány mikrosatelitní nestabilitou a mají často medulární fenotyp (35).
Familiární adenomatózní polypóza (FAP)
autozomálně dominantní onemocnění s téměř kompletní penetrancí, charakterizované mnohočetnými polypy kolon s maligním potenciálem a se zvýšeným rizikem extrakolonických malignit. Riziko rozvoje pankreatické malignity je 4,5 krát zvýšené oproti běžné populaci (26).
Hereditární pankreatitida
klíčové jsou vrozené mutace v PRSS1 genu. Mechanismus rezistence k inaktivaci vede k rozvoji autodigesce a zánětlivé odpovědi. Předpokládá se, že karcinom vzniká jako důsledek zvýšeného obratu epiteliálních buněk duktů, jako výsledek chronického zánětu. Toto představuje jedinečný příklad, kde vrozeně zvýšené riziko karcinomu není způsobené žádným ze 3 základních typů tumorigenních genů. Některé rodiny mají až 60-ti násobné riziko vzniku karcinomu pankreatu (36).
Ataxia teleangiectasia
syndrom spojený se ztrátou ATM genu, jehož proteinový produkt hraje důležitou roli v buněčné odpovědi na genetický stres. Ačkoli mechanismus není jasný, ATM indukuje zástavu buněčného cyklu a DNA opravu nebo apoptózu skrze p53. Pacienti s ATM mutací mají trojnásobně vyšší riziko rozvoje karcinomu pankreatu (26).
Li-Fraumeni syndrom
jedná se o vrozenou mutaci p53, která postiženeného predisponuje k mnoha neoplaziím, přičemž pankreatické jsou poměrně vzácné.
Familiární karcinom pankreatu
byl popsán v roce 1987 a dosud není zcela jednoznačně definován. Většinou se popisuje jako přítomnost alespoň 2 příbuzných prvního stupně s karcinomem pankreatu bez akumulace jiných nádorových a familiárních onemocnění nebo alespoň 3 příbuzných jakéhokoliv stupně s pankreatickým karcinomem nebo 2 příbuzných jakéhokoliv stupně s karcinomem pankreatu, jejichž součet věku v čase diagnózy je menší než 110 let (37).
Kauzální mutace zůstává neznámá, ale asi 1 ze 6 rodin s familiárním karcinomem s mnohočetným postižením členů (3 a více) je nositelem vrozené BRCA2 mutace (38). Riziko rozvoje karcinomu pankreatu u osob s 2 příbuznými 1. stupně postiženými tímto karcinomem je 18 krát vyšší, při postižení 3 příbuzných 1. stupně je riziko 57 krát vyšší (37).
Prekurzory karcinomu pankreatu
Většinu nádorů pankreatu představuje duktální adenokarcinom. Podobně jako jiné epitelové karcinomy i duktální adenokarcinom pankreatu vzniká postupnou progresí z neinvazivních prekurzorů. V současné době jsou definovány celkem 3 léze s maligním potenciálem:
- PanIN ( Pancreatic Intraepithelial Neoplasia)
- IPMN ( Intraductal Papillary Mucinous Neoplasms)
- MCN ( Mucinous Cystic Neoplasms)
PanIN, neboli pankreatická intraepiteliální neoplazie, bývá nejčastěji lokalizována v hlavě pankreatu, vzniká především v menších pankreatických vývodech a je klasifikována do tří stupňů.
Nejnižším je PanIN-1; její výskyt se popisuje až u 1/2 starých osob a přibližně 1 z 500 dospěje do stádia invaze. PanIN-1A je charakterizována náhradou kuboidních buněk pankreatických vývodů vysokými protáhlými buňkami s malými jádry lokalizovanými při bazi. V případě, že tyto buňky vytvoří papilární strukturu, hodnotí se jako PanIN-1B.
PanIN-2 jsou méně časté, nalézáné u asi 10 % starých osob, mají však vyšší maligní potenciál a typické jsou pro ně abnormality jader, např. ztráta polarity, zvětšení či kondenzace chromatinu.
U PanIN-3 lézí nacházíme kribriformí shluky epiteliálních buněk oddělující se do lumen společně s nekrózami a těžšími jadernými atypiemi. PanIN-3 léze se většinou vyskytují společně s jednoznačným invazivním karcinomem (39).
Progrese morfologická je důsledkem progrese genetické (40–43) (Obr. 1). Chromozomální nestabilita je považována za časné poškození v průběhu tumorogeneze. Je přítomná téměř u všech PanIN-1 (23).
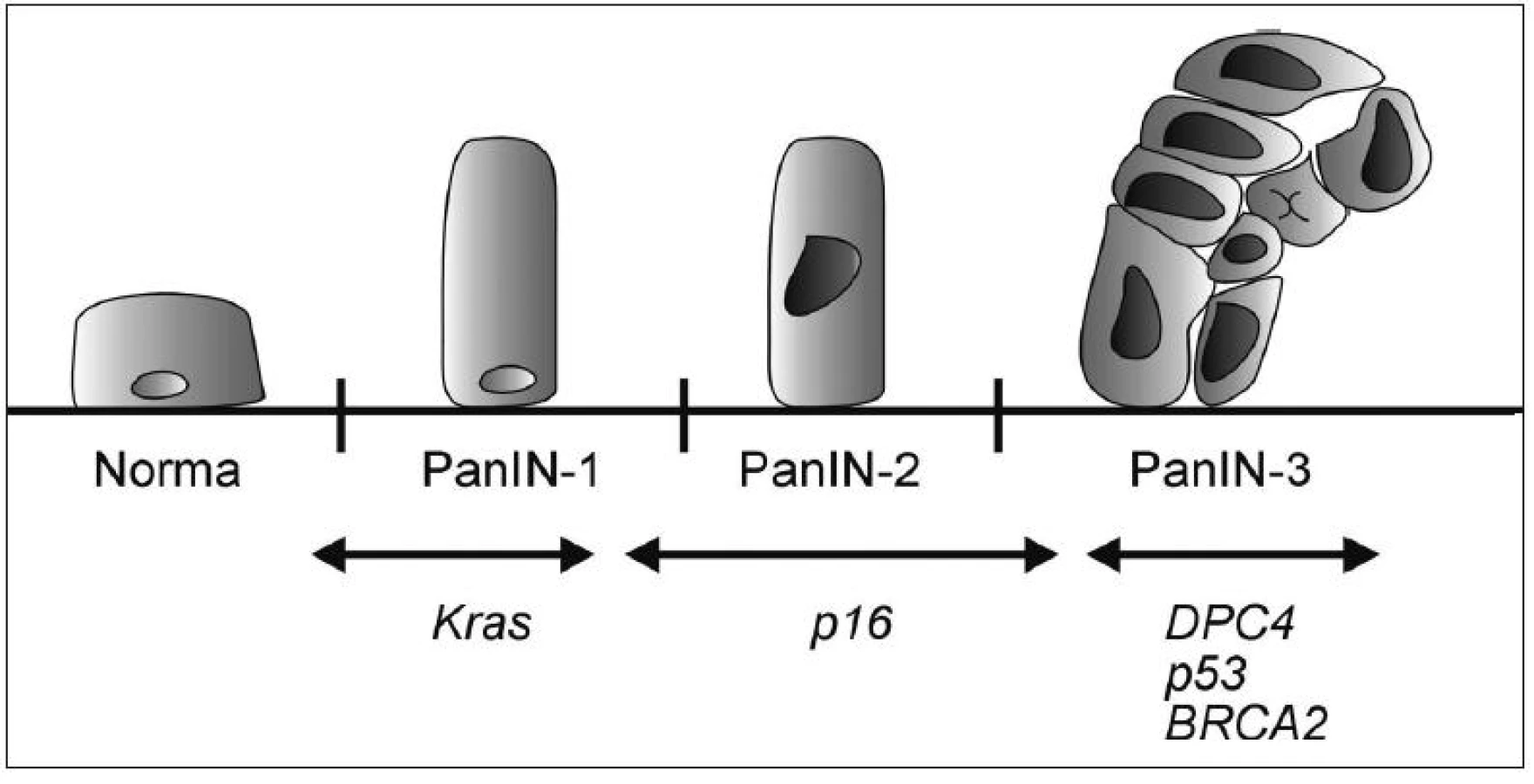
Mutace Kras2 protoonkogenu se vyskytuje asi u poloviny PanIN-1 lézí (24). Tato frekvence se zvyšuje u PanIN-2, kdy se navíc přidávají CDKN2A (p16) abnormality (44). Inaktivace proteinu p53 je považována za pozdější a objevuje se u vyšších stádií PanIN. Ztráta MADH4 proteinu je charakteristická pro PanIN-3 léze (45). Zdá se pravděpodobné, že tato uspořádaná progrese intraduktální neoplazie vede k takové konstelaci genetického poškození, které je nakonec odpovědné za invazivní a metastatický charakter tumoru (39, 40).
Jinou abnormalitou pankreatických vývodů s maligním potenciálem jsou IPMN (Intraductal Papillary Mucinous Neoplasms). Jejich průměrná velikost většinou nepřesahuje 1 centimetr a lze je zachytit zobrazovacími metodami. IPMN vycházející z hlavního pankreatického vývodu (main duct type) mají větší maligní potenciál a představují ve většině případů indikaci k chirurgickému řešení. Rozhodnutí o léčbě IPMN, vycházejících z větví hlavního vývodu (branch duct type), je složitější a záleží na více faktorech, kterými jsou např. přítomnost obtíží nebo vztah k hlavnímu vývodu (46-48).
MCN – mucinózní cystické nádory – tvoří 40 až 50 % všech cystických lézí pankreatu a nekomunikují s pankreatickými vývody. Jsou nejčastěji lokalizované v těle a ocasu, obsahují stroma ovariálního typu a vyskytují se především u žen. Většina z nich obsahuje adenomové struktury, v části případů je přítomen duktální adenokarcinom (46–48). Jsou indikací k chirurgické resekci (49).
Prevence
Základním preventivním opatřením je eliminace ovlivnitelných rizikových faktorů. Dosud bylo identifikováno několik faktorů, které zvyšují riziko rozvoje karcinomu pankreatu. Věk, kouření, obezita, chronický zánět pankreatu, rasa, diabetes mellitus či přítomnost některých nádorových onemocnění v rodině riziko vzniku karcinomu významně zvyšují (50).
Role diety bohaté na tuky a maso zůstává diskutabilní (51). Asi 25 % karcinomů pankreatu je spojováno s kouřením cigaret, kouření déle jak 20 let dvojnásobně zvyšuje riziko jeho vzniku, navíc u kuřáků, jejichž rodinná anamnéza je pozitivní ve smyslu karcinomu pankreatu, je riziko 3,7 krát vyšší (52) nebo se onemocnění projeví o jednu až dvě dekády dříve (53). Znalost těchto faktorů a prevence těch ovlivnitelných dávají určitou naději na snížení výskytu tohoto onemocnění.
Screening a časná diagnostika
Cílem screeningu je zachytit chorobu v časné neinvazivní fázi, kdy je ještě léčitelná. Prvním krokem je identifikace pacientů s vysokým rizikem. Kandidáty screeningu jsou pacienti s
- hereditární pankreatitidou,
- Peutz-Jeghers syndromem,
- FAMMM syndromem,
- BRCA syndromem,
- příbuzní starší 55 let v rodině s HNPCC s výskytem kolorektálního a endometriálního karcinomu,
- rodiny s familiárním výskytem karcinomu pankreatu při postižení alespoň 2 příbuzných prvního stupně (54).
Další diskutovanou skupinou, která je cílem mnohých probíhajících studií, jsou pacienti s nově vzniklým diabetes mellitus. Mnoho pacientů s karcinomem pankreatu rozvine diabetes mellitus již v asymptomatickém období onemocnění. Existují i názory, že pacienti s nově vzniklým diabetem bez pozitivní rodinné anamnézy by měli podstoupit screeninové vyšetření (55).
Vyvstává otázka: kdy screening u rizikových pacientů zahájit?
Karcinom pankreatu je neobvyklý u pacientů mladších 45 let a zcela vzácný ve věku pod 25 let (56). U familiárního karcinomu pankreatu riziko spojené s věkem závisí na věku v čase diagnózy jednotlivých postižených v rámci konkrétní rodiny. U následujících generací je tendence k časnějšímu rozvoji onemocnění (57), riziko se významně zvyšuje ve věku v čase diagnózy postiženého sourozence a v nižším věku, než byl věk v čase diagnózy u postiženého rodiče. Průměrný věk rozvoje onemocnění je v 6. dekádě, screening by měl začít ve 40 letech anebo o 10 let dříve, než byl věk nejmladšího postiženého v rodině (2). Postižení Peutz-Jeghers syndromem onemocní karcinomem pankreatu nejčastěji ve 3. a 4. dekádě, proto by měli být pravidelně vyšetřováni od 30 let (2).
Prozatím neexistuje doporučení, jak často vysoce rizikové pacienty podrobit screeningovému vyšetření, obecně je však doporučováno rozmezí jeden až tři roky.
Jednoduchá screeningová metoda pro adenokarcinom pankreatu prozatím neexistuje, avšak nadějnou se do budoucna zdá být kombinace biomarkeru v plazmě nebo pankreatické šťávě se zobrazovací metodou.
Mezi nejvíce prozkoumané biochemické markery patří CA 19-9 a postprandiální glykémie. Vyšetřování tumor markeru CA19-9 se neukázalo jako efektivní (58), jen 50 % karcinomů pod 2 cm má současně elevovanou hladinu CA 19-9. Postprandiální a lačná glykémie jsou další možné modality, neboť rozvoj inzulinové rezistence je asociován se vznikem karcinomu pankreatu (59).
Ze zobrazovacích metod má pro ložiska v pankreatu vysokou senzitivitu (více jak 90 %) endoskopická sonografie (EUS), umožňuje identifikovat i velmi časné tumory a prekancerózní léze, především IPMN (58). Další výhodou je možnost odběru aspirátu tenkou jehlou. Nelze však pomocí EUS rozlišit maligní a benigní charakter léze.
Vyšetření počítačovou tomografií (CT) je běžně preferováno k zobrazení a stagingu pankreatických onemocnění, avšak nevýhodou je nemožnost záchytu velmi malých neinvazivních lézí a expozice záření. Jako screeningová metoda je tedy méně vhodné než EUS.
Magnetická rezonance (MR) je alternativní metoda bez přítomnosti radiace, může zachytit i poměrně malé léze, ale limitující je malé rozlišení a pohybové artefakty (60).
Lze též aplikovat endoskopickou retrográdní cholangiopankreatikografii (ERCP) s využitím současného odběru vzorku pankreatické šťávy, benefit však musí převážit riziko vzniku možných komplikací.
Několik center zabývajících se touto problematikou vytvořilo screeningové protokoly. The European Registry of Hereditary Pancreatitis and Familiar Pancreatic Cancer (EUROPAC) využívá EUS zobrazení a CT společně s hladinou CA19-9 a glykémií nalačno. Pacienti bez potenciálně maligní léze podstupují ještě ERCP společně s analýzou pankreatické šťávy, ve které se zkoumá přítomnost p53 a Kras mutace a CDKN2A hypermethylace. Výsledky studie za použití tohoto protokolu dosud nebyly publikovány.
University of Washington podrobuje rizikové pacienty EUS, při nálezu abnormality následuje ERCP vyšetření. Ve studii využívající tento protokol bylo 75 pacientů, u 15 z nich byla abnormalita na obou použitých zobrazovacích metodách, v 10 případech se jednalo o PanIN-3 lézi, v 5 případech PanIN-2, nebyl detekován žádný karcinom. Jeden pacient rozvinul inoperabilní malignitu pankreatu v době sledování.
Moffitt Cancer Centre využívá protokol, ve kterém jsou vysoce rizikoví pacienti vyšetřeni EUS, eventuálně s aspirační biopsií tenkou jehlou (FNAB). Dále navíc podstoupí CT vyšetření. Výsledky této studie dosud nebyly publikovány.
Jinou strategií je použití EUS a MR v ročních intervalech, v případě zjištění abnormality doplnění FNAB. Teprve výsledky výše popsaných protokolů určí budoucí standard.
Cílená terapie
Cílená terapie znamená využití znalostí specifických modulačních alterací určitého typu nádorového onemocnění k rozvoji nových specifických terapeutických postupů. Aberace nezbytné pro růst nádorové buňky se stávají terčem protinádorové léčby. Jednou z možností je inhibice aktivované signální dráhy. Vysoký výskyt mutací Kras2 u karcinomu pankreatu nabízel naději při užití farnesyl transferázových inhibitorů či jiných inibitorů funkce Ras proteinů. Alternativně lze využít absolutního biochemického rozdílu mezi nádorovými a nenádorovými buňkami. V tomto případě je cílem terapie absolutní ztráta genové funkce, tudíž rakovinová buňka nemůže vyvinout rezistenci k léčbě. Inaktivované geny tak mohou být vhodné jako cíl nových terapeutik vysoce specifických pro nádorové buňky. Např. buňky postrádající BRCA2 protein a buňky defektní v homologní rekombinaci poškozené DNA jsou zvýšeně citlivé k mitomycinu C či jiným preparátům způsobujícím meziřetězcové můstky nebo jiným látkám způsobujícím poškození DNA (60).
Závěr
K racionalizaci screeningu, diagnostiky a terapie karcinomu pankreatu je nutné zohlednění nově objevených patogenetických principů. Vzhledem k vyčerpání možností chirurgické či standardní onkologické léčby to představuje jediný potenciál ke zlepšení prognózy takto závažného onemocnění.
MUDr. Tomáš Hucl, Ph.D.
Klinika
hepatogastroenterologie IKEM,
Vídeňská
1958/9,
140
21 Praha 4
adela.saglova@ikem.cz
tomas.hucl@ikem.cz
Sources
1. Ries, L.A., Eisner, M.P., Kosary, C.L. et al. SEER Cancer Statistics Review, 1973-1996. Bethesda: National Cancer Institute, 2000.
2. Canto, M.I. Strategies for screening for pancreatic adenocarcinoma in high-risk patients. Semin. Oncol. 2007, 34, p. 295-302.
3. Beger, H.G., Rau, B., Gansauge F. et al. Treatment of pancreatic cancer: challenge of the facts. World J. Surg. 2003, 27, p.1075-1084.
4. Almoguera, C., Shibata, D., Forrester, K. et al. Most human carcinomas of the exocrine pankreas contain mutant c-Kras genes. Cell 1988, 53, p. 549-554.
5. Wilentz, R.E., Goggins, M., Redston, M. et al. Genetic, immunohistochemical, and clinical feature sof medullary carcinomas of the pancreas: a newly described and characterized entity. Am. J. Pathol. 2000,156, p. 1641-1651.
6. Pellegata, S., Sessa, F., Renault, B. et al. K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994, 54, p. 1556-1560.
7. Rozenblum, E., Schutte, M., Goggins, M. et al. Tumor-supressive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, p. 1731-1734.
8. Hahn, S.A., Schutte , M., Hoque, A.T. et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, p. 350-353.
9. Goggins, M., Shekher, M., Turnacioglu, K. et al. Genetic alterations of the TGF beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998, 58, p. 5329-5332.
10. Su, G.H., Bansal, R., Montgomery, E. et al. ACVR1B (ALK4) gene mutations in pancreatic carcinoma. Proc. Natl. Acad. Sci. USA, 2001, 98, p. 3254-3257.
11. Su, G.H., Hruban, R.H., Bansal, R.K. et al. Germline and static mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol. 1999, 154, p. 1835-1840.
12. Ruggeri, B., Zhang, S.Y., Caamano, J. et al. Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and RB-1 tumor-supressor genes. Oncogene 1992, 7, p. 1503-1511.
13. Su, G.H., Hilgers, W., Shekher, M. et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor-supressor gene. Cancer Res. 1998, 58, p. 2339-2342.
14. Kern, S.E., Hruban, R.H., Hidalgo, M. et al. An introduction to pancreatic adenocarcinoma genetics, pathology and therapy. Cancer Biol. Ther. 2002, 1, p. 607-613.
15. Calhoun, E.S., Jones, J.B., Ashfaq, R. et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am. J. Pathol. 2003, 163, p. 1255-1260.
16. Hempen, P.M., Kurpad, H., Calhoun, E.S. et al. A double missense variation of the BUB1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum. Mutat. 2003, 21, p. 445.
17. Van der Heijden, M.S., Yeo, C.J., Hruban, R.H. et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003, 15, p. 2585-2588.
18. Goggins, M., Schutte, M., Lu, J. et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996, 56, p. 5360-5364.
19. Maitra, A., Kern, S.E., Hruban, R.H. Molecular pathogenesis of pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, p. 211-226.
20. Wallrapp, C., Muller-Pillasch, F., Solinas-Toldo, S. et al. Characterization of a high copy number amplification at 6q24 in pancreatic cancer identifies c-MYB as a candidate oncogene. Cancer Res. 1997, 57, p. 3135-3139.
21. Ruggeri, B.A., Huang, L., Wood, M. et al. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 1998, 21, p. 81-86.
22. Abramson, M.A., Jazag, A., van der Zee, J.A. et al. The molecular biology od pancreatic cancer. Gastrointest. Cancer Res. 2007, 1(suppl 2), S7-S12.
23. Van Heek, N.T., Meeker, A.K., Kern, S.E. et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am. J. Pathol. 2002, 161, p. 1541-1547.
24. Caldas, C., Hahn, S.A., Hruban, R.H. et al. Detection of K-ras mutations in the stool of patiens with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 1994, 54, p. 3568-3573.
25. Jones, J.B., Song, J.J., Hempen, P.M. et al. Detection of mitochondrial DNA mutations in pancreatic cancer offers a mass-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001, 61, p. 1299-1304.
26. Vimalachandran, D. Genetics and prevention of pancreatic cancer [on line]. 2004-11. Dostupný z WWW: http://www.medscape.com/viewarticle/ 468132_5.
27. Lynch, H.T., Smyrk, T., Kern, S.E. et al. Familial pancreatic cancer: a review. Semin. Oncol. 1996, 23, p. 251-275.
28. Klein, A.P., Hruban, R.H., Brune, K.A. et al. Familial pancreatic cancer. Cancer J. 2001, 7, p. 266-273.
29. Hucl, T. Molekulární patogeneze karcinomu pankreatu. Čes a Slov Ganstroent. Hepatol. 2009, 63(2), s. 58-64.
30. Lal, G., Lui, G., Schmocker, B. et al. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000, 60(2), p. 409-416.
31. Goldstein, A.M., Fraser, M.C., Struewing, J.P. et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N. Engl. J. Med. 1995, 333, p. 970-974.
32. Flander, T.Y. Pancreatic adenocarcinoma: epidemiology and genetics. Med. Genet. 1996, 33, p. 889-898.
33. Bartsch, D.K., Sina-Frey, M., Lang, S. et al. CDKN2A germline mutations in familial pancreatic cancer. Ann. Surg. 2002, 236(6), p. 730-737.
34. Giardiello, F.M., Brensinger, J.D., Tersmette, A.C. et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, p. 1447-1453.
35. Lynch, H.T., Voorhees, G.H., Lanspa, S.J. et al. Pancreatic carcinoma and hereditary nonpolyposis colorectal cancer: a family study. Br. J. Cancer. 1985, 52, p. 271-273.
36. Lowenfels, A.B., Maisonneuve, P., DiMagno, E.P. et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J. Natl. Cancer Inst. 1997, 89, p. 442-446.
37. Tersmette, A.C., Petersen, G.M., Offerhaus, G.J. et al. Increased risk od incident pancreatic cancer among first-degree relatives of patient with familial pancreatic cancer. Clin. Cancer Res. 2001, 7(3), p. 738-744.
38. Murphy, K.M., Brune, K.A., Griffin, C. et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17 %. Cancer Res. 2002, 62, p. 3789-3793.
39. Hruban, R.H., Adsaz, N.V., Albores-Saavedra, J. et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, p. 579-586.
40. Hruban, R.H., Maitra, A., Kern, S.E. et al. Precursors to pancreatic cancer. Gastroenterol. Clin. North. Am. 2007, 36, p. 831-849.
41. Kern, S.E. Advances from genetic clues in pancreatic cancer. Curr. Opin. Onc. 1998, 10, p. 74-80.
42. Hruban, R.H., Wilentz, R., Kern, S.E. Genetic progression in the pancreatic ducts. Am. J. Pathol. 2000, 156, p. 1821-1825.
43. Hruban, R.H., Adsay, N.V., Albores-Saavedra, J. et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, p. 579-586.
44. Moskaluk, C.A., Hruban, R.H., Kern, S.E. p16 and K-ras mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, p. 2140-2143.
45. Wilentz, R.E., Iacobuzio-Donahue, C.A., Argani, P. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, p. 2002-2005.
46. Sohn, T.A., Yeo, C.J., Cameron, J.L. et al. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann. Surg. 2004, 239, p. 788-797.
47. Hruban, R.H., Maitra, A., Kern, S.E. et al. Precursors to pancreatic cancer. Gastroenterol. Clin. North. Am. 2007, 36, p. 831-849.
48. Bassi, C., Sarr, M.G., Lillemoe, K.D. et al. Natural history of intraductal papillary mucinous neoplasms (IPMN): current evidence and implications for management. J. Gastrointest. Surg. 2008, 12, p. 645-650.
49. Tanaka, M., Chari, S., Adsay, V. et al. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology 2006, 6, p. 17–32.
50. Gold, E.B. Epidemiology of and risk factors for pancreatic cancer. Surg. Clin. North Am. 1995, 75, p. 819-843.
51. Michaud, D.S. Dietary meat, dairy products, fat, and cholesterol and pancreatic cancer risk in a prospective study. Am. J. Epidemiol. 2003, 157, p. 1115-1125.
52. Fuchs, G.S., Colditz, G.A., Stamfer, M.J. et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch. Intern. Med. 1996, 156(19), p. 2255-2260.
53. Lowenfels, A.B., Maisonnevue, P., Whitcomb, D.C. et al. Cigarette smoking as a risk factor of pancreatic patients with hereditary pancreatitis. JAMA 2001, 286(2), p. 169-170.
54. Steinberg, W.M., Barkin, J.S., Bradley, E.L. III. et al. Should patients with a strong family history of pancreatic cancer be screened on a periodic basis for cancer of pancreas? Pancreas 2009. 38(5). p. e137-e150.
55. Chiari, T.S, Leibson, C.L., Rabe, K.G. et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008. 134(1). p. 95-101.
56. Howes, N., Lerch, M.N., Greenhalf, W. et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, p. 252-261.
57. McFaul, C., Greenhalf, W., Earl, J. et al. Anticipation in familial pancreatic cancer. Gut 2006, 55, p. 252-258.
58. Kim, J.E., Lee, K.T., Lee, J.K. et al. Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J. Gastroenterol. Hepatol. 2004, 19, p. 182-186.
59. Greenhalf, W., Grocock, C., Harcus, M. et al. Screening of high-risk families for pancreatic cancer. Pancreatology 2009, 9, p. 215-222.
60. Van der Heijden, M.S., Brody, J.R., Dezentje, D.A. et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin. Cancer Res. 2005; 11, p. 7508-7515.
Labels
General practitioner for children and adolescents General practitioner for adultsArticle was published in
General Practitioner

2010 Issue 8
- Memantine Eases Daily Life for Patients and Caregivers
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
Most read in this issue
- Spontánní intrakraniální hypotenze
- Zinek a jeho vztah k nádorům prostaty
- Biomonitoring – význam a použití pro hodnocení expozice populace chemickým (toxickým) látkám z prostředí
- Pohybové aktivity jako součást prevence kardiovaskulárních onemocnění v ordinaci praktického lékaře