
Idiopatický hypereozinofilní syndrom a chronická eozinofilní leukemie (diferenciální diagnóza a terapie ve světle nových poznatků)
The idiopathic hypereosinophilic syndrome and chronic eosinophilic leukemia
Idiopathic hypereosinophilic syndrome is a heterogenous group of hematological disorders characterized by eosinophilia (> 1.5 x 109/l) persistent for more than 6 months, exclusion of reactive eosinophilia from other causes, such as parasitic infections or allergy, and evidence of end-organ damage [22]. According to World Health Organization the exclusion includes all neoplastic disorders in which eosinophils are part of the neoplastic clone. Excluded should be also T cell population with aberant phenotype and abnormal cytokine production [7], recently considert also as „lymphocytic“ variants of the HES [42]. HES has to be reclassified as chronic eosinophilic leukemia (CEL) when there is evidence for clonality based on the presence of chromosomal abnormalities or inactivation in female patients [7]. The successful empiric treatment of patients with tyrosine kinase inhibitor imatinib (Glivec) suggested the presence of an imatinib-sensitive tyrosine kinase inhibitor. The identification of a specific intersticial chromosoma deletion del(4)(q12;q12) creating the FIP1L1-PDGFRA fusion gene confirmed this hypothesis. Patients carrying this gene should be reclassified as CEL and detection of this gene is a positive predictor for response to imatinib therapy [11,45]. Effective doses of imatinib are 100 mg/day. The side effects are minimal. The only exception is an acute left ventricular dysfunction which has been reported in three patients within the first week of treatment with imatinib [36,38]. Imatinib has been successfully used also in some patients with the constitutively activated thyrosine kinase ETV6-PDGFRβ [1] and in systemic mast cell disease associated with eosinophilia [35]. Other therapeutical options for HES/CEL have been mentioned. The resistence to imatinib and the possibilities how to overcome it are discussed.
Key words:
hypereosinophilia - chronic eosinophilic leukemia - imatinib
:
L. Chrobák; J. Voglová
:
Oddělení klinické hematologie II. interní kliniky Lékařské fakulty UK a FN, Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
:
Vnitř Lék 2005; 51(12): 1385-1393
:
Review
Předneseno na XIX. olomouckých dnech s mezinárodní účastí ve dnech 15. - 18. června 2005.
Idiopatický hypereozinofilní syndrom (HES) je heterogenní skupina hematologických stavů charakterizovaná eozinofilií (> 1,5 x 109/l) přetrvávající po dobu delší než 6 měsíců, vyloučením reaktivní eozinofilie z jiných příčin, jako parazitárních onemocnění nebo alergie, a průkazem orgánového poškození [22]. Podle Světové zdravotnické organizace vyloučení zahrnuje také nádorové procesy se sekundární reaktivní eozinofilií a jiná nádorová onemocnění, u nichž je eozinofilie součástí nádorového klonu. Vyloučit je nutno také populace T lymfocytů s aberantním fenotypem a abnormální produkcí cytokinů [7], nově označené také jako „lymfocytární“ varianty HES [42]. HES je nutno reklasifikovat jako chronickou eozinofilní leukemii (CEL), pokud jsou známky svědčící pro klonalitu procesu spočívající na přítomnosti chromozomálních abnormalit nebo inaktivace X-chromozomu u žen [7]. Úspěšná empirická léčba HES nemocných inhibitorem tyrozinkinázy imatinibem (Glivec) nasvědčovala přítomnosti tyrozinkinázy citlivé na imatinib. Průkaz specifické intersticiální delece del(4)(q12;q12) s tvorbou fúzního genu FIP1L1-PDGFRA potvrdil tuto hypotézu [11]. Nemocné s tímto fúzním genem je nutno překlasifikovat jako CEL a jeho detekce je indikátorem pozitivní odpovědi na imatinib [11,45]. Účinné dávky jsou 100 mg/den a nežádoucí účinky jsou minimální. Jedinou výjimkou je akutní insuficience levé komory, která vznikla u 3 nemocných během prvního týdne léčby imatinibem [36,38]. Imatinib byl také úspěšně použit u nemocných s konstitivně aktivovanou tyrozinkinázou ETV6-PDGFRβ [1] a u systémové mastocytózy sdružené s eozinofilií [35]. Je diskutována otázka rezistence na imatinib a možnosti jejího překonání.
Klíčová slova:
hypereozinofilní syndrom - chronická eozinofilní leukemie - terapie imatinibem
Úvod
Idiopatický hypereozinofilní syndrom (HES) a chronická eozinofilní leukemie (CEL) představují crux medicorum z hlediska diagnostického i terapeutického. V posledních dvou letech došlo k podstatnému obohacení našich poznatků o tomto syndromu, pokud jde o jeho diagnostiku a léčbu.
Fyziologie a patofyziologie eozinofilů
Eozinofily tvoří normálně jen malou část leukocytů obvodové krve. Normální počet eozinofilů se udává mezi 0,4 - 0,6 x 109/l. Po krátkém pobytu v obvodové krvi přestupují eozinofily do tkání, především do střeva, bronchiální sliznice a do kůže. Produkce, maturace a zánik eozinofilů kontrolují cytokiny (IL-3, IL-5, IL-13) a granulocyty a makrofágy kolonie stimulující faktor (GM-CS F) [10,27]. Hlavní úloha připadá IL-5, který je produkován podskupinou tzv. Th2 lymfocytů, funkčně odpovídajících pomahačským lymfocytům. IL-5 ovlivňuje terminální diferenciaci eozinofilů.
Zvýšený počet eozinofilů může mít nejčastěji dvojí příčinu: 1. Hypereozinofilie je reaktivní odpovědí na sekreci cytokinů aberantními T lymfocyty s průkazem jejich klonality nebo i bez ní. 2. Jde o klonální proliferaci a poruchu kmenové krvetvorné buňky v kostní dřeni, kde je zvýšený počet eozinofilů součástí neoplastického klonu.
Nejčastější příčinou výrazně zvýšeného počtu eozinofilů na světě jsou helmintiázy [9] a v industrializovaných krajinách alergická onemocnění [41]. Na klonální procesy v kostní dřeni připadá méně než 1 % eozinofilních syndromů a na idiopatický hypereozinofilní syndrom ještě méně [10].
Poškození orgánů eozinofily
Ať je příčina eozinofilie jakákoliv, eozinofily poškozují různé orgány, tzv. terminální orgány eozinofilního poškození, především srdce, plíce, kůži, centrální a periferní nervový systém. Poškození je důsledkem uvolnění obsahu granul eozinofilů obsahujících kationické proteiny - MBP (Major Basic Protein), ECP (Eosinophil Cationic Protein), EDN (Eosinophil Derived Neurotoxin) a EPO (Eosinophil PerOxydase) [27]. K projevům HES patří také tromboembolické komplikace, kde zdrojem embolů je nejčastěji srdce [48].
Na druhé straně mají toxické proteiny uvolňované z eozinofilních granul blahodárný účinek při potírání parazitů.
Hypereozinofilní syndrom (HES)
Hypereozinofilní syndrom a idiopatický hypereozinofilní syndrom jsou synonyma, kde název idiopatický hypereozinofilní syndrom vyjadřuje přesněji obsah pojmu. HES je vzácné hematologické onemocnění s abnormálně zvýšenou produkcí eozinofilů. Postihuje daleko častěji muže než ženy (9 : 1). Onemocnění začíná nejčastěji mezi 20 až 50 léty, ale vyskytuje se vzácně i u dětí a bylo popsáno i u 5měsíčního kojence [28]. Nejčastějšími příznaky jsou horečka, únavnost, úbytek na váze, kašel, bolesti ve svalech, svědění kůže a průjmy [7].
Název hypereozinofilní syndrom použili poprvé v roce 1968 Hardy a Anderson [21], kteří pozorovali 3 nemocné s hypereozinofilií, hepatosplenomegalií, kardiálními a plicními symptomy a usoudili, že nejde o maligní onemocnění. Chusid et al [22] v roce 1975 na podkladě 14 vlastních pozorování a literárních sdělení definovali 3 základní znaky HES:
- Počet eozinofilů ≥ 1,5 x 109/l přetrvávající po dobu 6 měsíců.
- Vyloučení všech známých příčin eozinofilie, jmenovitě parazitárních a alergických onemocnění.
- Přítomnost orgánového poškození.
Častost orgánového postižení u HES uvádí tab. 1, která zahrnuje 105 nemocných ze 3 velkých souborů [48]. Nejčastějším poškozením, které je nejčastější příčinou smrti u HES, je postižení srdce.
Izolované postižení orgánů bez periferní eozinofilie
Vedle postižení orgánů v rámci HES existuje infiltrace jednotlivých orgánů eozinofily s orgánovým poškozením bez eozinofilie v obvodové krvi. Patří sem eozinofilní celulitida (Wellsova nemoc), eozinofilní pneumonie (Löfflerův syndrom), eozinofilní fasciitida (Schulmanův syndrom), eozinofilní pankreatitida, eozinofilní synovitida, eozinofilní ascites, eozinofilní gastritida. Stejné orgány mohou být infiltrovány i při klonálním procesu dřeně s eozinofilií. Jindy se původně jednoorgánové postižení může rozšířit i na další orgány s eozinofilií v obvodové krvi [10].
Chronická eozinofilní leukemie (CEL)
V roce 2001 podala Bainová et al definici CEL, která se stala i definicí SZO [7].
Při definici se ztotožnila se 3 základními kritérii podle Chisuda et al [22], rozšířila však spektrum nemocí a stavů, které je nutno vyloučit. U maligních onemocnění je nutno vyloučit procesy s reaktivní eozinofilií, především Hodgkinův lymfom, NHL - zvláště T-NHL, akutní lymfoblastickou leukemii, angioimunoblastickou lymfadenopatii, vzácně i karcinomy [8] a dále stavy, při kterých je eozinofilie součástí neoplastického klonu, jakým je AML s inverzí (16),t(16;16)(p13;q22) a známé chronické myeloproliferativní stavy (CML, PV, ET, MF). Mezi stavy, které je nutno rovněž vyloučit, patří i stavy s abnormální populací T lymfocytů se zvýšenou produkcí IL-5 a s průkazem klonality nebo i bez tohoto průkazu. Jde hlavně o fenotyp CD3-, CD4+, který je jednoznačně patologický a vzácněji o fenotyp CD3+, CD4-, CD8-, který se vyskytuje v rané fázi vývoje. Roufosse navrhl nově pro stavy charakterizované nemaligní expanzí T lymfocytů produkujících IL-5 název lymfocytární varianta HES [42].
Pokud nelze prokázat žádné onemocnění, které může způsobit eozinofilii, není přítomno známé klonální myeloidní onemocnění a není přítomna abnormální populace T lymfocvtů, lze stav pokládat za idiopatický hypereozinofilní syndrom.
Diagnóza HES je diagnóza per exclusionem. Diagnóza CEL vyžaduje navíc:
- a) buď zvýšení blastů v obvodové krvi nad 2 % nebo ve dřeni na 5 - 19 %
- b) nebo průkaz klonálního chromozomálního postižení nebo klonálního postižení doloženého jinak.
Průkaz klonality eozinofilů však narážel na poměrně velké obtíže pro malý počet chromozomálních abnormalit prokazatelných standardním cytogenetickým vyšetřením. Výrazná převaha mužů (9 : 1) vylučovala možnost využít inaktivaci X-chromozomu [29] nebo průkaz jednoho typu izoenzymu G6PD (A nebo B) u heterozygotních žen.
Chromozomální abnormality u CEL známé do doby průkazu fúzního genu FIP1L1-PDGFRA
1. Myeloproliferativní syndrom 8p11 (EMS - eozinofilní myeloproliferativní syndrom). V roce 1995 popsali MacDonald et al [30] a Inhorn et al [23] na podkladě vlastních pozorování a několika sdělení v literatuře novou jednotku charakterizovanou periferní lymfadenopatií typu T-lymfoblastického lymfomu, eozinofilií a myeloproliferací. Jednotku označili MacDonald et al jako myeloproliferativní syndrom 8p11 (EMS). Maligní proces vychází z pluripotentní kmenové buňky s další diferenciací směrem lymfoidním a myeloidním. Syndrom je cytogeneticky charakterizován reciprokou translokací postihující pruh 8p11 kódující FGFR1-receptor tyrozinkinázy pro růstový faktor fibroblastů a několik dalších chromozomů, z nichž 4 jsou známé. Tím vznikají fúzní geny mající aktivitu konstitutivní tyrozinkinázy. U více než 60 % nemocných je přítomná splenomegalie. Onemocnění má velmi agresivní průběh a končí zpravidla jako AML. Jedinou účinnou terapií je alogenní transplantace krvetvorných buněk.
2. Chromozomální translokace s q5(31-35). Bainová [5] označila stavy sdružené s touto translokací jako CEL překrývající se s chronickou myelocytární leukémií, Apperley et al [1] jako myeloproliferativní choroby s aktivací PDGFRβ. Dosud bylo popsáno přes 30 případů [45]. Nejčastější je translokace t(5;12)(q33;p13) s více než 20 pozorováními, kdy jde o fúzi PDGFRB s genem původně označeným Golubem jako TEL [18]. Současně se používá označení ETV6. PDGFRB je receptorem tyrozinkinázy a hraje významnou roli v hematopoeze. Steerová a Cross [45] soudí že nemocní s přestavbou PDGFRB by mohli být nejlépe klasifikování jako podskupina MPD/MDS (myeloproliferativního/myelodysplastického) syndromu.
Apperley et al [1] použili v roce 2004 u 4 nemocných s touto translokací imatinib mesylát v dávce 400 mg/denně. U 3 nemocných s eozinofilií a leukocytózou šlo o fúzní gen ETV6-PDGFRB, u čtvrtého nemocného partnerský gen nebyl znám. U tohoto 20letého muže, u kterého onemocnění trvalo 14 let, byly přítomny vedle eozinofilie erytematózní kožní léze s rozsáhlými ulceracemi. Kožní léze se začaly hojit krátce po nasazení imatinibu. Krevní obraz se normalizoval u všech nemocných do 4 týdnů. Translokace t(5;12)(q33;p13) nebyla prokazatelná u 3 nemocných po 12 týdnech, u čtvrtého nemocného po 36 týdnech léčby imatinibem.
3. Dalšími známými a nejčastěji postiženými chromozomy u CEL jsou chromozomy: 4, 6, 7, 8, 10, 12, 15, 17, -7, +8, t(7;12), t(14;16) a trisomie 10 [6].
CEL s fúzním genem FIP1L1-PDGFRA
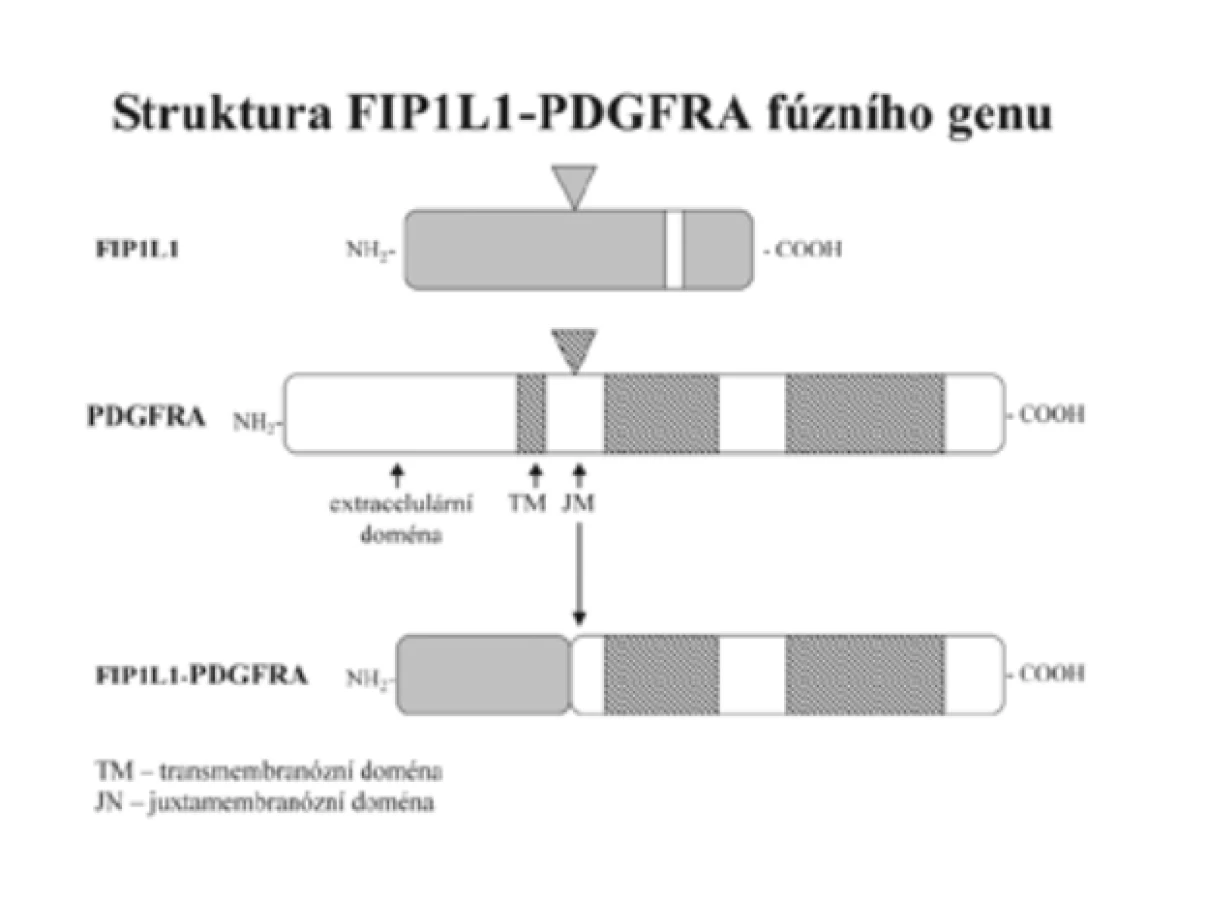
Diagnostiku CEL usnadnil převratným způsobem průkaz fúzního genu FIP1L1-PDGFRA. V letech 2001 až 2002 podalo několik autorů vedeno příznivými výsledky léčby CML imatinib mesylátem (dále imatinib) experimentálně empiricky tento účinný inhibitor tyrozinkinázy BCR-ABL několika nemocným pokládaným do té doby za HES [3,14,20,44]. Výsledek byl překvapivý. Při dávce 100 mg/den, tedy podstatně nižší než u CML (400 mg/den), se počet eozinofilů normalizoval během jednoho až dvou týdnů. [3,11,14,20]. Cools et al [11] dospěli k závěru, že ve hře je inhibice dosud neidentifikované tyrozinkinázy. To ho vedlo k odkrytí nového fúzního genu FIP1L1-PDGFRA, který nevzniká translokací, jak je tomu u BCR/ABL, ale intersticiální delecí malého úseku chromozomu 4 - (del)(q12;q12) odpovídajícího 800 kb (obr. 1 a 2). Delece není prokazatelná klasickou cytogenetickou technikou, ale pomocí techniky FISH nebo RT-PCR. Autoři [11] následně vyšetřili soubor 16 nemocných původně diagnostikovaných jako HES a u 9 z nich prokázali fúzní gen FIP1L1-PDGFRA, to je v 56 %. Všichni nemocní s tímto fúzním genem, kterým byl dosud podán imatinib, zareagovali příznivě. K příznivé odpovědi však došlo i u 4 nemocných, u kterých gen FIP1L1-PDGFRA přítomen nebyl [11]. Cools et al [11] usoudili, že by mohlo jít o jiný, dosud neidentifikovaný gen, který tvoří fúzní gen s PDGFRA s aktivitou tyrozinkinázy citlivé na imatinib. Počet nemocných s příznivou odpovědí na imatinib tvoří asi 40 % z těch, u nichž FIP1L1-PDGFRA nebyl prokázán [11].

Klinické a laboratorní nálezy u HES a CEL
S průkazem fúzního genu FIP1L1-PDGFRA bylo nutno poměrně velkou část nemocných překlasifikovat z HES na CEL [11]. Splenomegalie, hepatomegalie, anémie a trombocytopenie byly dříve pokládány za projevy, které svědčí spíše pro CEL než HES [4], ale nesnadný průkaz klonality tento závěr oslaboval. Vzhledem k tomu, že fúzní gen FIP1L1-PDGFRA byl prokázán teprve v roce 2003, neexistuje větší série nemocných, která by srovnávala klinické a laboratorní nálezy u HES s nálezy u CEL. Jedna menší retrospektivní studie Vandenberghea et al z roku 2004 [47] zahrnuje celkem 17 nemocných. U 8 šlo o CEL FIP1L1-PDGFRA (+), u 3 o CEL FIP1L1-PDGFRA (-) a u 6 o HES. Z klinických nálezů byl rozdíl v přítomnosti splenomegalie, která byla zjištěna u 73 % nemocných (u 8 z 11) s CEL, ale jen u jednoho nemocného s HES. Hladina hemoglobinu, počet leukocytů a destiček nebyl u nemocných s CEL a HES podstatně rozdílný. Rovněž počet eozinofilů neměl diskriminační význam. Překrývání hodnot u HES a CEL je značné. V dřívějších pracích je uváděn velký počet morfologických abnormalit eozinofilů, jako chudá granulace s oblastmi čiré cytoplazmy, vakuolizace cytoplazmy, hypersegmentace i hyposegmentace. Tyto změny lze však pozorovat jak u reaktivních, tak i u leukemických eozinofilií [7]. Vanderberghe [47] však zjistil velmi výrazný rozdíl v sérové hladině vitaminu B12, která byla u všech nemocných s CEL výrazně zvýšena nad 1 800-2 000 ng, ale normální u všech nemocných s HES. V dřívějších pracích zahrnujících nemocné s HES i CEL byla zvýšená hodnota vitaminu B12 uváděna přibližně u poloviny nemocných [5,49] a neměla diferenciálně diagnostický význam. Práce Vanderberghea et al [47] s použitím nových diagnostických kriterií pro CEL však přesvědčivě ukazuje, že výrazně zvýšená hodnota vitaminu B12 je charakteristickým nálezem, a to jak u FIP1L1-PDGFRA (+) CEL, tak i FIP1L1-PDGFRA (-) CEL.
Vyšetření indikovaná u HES a CEL
U všech nemocných splňujících kritéria HES, u nichž byly vyloučeny známé příčiny eozinofilie včetně abnormální populace T-lymfocytů, je nutno se zaměřit na vyloučení CEL nebo stavu překrývajícího se s obrazem CEL [1,5,45].
U všech nemocných je proto nutné klasické cytogenetické vyšetření a vyšetření zaměřené na průkaz fúzního genu FIP1L1-PDGFRA s použitím FISH (fluorescence in situ hybridization), analýzy nebo RT-PCR (reverse-trancriptase polymerase chain reaction). Nejzávažnějším orgánovým poškozením u HES a CEL je postižení srdce s endomyokardiální fibrózou a s poškozením šlašinek se vznikem mitrální a trikuspidální insuficience. Aortální chlopeň je poškozena vzácně [19]. Na poškozeném endokardu a na chlopních vznikají tromby s embolizací do velkého oběhu a do plic. Většina nemocných umírá na městnavé srdeční selhání a tromboembolické komplikace [48]. Pečlivé vyšetření srdce je proto indikováno u každého nemocného s HES a CEL. Postižení srdce nekoreluje s hladinou eozinofilů, takže pečlivé sledování nemocného je nezbytné. Echokardiografie, magnetická rezonance a katetrizace jsou schopny prokázat jen pokročilá stadia srdečního poškození, časné stadium prokáže pouze endomyokardiální biopsie [48].
Nově byl fúzní gen FIP1L1-PDGFRA prokázán také u varianty systémové mastocytózy sdružené s eozinofilií [35]. Od CEL je ji možno odlišit histopatologickým vyšetřením kostní dřeně a zřetelně zvýšenou hladinou sérové tryptázy [35].
Terapie hypereozinofilie, HES a CEL
V minulosti měl HES velmi špatnou prognózu. V sestavě 57 nemocných z roku 1978 [37] byla průměrná doba přežití 9 měsíců a pouhých 12 % nemocných přežívalo 3 roky.
Moderní diagnostické metody a s tím související zavedení imatinibu do terapie pravděpodobně přispěje ke zlepšení přežití.
U nemocných s hypereozinofilií bez známek orgánového poškození není terapie nutná. Tito nemocní mohou mít dlouhodobý benigní průběh bez nutností terapie. Vzhledem k tomu, že se však závažné poškození srdce může vyvinout nepředpokládaně a nekoreluje s žádnou hodnotou periferní eozinofilie, tito nemocní by měli být pečlivě sledováni klinicky a echograficky nejméně v 6měsíčních intervalech [48].
Terapeutický přístup se v průběhu let měnil. Vstupní terapií byly zpravidla glukokortikoidy, které se používají i dnes, někdy s překvapivě pozitivním výsledkem [32,33]. Pokud onemocnění progredovalo, bylo to indikací pro nasazení hlavně hydroxyurey nebo nověji INF-α [48]. Racionální indikací pro použití hydroxyurey a IFN-α byla určitá podobnost s CML. U některých nemocných lze tuto terapii použít i dnes, když není indikace pro léčbu imatinibem.
Ačkoliv mezi nemocnými, kteří splňují kritéria HES, je značná klinická rozdílnost, je užitečné pokusit se vyčlenit dvě skupiny - nemocné s abnormální populací T-lymfocytů a nemocné splňující spíše kritéria myeloproliferace, kteří mají často splenomegalii a vysokou hodnotu sérové hladiny vitaminu B12.
Terapie u nemocných s aberantní populací T-lymfocytů
Nemocní s abnormální populací T-lymfocytů, kteří produkují IL-5, mají většinou nápadně shodný klinický obraz: hlavní klinickou manifestací jsou především kožní projevy včetně ekzému, erytrodemie, kopřivky, pruritu a angioedému a relativně vzácné poškození dalších orgánů. U nemocných bývá zvýšená sérová hladina IgE a někdy i polyklonální hypergamaglobulinemie [42].
U těchto nemocných je užitečné začít s terapií kortikosteroidy, které snižují hladinu eozinofilů v krvi a uvolňování obsahu eozinofilních granul [10]. Z dalších léků byly použity 2-chlorodeoxyadenosin, cyklosporin a chlorambucil [48], zatímco INF-α jako monoerapie se nedoporučuje. Pitini et al [39] použili u nemocného s fenotypem CD-3, CD4+, CD8+ a vysokou hladinou IL-5 (280 pg/mol) po neúspěchu hydroxycarbamidu, interferonu-α a imatinibu (400 mg/d.) monoklonální protilátku alemtuzumab namířenou proti CD52. CD52 je přítomen na lymfocytech, eozinofilech a monocytech. Výrazné kožní léze se u nemocného zmenšily, ejekční frakce levé komory stoupla z 48 % na 61 % a sérová hladina IL-5 poklesla na 9 pg/mol. Nemocnému se při dávce alemtuzumabu 30 mg každé 3 týdny daří po 6 měsících dobře, počet eozinofilů je 0,3 x 109 /l. Selfick [43] dosáhl u nemocného s kožními projevy při udržovací dávce 30 mg každé 3 týdny zřetelný ústup kožního nálezu a zlepšení kvality života s přechodným zhoršováním a zlepšováním počtu eozinofilů vždy před infuzí a po ní.
Angioedém s HES může být způsoben poruchou regulace T-lymfocytů nebo poruchou funkce eozinofilů. Orson [34] dosáhl protahované remise po intravenózním podání imunoglobulinu za současného snižování kortikoidů.
Terapie imatinibem
Kritický obrat v terapii HES znamenal nečekaně příznivý účinek imatinibu [3,14,20,44] podložený průkazem fúzního genu vzniklého intersticiální delecí - del(4)(q12;q12), FIP1L1-PDGFRA, který vede ke konstitutivní aktivaci tyrozinkinázy produkované PDGFRA [11]. Všichni nemocní s tímto fúzním genem jsou jednoznačně vnímaví na léčbu imatinibem, a to i ti, kteří neodpověděli na léčbu steroidy, hydroxyureou, IFN-α i 2-chlorodeoxyadenosinem a cytarabinem [3]. Imatinib se ukázal účinný i u varianty systémové mastocytózy s FIP1L1-PDGFRA a eozinofilií, pokud nebyla přítomna mutace c-kit (Asp 816 Val) [35]. Imatinib je účinný také u 40 % nemocných s HES, kde fúzní gen prokázán nebyl [11]. Mimoto imatinib byl použit s úspěchem i u CEL vzniklé mutací genu PDGFRB lokalizovaného na chromozomu 5q33.
Účinná dávka imatinibu je 100 mg/den a je podstatně nižší než u CML. Nástup účinku je rychlý a trvalý. K úpravě krevního obrazu a normalizaci počtu eozinofilů dochází do jednoho až dvou týdnů. Jen ojediněle bylo nutno dávku imatinibu zvýšit ze 100 mg na 400 mg [11,14]. Zároveň s úpravou krevního obrazu se zlepšuje i objektivní nález a ustupují subjektivní obtíže. Klion et al [26] však nezaznamenali zlepšení srdeční funkce u 3 nemocných s endokardiální fibrózou a městnavým srdečním selháním, zatímco došlo k úplné hematologické odpovědi a zlepšení v postižení ostatních orgánů. Autor to přisuzuje ireparabilnímu strukturálnímu poškození myokardu, kterému by se pravděpodobně dalo zabránit časněji zahájenou terapií, jak pro to svědčí jiná pozorování [2]. Nedořešenou otázkou je udržovací terapie. Někteří pokračují se vstupní dávkou 100 mg denně. Cools et al [13] udrželi nemocné v remisi při dávce 100 mg/týdně. Zda je možné terapii imatinibem po dosažení molekulární remise trvale přerušit, je otázka. Zkušenosti získané s léčbou CML pro to nesvědčí [15].
Nežádoucí účinky terapie imatinibem
Terapie imatinibem při nízkých dávkách 100 mg/den je velmi dobře snášena a nežádoucí účinky jsou minimální. Nejzávažnější komplikací je selhání levé komory s kardiogenním šokem, ke kterému došlo během prvního týdne léčby u 1 nemocného Pardananiho et al [36] a u 2 z 5 nemocných Pitiniho et al [38]. Pardanani et al [36] prokázali biopsií myokardu infiltraci eozinofily s degranulací. U všech 3 nemocných byla prokázána vyšší hladina troponinu cTnT již před léčbou nebo byl zaznamenán její vzestup v průběhu léčby. Všichni nemocní odpověděli promptně na vysoké dávky kortikoidů. Příčina náhlé dysfunkce levé komory není plně objasněna. Pitini et al [38] ji vidí v zánětlivé odpovědi na degranulaci infiltrujících eozinofilů. Každý nemocný s hypereozinofilií by proto měl být před zahájením terapie imatinibem vyšetřen echokardiograficky. Tefferi a Pardanani [46] doporučují monitorování hladiny cTnT před zahájením terapie a v jejím průběhu a podávání kortikosteroidů před zahájením terapie a v prvních 2 týdnech léčby.
Rezistence na imatinib
Cools et al [11] pozoroval u jednoho nemocného po 5 měsících léčby relaps onemocnění. Příčinou rezistence byla získaná mutace T674I v PDGFRA kinázové doméně. Obdobou se vznikem rezistencí u CML nutno předpokládat, že rovněž u CEL a dalších stavů léčených imatinibem bude počet rezistencí postupně přibývat. Multifaktoriální charakter rezistence nás staví před problém, jak tuto rezistenci překonat. Jednou z možností by mohla být kombinace imatinibu s konvenční cytostatickou terapií. Další možností je použití jiného inhibitoru. Experimentálně u myší byl testován účinek PKC 412 (N-benzoyltaurosporin). PKC 412 byl účinný nejen u stavů s fúzním genem FIP1L1-PDGFRA, ale i při přítomnosti mutace T674I, zatímco imatinib byl v druhém případě neúčinný [12].
Imatinib u nemocných s negativním FIP1L1-PDGFRA genem
Skutečnost, že 40 % nemocných s negativním fúzním genem FIP1L1-PDGFRA a negativním standardním cytogenetickým nálezem zareagovalo pozitivně na imatinib, vede k závěru, že by se imatinib měl u těchto nemocných zkusit, aby se předešlo postižení srdce s fatálními následky [48]. Tento závěr podporuje i skutečnost, že u nemocných citlivých na imatinib se výsledek dostavuje záhy a po malé dávce.
Transplantace krvetvorných buněk
U velmi agresivního myeloproliferativního syndromu 8p 11 (EMS) je jedinou účinnou terapií alogenní transplantace. Ze 7 nemocných s alogenní transplantací 3 zemřeli na komplikace související s transplantací a 4 jsou v úplné remisi [31]. Imatinib není aktivní proti FGFR1, který je součástí fúzního genu u EMS, a je proto málo pravděpodobné, že by u EMS byl účinný. Žádný nemocný s EMS však dosud imatinibem léčen nebyl.
Alogenní transplantace kmenových buněk byla provedena s úspěchem i u dalších nemocných mimo EMS [16].
Terapie naměřená proti IL-5
Interleukinu-5 je připisována významná role v ovlivnění diferenciace, aktivace a přežívání eozinofilů. Terapie namířená proti IL-5 přichází v úvahu tam, kde na podkladě dnešních znalostí není indikován imatinib. Několik autorů použilo z této indikace humanizovanou protilátku anti-IL-5 (SCH55700, Shering-Plough, mepolizumab) [40,17,25].
Plötz et al [40] podali mepolizumab u 3 nemocných s HES a kožními manifestacemi v dávce 750 mg v i.v. infuzi s dobrým výsledkem. Klonální T-lymfocyty nebyly prokázány. Kim et al [24] dosáhli u 6 z 8 nemocných s hypereozinofilním syndromem nebo eozinofilní gastritidou s periferní eozinofilií pokles eozinofilů a zlepšení klinických symptomů. Garret et al [17] použili s úspěchem mepolizumab u 4 nemocných s HES v dávce 10 mg/kg ve 3 i.v. dávkách ve 4 týdenních intervalech. Remise po poslední dávce trvala 3 měsíce. Tolerance protilátky je dobrá, pokles eozinofilů byl pozorován za 24-48 hod. [25,40]. Po vysazení však došlo k vzestupu eozinofilů a exacerbaci symptomů [24]. Po opětovném nasazení došlo sice k poklesu eozinofilů a zlepšení subjektivních obtíží, ale v menší míře než po vstupní léčbě [25].
Dosud nevyřešené otázky léčby HES a CEL
Zavedení terapie imatinibem u CEL a HES znamenalo značný pokrok v léčbě těchto stavů. Některé otázky však zůstávají nedořešeny. Je to především určení spektra nemocných, u kterých má terapie imatinibem výhled na úspěch, včetně zjištění podkladu příznivé odpovědi na molekulární úrovni. Mohlo by k tomu vést další prohloubení cytogenetických a molekulárně genetických analýz. Dosud se nelze rovněž vyjádřit k délce trvání remisí a k celkovému přežití. Otevřena zůstává otázka udržovací terapie a problematika prevence a terapie relapsu.
prof. MUDr. Ladislav Chrobák, CSc.
www.fnhk.cz
e-mail: ladislavchrobak@seznam.cz
Doručeno do redakce: 4. 8. 2005
Přijato po recenzi: 5. 9. 2005
Sources
1. Apperley JF, Gardebas M, Melo JV et al. Response to imatinib mesylate in patients with chronic myeloproliferative disease with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 2002; 347: 481-487.
2. Ascione L, DeMichele M, Accadia M et al. Reversal of cardiac abnormalities in a young man with idiopathic hypereosinophilic syndrome using a tyrosine kinase inhibitor. Eur J Echocardiogr 2004; 5: 386-390.
3. Ault P, Cortes J, Koller C et al. Response of idiopathic hypereosinophilic syndrome to treatment with imatinib mesylate. Leuk Res 2002; 26: 881-884.
4. Bain BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol 1996; 95: 2-9.
5. Bain BJ. Hypereosinophilia. Curr Opin Hematol 2000; 7: 21-25.
6. Bain BJ. Cytogenetic and molecular genetic aspects of eosinophilic leukaemias. Br J Haematol 2003; 122: 173-179.
7. Bain BJ, Pierre R, Imbert M et al. Chronic eosinophilic leukaemia and the hypereosinophilic syndrome. In: Jaffe ES, Harris NL, Stein H et al. World Health Organization classification of tumors: Pathology and genetics of tumours of the haematopoietic and lymphoid tissues. Lyon, France: IARC Press 2001: 1201-1214.
8. Bonaventure C, Mastroianni B, Alessio A et al. Hypereosinophilie paraneoplasique associée à un carcinome hepatocellulaire. Gastroenterol Clin Biol 2003; 27: 1167-1169.
9. Brigden ML. A practical workup for eosinophilia. You can investigate the most likely causes right in your office. Postgrad Med 1999; 105: 207-210.
10. Brito-Babapulle F. The eosinophilias, including the idiopathic hypereosinophilic syndrome. Br J Haematol 2003; 121: 203-223.
11. Cools J, DeAngelo D, Gotlib J et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348: 1201-1214.
12. Cools J, Stover EH, Boulton CL et al. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRα-induced myeloproliferative disease. Cancer Cell 2003; 3: 459-469.
13. Cools J, Stover EH, Wlodarska I et al. The FIP1L1-PDGFRα kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia. Curr Opin Hematol 2003; 11: 51-57.
14. Cortes J, Ault P, Koller C et al. Efficacy of imatinib mesylate in treatment of idiopathic hypereosinophilic syndrome Blood 2003; 101: 4714-4716.
15. Cortes J, O´Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood 2004; 104: 2204-2205.
16. Cooper MA, Akard LP, Thompson JM et al. Hypereosinophilic syndrome: long-term remission following allogeneic stem cell transplant in spite of transient eosinophilic post-transplant. Am J Hematol 2005; 78: 33-36.
17. Garret JK, Jameson SC, Thomson B et al. Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic syndromes. J Allergy Clin Immunol 2004; 113: 115-119.
18. Golub TR, Barker GF, Lovett M et al. Fusion of PDGF receptor beta to a novel els-like gene, tel in chronic myelomonocytic leukemia with t (5;12) chromosomal translocation. Cell 1994; 77: 307-316.
19. Gudmondsson GS, Ohr J, Leya F et al. An unusual case of recurrent Löffler endomyocarditis of the aortic valve. Arch Pathol Lab Med 2003; 127: 606-609.
20. Gleich GJ, Leiferman KM, Pardanani A et al. Treatment of hypereosinophilic syndrome with imatinib mesylate. Lancet 2002; 359: 1577-1578.
21. Hardy WR, Anderson RE The hypereosinophilic syndromes. Ann Intern Med 1968; 68: 1220-1229.
22. Chusid MJ, Dale DC, West BC et al. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975; 54: 1-27.
23. Inhorn RC, Aster JC, Roach SA et al. A syndrome of lymphoblastic lymphoma, eosinophilia and myeloid hyperplasia/malignancy associated with t (8;13) (p 11; q 11): description of distinctive clinico-pathologic entity. Blood 1995; 85: 1881-1887.
24. Kim YJ, Prussin C, Martin B et al. Rebound eosinophilia after treatment of hypereosinophilic syndrome and eosinophilic gastroenteritis with monoclonal anti-IL-5 antibody SCH55700. J Allergy Clin Immunol 2004; 114: 1449-1455.
25. Klion AD, Law MA, Noel P et al. Safety and efficacy of the monoclonal anti-interleukin-5 antibody SCH55700 in the treatment of patients with hypereosinophilic syndrome. Blood 2004; 103: 2939-2941.
26. Klion AD, Robyn J, Akin C et al. Molecular remission and reversal of myelofibrosis in response to imatinib mesylate treatment in patients with the myeloproliferative variants of hypereosinophilic syndrome. Blood 2004; 103: 473-478.
27. Krejsek J, Kopecký O. Klinická imunologie. Hradec Králové: Nucleus 2004.
28. Leblond P, Lepers S, Thebaud E et al. Le syndrome d´hypereosinophilie essentielle: à propos d´un cas chez un nourrisson. Arch Pediatr 2004; 11: 219-222.
29. Luppi M, Marasca R, Morselli M et al. Clonal nature of hypereosinophilic syndrome. Blood 1994; 84: 349-350.
30. MacDonald D, Aguiar RCT, Mason PJ et al. A new myeloproliferative disorder associated with chromosomal translocation involving 8p11: A review. Leukemia 1995; 9: 1628-1630.
31. MacDonald D, Reiter A, Cross NCP. The 8p11 myeloproliferative syndrome: a distinct clinical entity caused by constitutive activation of FGFR1. Acta Haematol 2002; 107: 101-107.
32. Maruyoshi H, Nakatani S, Yasumura Y et al. Löffler´s endocarditis associated with unusual ECG change mimicking posterior myocardial infarction. Heart-Vessels 2003; 18: 43-46.
33. Nishie M, Tomiyama M, Kamijo M et al. Acute cholecystitis and duodenitis associated with Churg-Strauss syndrome. Hepatogastroenterology 2003; 50: 998-1002.
34. Orson FM. Intravenous immunoglobulin therapy supresses manifestations of the angioedema with hypereosinophilic syndrome. Am J Med Sci 2003; 326: 94-97.
35. Pardanani A, Elliot M, Li CY et al. Imatinib for systemic mast-cell disease. Lancet 2003; 362: 535-537.
36. Pardanani A, Reeder T, Porrata LF et al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood 2003; 101: 3391-3397.
37. Parrillo JE, Fauci AS, Wolff SM. Therapy of hypereosinophilic syndrome. Ann Intern Med 1978; 89: 167-172.
38. Pitini V, Arrigo C, Azzarello D et al. Serum concentration of cardiac Troponin T in patients with hypereosinophilic syndrome treated with imatinib is predictive of adverse outcomes. Blood 2003; 102: 3456-3457.
39. Pitini V, Teti D, Arrigo C et al. Alemtuzumab therapy for idiopathic hypereosinophilic syndrome with abnormal T cells: a case report. Br J Haematol 2004; 127: 474.
40. Plötz SG, Simon HU, Darson V et al. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome with eosinophilic dermatitis. N Engl J Med 2003; 349: 2334-2339.
41. Rosenberg ME. Mechanisms of disease: eosinophilia. N Engl J Med 1985; 313: 1485-1492.
42. Roufosse F, Cogan E, Goldman M. The hypereosinophilic syndrome revisited. Annu Rev Med 2003; 54: 169-184.
43. Sefcick A, Sowter D, DasGusta E et al. Alemtuzumab therapy for refractory idiopathic hypereosinophilic syndrome. Br J Haematol 2004; 124: 558-559.
44. Schaller JL, Burkland GA. Rapid and complete control of idiopathic hypereosinophilia with imatinib mesylate. Med Gen Med 2001; 3: 9.
45. Steer EJ, Cross NCP. Myeloproliferative disorders with translocations of chromosome 5q 31-35; role of the platelet-derived growth factor receptor beta. Acta Haematol 2002; 107: 113-122.
46. Tefferi A, Pardanani A. Imatinib treatment-induced cardiomyopathy in hypereosinophilic syndrome. Blood 2003; 102: 3457.
47. Vandenberghe P, Wlodarska J, Michaux L et al. Clinical and molecular features of FIP1L1-PDGFRA(+) chronic eosinophilic leukemia. Leukemia 2004; 18: 734-742.
48. Weller PF, Bubley G. The idiopathic hypereosinophilic syndrome. Blood 1994; 83: 2759-2779.
49. Zittoun J, Farcet JP, Marquet J et al. Cobalamin (vitamin B12) and B12 binding proteins in hypereosinophilic syndromes and secondary eosinophilia. Blood 1984; 63: 779-783.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 12
Most read in this issue
- The idiopathic hypereosinophilic syndrome and chronic eosinophilic leukemia
- Possibilities of ultrasonographic differentiation of neck and axillary lymphadenopathy
- The relation of GERD, bronchial asthma and symptomatology from ear, nose and throught regions
- Idiopathic pulmonary fibrosis