
Doporučený postup diagnostiky a terapie esenciální trombocytemie a trombocytemie provázející jiné myeloproliferativní choroby
Practice guidelines for diagnosis and therapy of essential thrombocytaemia and thrombocytaemia associated with other myeloproliferative diseases
Essential thrombocytaemia (ET) is not a frequent disease (incidence rate is about 0.1–1.5 cases per 100,000 persons) but as new approaches and possibilities advancing the standard care but contrariwise requiring responsible and systematic approach occur both in relationship with diagnosis and with treatment, it is necessary to deal appropriately with this problem. There is a variety of aspects, which evoke controversy within the scope of diagnosis and differential diagnosis – the most important thing is whether the histological examination of bone marrow is required or not and how to organise care of patients. All this relates not only to essential thrombocytopenia but also to myeloproliferative diseases associated with other myeloproliferative diseases at all (MPO-T). Enclosed recommended practice guidelines could help to serve as a guide in problems given without limiting the space for disputation.
Key words:
myeloproliferation – (essential) thrombocytaemia – megakaryopoesis – anagrelide – interferon – hydroxyurea
:
M. Penka 1; J. Schwarz 2; R. Pytlík 3; M. Doubek 4; Y. Brychtová 4; P. Ďulíček 5
:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
1; Ústav hematologie a krevní transfuze, Praha, ředitel prof. MUDr. Pavel Klener, DrSc.
2; I. interní klinika 1. lékařské fakulty UK a VFN, Praha, přednosta prof. MUDr. Pavel Klener, DrSc.
3; Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
4; Oddělení klinické hematologie – II. interní klinika Lékařské fakulty UK a FN, Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
5
:
Vnitř Lék 2005; 51(6): 741-751
:
Guidelines
v souvislosti s diagnostikou, tak i léčbou nové přístupy a možnosti, které posouvají standardní péči, ale na druhou stranu vyžadují uvážlivý a cílevědomý přístup, je třeba se tímto problémem odpovídajícím způsobem zabývat. Je celá řada aspektů, které v rámci diagnostiky a diferenciální diagnostiky vyvolávají polemiku – nejvýznamnější je asi, zda je nutné či není histologické vyšetření kostní dřeně a v rámci léčby, kdy léčbu započít, jakým lékem a jak péči o nemocné organizovat. Vše se dotýká nejen esenciální trombocytemie, ale i myeloproliferativních onemocnění provázených trombocytemií vůbec (MPO-T). Přiložený doporučený postup by mohl posloužit jako vodítko v dané problematice, aniž by omezoval prostor k polemice.
Klíčová slova:
myeloproliferace – (esenciální) trombocytemie – megakaryopoéza – anagrelid – interferon – hydroxyurea
Definice esenciální trombocytemie (ET) a trombocytemie provázející další myeloproliferativní choroby (MPO−T)
Esenciální trombocytemie (ET) je onemocnění projevující se zvýšeným počtem krevních destiček, pro něž nelze uplatnit kritéria platná pro jiné myeloproliferativní choroby nebo reaktivní zvýšení krevních destiček. Trombocytemie mohou tedy provázet idiopatickou myelofibrózu (IMF) – nejčastěji její prefibrotické (proliferativní) stadium, pravou polycytemii (polycytemia vera – PV) a chronické myeloidní leukemie (CML). Z hlediska průkazu Ph1 chromozomu lze rozdělit zmíněné myeloproliferativní choroby na Ph1 negativní (IMF, PV a sama ET) a Ph1 pozitivní (CML). S trombocytemií se setkáváme také v souvislosti s infekcemi, záněty, malignitami, krvácením, hemolytickou anémií nebo po splenektomii či v důsledku účinku některých léků (viz níže). V těchto případech mluvíváme častěji o trombocytóze a její klinická relevance nebývá tak závažná.
Patogeneza Ph1 negativních myeloproliferativních onemocnění
Mezi klasická Ph1 negativní onemocnění patří tedy polycytemia vera (PV), idiopatická myelofibróza (IMF) a esenciální trombocytemie (ET). Příčina těchto onemocnění zůstává neznámá, ale jsou klasifikována společně, protože splňují pět základních charakteristik:
- původ v pluripotentní hematopoetické progenitorové buňce;
- dominance klonu nad normálními hematopoetickými progenitorovými buňkami;
- zvýšená produkce jedné nebo více řad krevních elementů i přes nepřítomnost fyziologického stimulu;
- extramedulární hematopoéza;
- transformace do akutní myeloidní leukemie (velmi variabilní u jednotlivých typů myeloproliferací [9].
V poslední době byly definovány nové funkční a molekulární charakteristiky těchto onemocnění. Většina myeloproliferativních onemocnění nemá rodinný výskyt a jejich příčinnou je somatická mutace na úrovni krvetvorné buňky. Kromě některých případů esenciální trombocytemie jsou myeloproliferativní nemoci klonálním onemocněním krvetvorné buňky [11].
Normální hematopoéza je regulovaná souhrou stimulačních (růstové faktory) a inhibičních signálů. U myeloproliferativních onemocnění vzniká somatická mutace na úrovni kmenové buňky a vede k minimálně dvěma různým typům poškozením: hypersenzitivita progenitorů ke stimulačním růstovým faktorům a rezistence k inhibičním signálům.
Fenotypickým projevem somatických mutací je porucha buněčné odpovědi a deregulace genové exprese. Poškozená odpověď hematopoetických progenitorů na cytokiny, kdy BFU-E kolonie rostou bez přidání růstových faktorů (endogenní růst erytroidních kolonií), je přítomna u všech pacientů s polycytemia vera [14,17,31] a u většiny případů esenciální trombocytemie. Tento fenomén je jedním z diagnostických kritérií dle WHO u polycytemia vera. Abnormální in vitro růst hematopoetických progenitorů u myeloproliferativních nemocí inspiroval studie zaměřené na poškození signalizace cytokinového receptoru. Nebyly nalezeny žádné mutace genů pro erytropoetin, pro erytropeotinový receptor, IGF-1 receptor nebo trombopoetinový receptor (c-MPL) [12].
Jak již bylo řečeno, u pacientů smyeloproliferativním onemocněním se nejedná o genovou mutaci trombopoetinového receptoru c-MPL, ale je významně snížena exprese MPL proteinu (trombopoetinového receptoru) na trombocytech u části pacientů. Snížená exprese MPL na trombocytech by se mohla stát novým diagnostickým markerem PV, ET a IMF. Snížená exprese c-MPL byla nalezena také u reaktivní trombocytózy a hereditární trombocytemie s genovou mutací TPO receptoru, ukazující, že tato změna může být způsobena různými molekulárním mechanizmy. Zvýšená exprese transkripčního koaktivátoru „high mobility group protein A2“ (HMGA2) byla nedávno popsána jako první molekulární marker pro IMF [1]. PRV-1 aHMGA2 jsou markery, které mohou být použitelné pro diagnózu PV a IMF.
Chromozomální aberace a jiné cytogenetické abnormality představují nález u menšiny pacientů. V době diagnózy u pacientů s PV v méně než 30 %, méně než 40 % u IMF, méně než 25 % u ET. Žádná chromozomová abnormalita není specifická jako u CML. Charakteristickou chromozomovou abnormalitou je delece 20q (10–15 % pacientů). Nejpravděpodobnějším genetickým mechanizmem souvisejícím s delecí 20q je inaktivace tumor supresorového genu, nemůžeme však vyloučit ani jiné mechanizmy. Další chromozomální abnormalitou je aberace 9p u 30 % pacientů s PV [2]. Zůstává nejasné, zda jde patogeneticky o primární změny nebo sekundární poškození při progresi nemoci.
Klasickým projevem polycytemia vera je trilineární hyperplazie, ale může se prezentovat jen izolovanou erytrocytózou, trombocytózou, leukocytózou a nebo jako myelofibróza smyeloidní metaplazií (MMM). Diagnostická kritéria PVSG [26] definují pouze pacienty, kteří dle konsenzu mají jistou diagnózu PV.
Idiopatická myelofibróza (IMF) (synonyma: myelofibróza s myeloidní metaplazií, chronická myelomonocytární metaplazie) je klonální onemocnění myeloidní a lymfoidní řady [34]. Myelofibróza je reaktivní proces a může být asociovaná s benigní nebo maligní nemocí včetně myeloproliferace. V kostní dřeni u IMF se tvoří depozita kolagenu, nové formace kosti (osteoskleróza) a angiogeneze. Tato stromální reakce je mediovaná cytokiny (fibroblast growth factor, IL-l, TGF-β). Zdrojem cytokinů jsou klonální megakaryocyty a monocyty [18,33]. Na druhé straně fibroblasty kostní dřeně jsou polyklonální [3]. Abnormální akumulace megakaryocytů v kostní dřeni a jejich tvorba cytokinů se zdá v patogenezi MMM klíčová.
V patogenezi ET dle současných poznatků hraje klíčovou roli klonální snížení exprese trombopoetinového receptoru na trombocytech a megakaryocytech. Mechanizmus snížené exprese c-MpL není znám. U zdravého pacienta probíhá regulace produkce trombocytů z megakaryocytů v kostní dřeni následovně: trombopoetin, který je tvořen v játrech, se naváže na trombopoetinové receptory na trombocytech v periferní krvi a volný plazmatický trombopoetin stimuluje megakaryopoézu a takto vede k produkci trombocytů. U sekundární (reaktivní) trombocytózy jiné onemocnění stimuluje zvýšenou syntézu trombopoetinu (pravděpodobně cestou cytokinů, např. IL-6), a takto vede ke zvýšení frakce volného plazmatického trombopoetinu a zvýšení megakaryopoézy. U klonální trombocytózy snížení exprese trombopoetinových receptorů (c-Mpl) na trombocytech vede ke zvýšení frakce volného plazmatického trombopoetinu a zvýšené megakaryopoézy. I přestože exprese trombopoetinových receptorů na megakaryocytech je snížená, megakaryocyty jsou hypersenzitivní na trombopoetin a dojde ke zvýšení produkce trombocytů [37].
Klinické příznaky trombocytemie při MPO
Klinické příznaky MPO-T jsou dány jednak typem základního onemocnění (zda jde o ET, PV, IMF nebo CML), jednak trombocytemií samotnou. Trombocytemie u MPO může být zcela asymptomatická (se stále častějšími vyšetřeními krevního obrazu s uvedením počtů trombocytů; těchto pacientů stále přibývá), mohou být přítomny (v době diagnózy i později) trombotické projevy na úrovni žilního systému (venózní tromboembolizmus – VTE), na úrovni arteriální i na úrovni mikrocirkulace (kardiovaskulární, neurologické a kožní projevy). Pacienti s trombocytemií mohou mít krvácivé projevy spojené s patologickou krvácivostí, které bývají následkem ne vždy prokazatelné poruchy funkce trombocytů. Při trombocytemiích nad 1 000 × 109/l jsou krvácivé projevy častější a někdy spojené s prodloužením aPTT, podle některých může jít o sekundární von Willebrandovu chorobu [28]. Další projevy trombocytemických MPO jsou svázány s možností progrese onemocnění do jiné klinické jednotky, nejzávažněji do sekundární akutní myeloidní leukemie (s-AML) s velmi špatnou prognózou [2,23].
Trombotické problémy jsou častější a quoad vitam závažnější než krvácivé. Proto také při sledování a léčbě pacientů s MPO-T klademe důraz především na prevenci závažných trombotických projevů.
Venózní trombózy se u pacientů s MPO-T vyskytují predilekčně v cévách dolních končetin (DK) a splanchniku, jsou provázeny možností plicní embolie. Mohou se vyskytnout případy plicních embolií, u kterých se neprokáže primární trombotické ložisko. Pokud se týká trombóz DK, vídáme jak povrchové tromboflebitidy, tak hluboké žilní trombózy (HŽT). Trombózy v oblasti splanchniku jsou někdy obtížně diagnostikovatelné a mohou být mylně pokládány za jiná algická onemocnění gastrointestinálního traktu, pakliže se na ně nemyslí. Časté jsou trombózy ve v. lienalis, v. portae, v. Mesenterica superior i inferior, nevzácně se může objevit trombóza jaterních žil s Budd-Chiariho syndromem. Pokud se týká arteriálního postižení, dominují trombotické cévní mozkové příhody (CMP), od mírné tranzitorní formy (TIA) až po devastující příhody potenciálně smrtelné. Dále se vyskytuje koronární syndrom v různých formách až po infarkty myokardu. Poškození malých tepének na DK může vést k syndromu modrých prstů až ke gangrénám prstů DK. Nejpestřejší klinické projevy mají poruchy mikrocirkulace s nejčastějšími projevy v oblasti CNS, které mohou mít velmi rozličnou symptomatologii neurologickou a oční. Patří sem bolesti hlavy, únava, mžitky před očima, rozostření vizu, skotomy a nejrůznější často velmi atypické stesky. Na DK, méně často na horních končetinách, se objevují červenofialová bolestivá ložiska erytromelalgie, která mají zvláštní histologický obraz se zánětlivými změnami s přítomností destičkových mikrotrombů a depozit fibrinu. Typická je pro ně až zázračně rychlá úleva od bolesti při léčbě kyselinou acetylosalicylovou (ASA) [23].
Diagnostika a diferenciální diagnostika esenciální trombocytemie a trombocytemií provázejících myeloproliferativní choroby
I. Diagnostika
Zatím neznáme znak, jenž by jednoznačně odlišil esenciální trombocytemii od trombocytemií při ostatních myeloproliferativních chorobách a dokonce ani od reaktivních trombocytemií.
K diagnostice esenciální trombocytemie bylo vytvořeno několik systémů, které používají vždy více různých kritérií. V tab. 1 jsou srovnána diagnostická kritéria esenciální trombocytemie vypracovaná „Polycytemia Vera Study Group“ (PVSG) [24,26] a „evropská“ či „rotterdamská“ diagnostická kritéria, „Thrombocythemia Vera Study Group“ (TVSG) [22]. Hematopatologická skupina z Kolína nad Rýnem definovala kritéria, na základě kterých je možno rozlišit jednotlivé Ph1-negativní myeloproliferace pomocí trepanobioptického vyšetření [25,43]. Kostní dřeň pacienta s esenciální trombocytemií má normální nebo pouze lehce zvýšenou buněčnost, obsahuje zmnožené zralé megakaryocyty s normálním poměrem jádro/cytoplazma a nenese dysplastické rysy. Základní rozdíly v interpretaci histopatologického nálezu (ve srovnání s kritérii PVSG) je založen na posouzení morfologických detailů megakaryocytů. Nalezneme absolutní převahu velkých, polyploidních megakaryocytů, s hutným jaderným chromatinem. Megakaryocyty jsou většinou ve skupinách, ale disperzní rozložení je možné. Erytropoéza a granulopoéza nejsou ani utlumené, ani hyperplastické. Oproti kritériím PVSG je v případě TVSG kritérií výslovně řečeno, že jakýkoli stupeň retikulinové fibrózy musí vést k úvahám o počátečním stadiu myeloidní metaplazie s myelofibrózou. Základním kritériem pro diagnózu myeloidní metaplazie s myelofibrózou (MMM, jinak též idiopatická myelofibróza – IMF, či agnogenní myeloidní metaplazie) není přítomnost retikulinových vláken, ale hyperproliferace dysplastických megakaryocytů v hypercelulární dřeni. Ve srovnání s ET nalezneme větší variabilitu ve velikosti megakaryocytů, od obrovských (většinou s jemnějším „obláčkovitým“ jaderným chromatinem), kterých je většina, až po malé formy, typické pro myelodysplastický syndrom. Tyto megakaryocyty tvoří shluky extendující směrem ke kostním trámcům a jsou výrazně dysplastické – s pyknotickým jádrem, abnormálním poměrem jádro/cytoplazma, často jsou patrná holá jádra bez cytoplazmy. Současně je přítomna hyperplazie neutrofilní řady. Tento obraz se označuje jako incipientní stadium myeloidní metaplazie, celulární stadium myelofibrózy, nebo jako esenciální megakaryocyticko-granulocytová metaplazie (EMGM) [44,45].
![Srovnání modifikovaných kritérií PVSG a kritérií TVSG pro diagnostiku esenciální trombocytemie [22,26].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/1f420db3a3b06826a6e148fbfee105b0.png)
Při diagnostice esenciální trombocytemie doporučujeme postupovat podle nejnovějších kritérii Světové zdravotnické organizace (WHO) [13], která jsou uvedena v tab. 2. „Rotterdamská“ („evropská“ či TVSG) a WHO kritéria jsou založena na stejné histopatologické klasifikaci, ale v jednom důležitém detailu se liší: počet trombocytů pro diagnózu ET začíná podle TVSG již nad 400, zatímco u WHO nad 600 × 109/l. Lze tedy pro praxi doporučit WHO kritéria, doplněná touto modifikací TVSG [13,21].
![Diagnostická kritéria esenciální trombocytemie podle Světové zdravotnické organizace (WHO kritéria) [13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/88f5db63b6f43e57ffd9f03f47ecab96.png)
II. Diferenciální diagnostika
Esenciální trombocytemie je vzácné onemocnění. Proto nepřekvapuje, že většina nemocných s vysokými počty krevních destiček má reaktivní trombocytózu. Podle Schafera [37] má reaktivní trombocytózu 88 % pacientů s počty trombocytů nad 500 × 109/l a 82 % pacientů s počtem trombocytů nad 1 000 × 109/l. Z tohoto faktu vyplývá, že diferenciální diagnostiku esenciální trombocytemie nelze v žádném případě podceňovat.
Trombocytóza je často náhodně zjištěný nález při vyšetření parametrů krevního obrazu. Jde o diagnostickou výzvu, která má tři hlavní příčiny: reaktivní proces (sekundární trombocytemie), klonální onemocnění kostní dřeně (esenciální trombocytemie, myeloproliferace a další) a familiární onemocnění. Diferenciální diagnostika trombocytemie by měl být proces o dvou krocích: v prvním kroku by měla být vyloučena reaktivní trombocytemie, ve druhém pak jiná myeloproliferace, případně další onemocnění kostní dřeně.
A) Reaktivní trombocytemie
Reaktivní (sekundární) trombocytemie může být způsobena následujícími faktory [37]:
- Faktory způsobující krátkodobé trombocytemie:
- a) akutní krevní ztráta;
- b) stav po trombocytopenii („rebound“);
- c) akutní zánět a infekce;
- d) fyzická zátěž.
- Faktory způsobující dlouhodobé trombocytemie:
- a) sideropenie;
- b) hemolytická anémie;
- c) splenektomie, asplenie – po splenektomii dochází ke zvýšené tvorbě trombopoetinu pravděpodobně vlivem vyšší produkce interleukinu-6 [7];
- d) malignity;
- e) chronické záněty (kolagenózy, pneumonitidy, tuberkulóza, chronické střevní záněty);
- f) léky (vinkristin, cytokiny, růstové faktory).
K odlišení reaktivní trombocytemie a esenciální trombocytemie nelze použít jen hodnotu počtu trombocytů v periferní krvi. Byly totiž popsány případy správně stanovené diagnózy esenciální trombocytemie u nemocných s počty krevních destiček v periferní krvi nižšími než 600 × 109/l [15,36]. Při obtížích s odlišením reaktivní trombocytemie je nutné histopatologické vyšetření. Při reaktivní trombocytemii se nalezne zmnožení morfologicky normálních megakaryocytů (výrazně menších než u MPO-T).
Tab. 3 shrnuje rozdíly onemocnění reaktivní (sekundární) trombocytemie a trombocytemie při myeloproliferativním onemocnění.
![Rozdíly mezi trombocytemií při myeloproliferativním onemocnění a sekundární trombocytemií [37].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/7015f6e7c4c44b19917207551ed6c56d.png)
B) Myeloproliferativní onemocnění
Diferenciálně diagnosticky je od esenciální trombocytemie nutné odlišit ostatní myeloproliferativní nemoci, u nichž jsou zjišťovány vyšší hodnoty krevních destiček. Kromě pravé polycytemie, chronické myeloidní leukemie a neklasifikovatelné chronické myeloproliferativní nemoci je nutné vyloučit především iniciální prefibrotické stadium idiopatické myelofibrózy [13,42]. Toto odlišení je možné jen histologicky a je důležité především z hlediska posouzení prognózy nemocného, a tedy i volby další terapie [20]. Nemocní s prefibrotickým stadiem idiopatické myelofibrózy mají prognózu významně horší než nemocní s esenciální trombocytemií [43] (obr. 1). U esenciální trombocytemie se v kostní dřeni nalézají zmnožené obrovské megakaryocyty s hyperploidními jádry a je patrné jejich abnormální shlukování. V případě myelofibrózy se v kostní dřeni také nachází zmnožené obrovské megakaryocyty s hyperploidními jádry a je patrné jejich abnormální shlukování. Na rozdíl od esenciální trombocytemie jsou megakaryocyty u myelofibrózy (esenciální megakaryocytární granulocytární metaplazie, myeloidní metaplazie) dysplastické, lze u nich pozorovat změny jaderného chromatinu a poruchy vyzrávání jádra a cytoplazmy [23,44] (blíže viz kapitola Diagnostika). V diferenciální diagnostice myeloproliferativních chorob je histologické vyšetření rozhodující [9,22,38]. Cytogenetické změny jsou přítomny jen u necelých 25 % nemocných s esenciální trombocytemií [41]. Molekulárně genetické vyšetření exprese receptoru pro trombopoetin ukazuje jeho sníženou expresi na trombocytech u všech myeloproliferativních chorob, i když tato exprese by měla být u esenciální trombocytemie nejnižší [41].
![Přežití a předpokládané zkrácení života u pacientů s esenciální trombocytemií a pacientů v časných fázích myeloidní metaplazie (MM) [45].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/46252f174cfc679b471f2bd188bd0d20.png)
C) Další onemocnění kostní dřeně
Z dalších onemocnění kostní dřeně je třeba na prvním místě zmínit myelodysplastický syndrom, zejména jeho variantu s delecí dlouhého raménka chromozomu 5 (5q–), pro niž je trombocytemie typická [42]. Odlišit je nutné i nemoci ze skupiny myelodysplastických/myeloproliferativních nemocí [13]. V prvním případě je pro diferenciální diagnostiku myelodysplastického syndromu 5q – nutné histologické a cytogenetické vyšetření, ve druhém případě je pro diferenciální diagnostiku rozhodující vyšetření histologické.
D) Familiární trombocytemie
Familiární trombocytemie jsou raritní onemocnění. Asi 20 % případů rodinných trombocytemií je způsobeno autozomálně dominantní mutací genu pro trombopoetin, která způsobuje jeho nadměrnou produkci. Zbylých 80 % případů má příčiny jiné a dosud neznámé. Byl popsán případ rodiny, u jejíchž členů byla zjištěna autozomálně dominantně dědičná trombocytemie způsobená bodovou mutací transmembránové domény receptoru pro trombopoetin c-MpL [37,39].
Doporučená vyšetření
Na základě výše uvedených skutečností navrhujeme následující paletu vyšetření, která by měla být použita v diagnostice esenciální trombocytemie a trombocytemií provázejících ostatní myeloproliferativní nemoci.
1. Základní diferenciální diagnostika
Tato vyšetření nemusí být prováděna na specializovaném hematologickém pracovišti. Nutnost provedení těchto vyšetření vyplývá z výše uvedeného textu.
- a) anamnéza;
- b) fyzikální vyšetření;
- c) vyšetření krevního obrazu, mikroskopické vyšetření diferenciálního rozpočtu leukocytů a vyšetření morfologie trombocytů a erytrocytů (poikilocytóza, drepanocytóza u IMF), přítomnost erytroblastů, alkalická fosfatáza leukocytů;
- d) základní koagulační vyšetření;
- e) základní biochemické vyšetření (sérová urea, kreatinin, ionty, jaterní enzymy včetně laktát dehydrogenázy);
- f) sérové železo a ferritin;
- g) C-reaktivní protein a sedimentace erytrocytů;
- h) vyšetření stolice na přítomnost okultního krvácení;
- i) RTG vyšetření srdce a plic, paranazálních dutin a panoramatický snímek zubů;
- j) revmatoidní faktor a antinukleární protilátky;
- k) ultrazvukové vyšetření břicha;
- l) ORL vyšetření;
- m)stomatologické vyšetření;
- n) gynekologické vyšetření u žen a urologické vyšetření u mužů.
2. Stanovení definitivní diagnózy a prognózy
a) Vyšetření nezbytná
- i) vyšetření kostní dřeně – trepanobioptické vyšetření s odběry dřeně na histologické, cytologické, cytogenetické (klasická cytogenetika a případně FISH na přítomnost translokace BCR-ABL) a molekulárně genetické vyšetření (BCR-ABL);
- ii) rozšířené koagulační vyšetření včetně vyšetření hladiny D-dimerů, antitrombinu a screeningu trombofilních stavů (protein C, protein S, APC rezistence, Leidenská mutace faktoru V, mutace genu pro protrombin 20210A, lupus anticoagulans, antikardiolipinové protilátky, protilátky anti-β2-glykorotein I) – nutné pro posouzení rizika trombózy, od něhož se odvíjí terapie.
b) Vyšetření doplňková
Mohou diagnózu a prognózu upřesnit, ale nejsou nezbytná pro její stanovení.
- i) vyšetření exprese genu PRV-l v granulocytech [40] a do budoucna vyšetření mutace JAK2;
- ii) vyšetření exprese trombopoetinového receptoru c-MpL na trombocytech;
- iii) vyšetření klonality u žen;
- iv) imunofenotypizační vyšetření kostní dřeně a periferní krve včetně imunofenotypizačního vyšetření trombocytů;
- v) vyšetření funkce trombocytů;
- vi) další koagulační vyšetření – hladina faktoru VIII, hladina homocysteinu, mutace MTHFR;
- vii) vyšetření růstu megakaryocytárních kolonií.

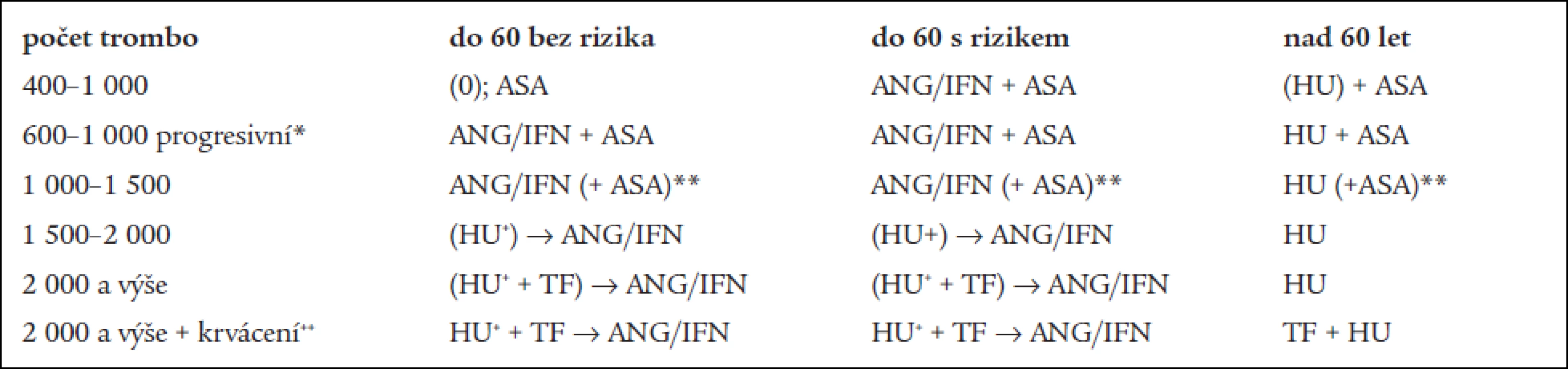

Terapie ET/MPO−T
Terapie:
- a) vychází ze zajištění diagnostiky ET (MPO-T) – dle definice WHO a její „rotterdamské“ či „evropské“ modifikace [21], která je provedena nebo potvrzena v Centru vysoce specializované hematologické péče (CVSHP) či v Centru pro trombózu a hemostázu (CTH – dále jen „Centra“);
- b) a ze stanovení individuální rizikovosti trombózy (ev. krvácení) daného pacienta. Jde tedy o stratifikaci pacientů podle rizika [37]. Rozhodující význam pro trombotické riziko mají: věk nad 60 let, předchozí trombózy, výše trombocytemie – jak vyplývá jednak zMichielsových prací [23] a jednak z poznatku, že cytoreduktivní léčba pomocí HU skutečně vede ke snížení incidence trombotických komplikací [4]. Mezi další rizikové faktory pro trombózu patří: přítomnost trombofilních markerů (deficit proteinů C a S, „leidenská“ mutace f. V, mutace protrombinového genu G20210A, deficit antitrombinu), přihlížíme i k ev. zjištění významného zvýšení hladin f. II a VIII nebo snížení f. XII, ke klinicky závažné formě aterosklerózy tepen srdce, mozku a dolních končetin, těžkému diabetu, malignitě, graviditě, hyperkoagulačnímu stavu při závažné infekci, stavu po závažném chirurgickém výkonu anebo stavu před ním;
- c) v rámci léčby lze použít léky s cyto - či tromboreduktivním účinkem: anagrelid (ANG), interferon α (IFN), hydroxyureu (HU), v krajních případech busulfan (BU), pipobroman (PB) a dále léky s antitrombocytárním, resp. Antiagregačním účinkem: kyselina acetylsalicylová (ASA), tiklopidin, klopidogrel, indobufen apod., případně doplňkové léky – antitrombotika (heparin, kumariny), symptomatika aj. [46];
- d) doporučení terapie ET (MPO-T) je většinou poskytováno v souvislosti se stanovením diagnózy Centrem ve spolupráci s ošetřujícím lékařem dle „Doporučeného postupu ČHS“. Léčba, pokud není pacient indikován k pouhému sledování a pokud se k ní rozhodneme, má v podstatě dvě alternativy, a to buď: 1) pouhé podávání antiagregancií (ASA) nebo 2) podávání léků s tromboreduktivním účinkem (viz výše). Cílem terapie má být korekce počtu trombocytů (u rizikových pacientů pod 400 ×
109/l, u nerizikových pod 600 ×
109/l) a zábrana vzniku klinických projevů krvácení či trombózy [8]. Lze ji podle základních aspektů věku a rizikových okolností rozčlenit následovně:
- ve věku pod 60 let – w&w (sledování bez léčby). V případě výskytu tromboembolických nebo krvácivých projevů nebo v případě jejich rizika včetně rizika kardiovaskulárního, či v případě vysokého počtu trombocytů (> 1 000 ×
109/l) – anagrelid nebo interferon α. Lékem první volby by zde mohl být anagrelid se svým cíleným tromboreduktivním účinkem [19,29,30,32]. Anagrelid nasazujeme v dávce 2 kapslí po 0,5 mg a zvyšujeme o 1 kapsli po 0,5 mg za týden. V případě, že je dosaženo odpovídajícího efektu při dobré toleranci léku, je indikováno pokračování zavedené léčby. Průměrná denní dávka se přitom pohybuje okolo 2,0 mg. Pokud není efektu dosahováno – nedochází k poklesu destiček, nebo je nutné k jeho dosažení použít dávky vyšší než 5 mg anagrelidu denně, popř. lék není dlouhodobě nemocným uspokojivě tolerován (bolesti hlavy, retence tekutin, otoky, tachykardie aj) – je vhodné zvážit záměnu anagrelidu – nejspíš za interferon α [6]. Léčbu interferonem α zahajujeme nejčastěji v dané indikaci dávkou 3 milionů jednotek subkutánně 3krát týdně. V případě, že by byl nasazen interferon α jako lék první volby (může to být výhodou při MPO-T s leukocytózou, kdy se jedná spíše o případy IMF), pak by se s jeho výměnou (za anagrelid či hydroxyureu) mělo počítat při nedosažení efektu léčby do 3 až 13 týdnů nebo nutnosti převýšení dávky 30 milionů jednotek interferonu týdně, či špatné snášenlivosti léku (těžko zvladatelný flu-like syndrom, anorexie, váhový úbytek apod.).
V případě nutnosti aplikace vyšších dávek je možno dle dosud nastíněných pravidel zvážit i kombinovanou léčbu (např. ANG/IFN, ANG/HU či IFN/HU) za účelem snížení dávek obou preparátů k co nejnižším dávkám zajišťujícím účinnost léčby při minimalizaci nežádoucích účinků [46].
U nemocných pod 60 let v celkově špatném zdravotním stavu lze ordinovat jako lék první volby HU (viz dále). Některými autory je doporučována vůbec jako lék volby [10]. - ve věku nad 60 let – pokud je přítomna klinická symptomatologie nebo dochází-li ke vzestupu destiček nad 1 000 × 109/l nebo progreduje-li trombocytemie rychle s nárůstem trombocytů o více než 200 × 109/l za méně než 2 měsíce, je indikována hydroxyurea [4,16]. Začínáme dávkou asi 1 000 až 1 500 mg denně a v následujících týdnech dávku dle kolísání počtu leukocytů a množství hemoglobinu upravujeme. Pokud zmíněné parametry umožní, řídí se další úprava dávky dle kolísání počtu destiček. Ve věku mezi 60–70 lety nemocného lze zvážit podávání ANG/IFN v případě zvláště dobrého zdravotního stavu a předpokladu dlouhodobé délky života v dobré kvalitě. Z důvodu možného leukemoidního účinku při dlouhodobém podávání HU zvláště v kombinaci s jinými cytostatiky, především alkylancii [47] se doporučuje ponechávat tuto léčebnou alternativu až ve zde zmíněné indikaci. U stavů s vysokou trombocytemií nad 1 500 × 109/l je možné léčbu zahájit u všech rizikových skupin HU (při vysokém riziku krvácení při velmi vysokých počtech trombocytů nebo při přítomnosti krvácivých projevů) a lze ji spojit s trombaferézou. HU doporučujeme v těchto případech proto, že má nejmenší rozpětí potřebných dávek a netratí se tolik času titrací vhodné dávky.
- v případě nežádoucích účinků hydroxyurey – podáváme anagrelid nebo interferon dle výše uvedených dávkovacích schémat, pouze u nemocných reagující špatně na běžné dávky nebo v případě celkově špatného zdravotního stavu – busulfan či jiná alkylancia.
- v případě nežádoucích účinků interferonu (např. závažný flu-like syndrom nereagující na paracetamol či jiná běžná analgetika/antipyretika) nebo v případě kontraindikací (jako např jaterní, ledvinné či srdeční onemocnění) – zvažujeme anagrelid či hydroxyureu.
- v případě nežádoucích účinků anagrelidu (cefalea, palpitace, otoky) nebo jeho kontraindikací (závažné onemocnění srdce, jaterní či renální selhání) – zvažujeme interfreon či hydroxyureu.
- v těhotenství (příp. také již i v době plánování těhotenství) – interferon α (viz výše), protože ANG není pro nedostatek zkušeností všeobecně doporučován, i když jsou zprávy i o úspěšně završených těhotenstvích za léčby anagrelidem [5,48].
- ve věku pod 60 let – w&w (sledování bez léčby). V případě výskytu tromboembolických nebo krvácivých projevů nebo v případě jejich rizika včetně rizika kardiovaskulárního, či v případě vysokého počtu trombocytů (> 1 000 ×
109/l) – anagrelid nebo interferon α. Lékem první volby by zde mohl být anagrelid se svým cíleným tromboreduktivním účinkem [19,29,30,32]. Anagrelid nasazujeme v dávce 2 kapslí po 0,5 mg a zvyšujeme o 1 kapsli po 0,5 mg za týden. V případě, že je dosaženo odpovídajícího efektu při dobré toleranci léku, je indikováno pokračování zavedené léčby. Průměrná denní dávka se přitom pohybuje okolo 2,0 mg. Pokud není efektu dosahováno – nedochází k poklesu destiček, nebo je nutné k jeho dosažení použít dávky vyšší než 5 mg anagrelidu denně, popř. lék není dlouhodobě nemocným uspokojivě tolerován (bolesti hlavy, retence tekutin, otoky, tachykardie aj) – je vhodné zvážit záměnu anagrelidu – nejspíš za interferon α [6]. Léčbu interferonem α zahajujeme nejčastěji v dané indikaci dávkou 3 milionů jednotek subkutánně 3krát týdně. V případě, že by byl nasazen interferon α jako lék první volby (může to být výhodou při MPO-T s leukocytózou, kdy se jedná spíše o případy IMF), pak by se s jeho výměnou (za anagrelid či hydroxyureu) mělo počítat při nedosažení efektu léčby do 3 až 13 týdnů nebo nutnosti převýšení dávky 30 milionů jednotek interferonu týdně, či špatné snášenlivosti léku (těžko zvladatelný flu-like syndrom, anorexie, váhový úbytek apod.).
- e) antiagregační léčba – nejčastěji ASA (většinou 50–100 mg denně) – je podávána v případě, kdy není dosaženo normálních počtů destiček, kdy však jejich počet nepřesahuje 1 200–1 500 × 109/l, dále v případě hrozby nebo výskytu trombotických projevů (kardiovaskulární riziko či symptomatologie a může být zvažována v souvislosti s některými trombofilními stavy – zejména antifosfolipidovým syndromem či u těhotných s trombofilní dispozicí). V některých případech se podává ASA samostatně bez léků s tromboreduktivním účinkem, v jiných případech společně s tromboredutkivními léky (viz výše léčebné schéma). Použít lze ale i jiné antiagregační preparáty (viz výše) – v obvyklém doporučeném dávkování.
- f) doplňková profylaxe a léčba zahrnuje především opatření řešící základní klinické projevy ET/MPO-T – tedy trombózu a krvácení. Vedle základní choroby k nim přispívají i choroby kardiovaskulární nebo jejich riziko a/nebo trombofilní vrozené a získané stavy přispívající především ke vzniku žilního tromboembolizmu nebo trombohemoragického syndromu, kdy se setkáváme současně s trombotickými i krvácivými příznaky. Krvácivé projevy mohou být však i důsledkem funkční poruchy trombocytů, které myeloproliferativní stavy provázejí.
V případech prodělaných trombóz je léčba doplněna o antitrombotika – heparin (nízkomolekulární nebo nefrakcionovaný) nebo k dlouhodobému režimu vhodnějšími kumariny. V případech prodělané trombózy a při současném výskytu dalších trombofilních dispozic, než je samotné základní onemocnění (ET/MPO-T), je indikována dlouhodobá či celoživotní perorální antikoagulační léčba.
V souvislosti s krvácením lze použít prostředky hemostyptické léčby používané při poruchách primární hemostázy (etamsylát, trombocytární koncentráty, plazmatické deriváty, nespecifická hemostyptika a případně a výjimečně antifibrinolytika).
Součástí doplňkové léčby může být léčba symptomatická, která představuje celou škálu dalších léčebných opatření v souvislosti s dalšími zdravotními těžkostmi nemocného s respektováním možné interakce s léky podávanými k léčbě ET/MPO-T (např. analgetika a antiflogistika, antibiotika apod.). - g) dispenzarizace pacientů (léčených i neléčených) – v Centru s kontaktem 1krát za rok.
- h) sledování pacientů spádovým hematologem ve spolupráci s Centrem, kde kontrola nejméně 1krát za půl roku.
- i) léčebný režim by měl být určen v rámci dohody ošetřujícího lékaře, centra a pacienta samotného. V případě nutnosti ustoupit od obvyklých pravidel – nežádoucí účinky, eskalace dávky léku nad obvyklou mez nebo výskyt dalších problémů (závažná onemocnění pacienta, obtížná komunikace s nemocným apod), je nutné řešit stav zajištěním individuálních podmínek péče ve spolupráci ošetřujícího lékaře s Centrem. Další úprava návrhu zajištění péče se odvíjí od dalšího vývoje diagnostiky a terapie.
prof. MUDr. Miroslav Penka, CSc.
www.fnbrno.cz
e-mail: m.penka@fnbrno.cz
Doručeno do redakce: 2. 5. 2005
Sources
1. Andrieux J, Demory JL, Dupriez B et al. Dysregulation and overexpression of HMGA2 in myelofibrosis with myeloid metaplasia. Genes Chormosomes Cancer 2004; 39 : 82–87.
2. Barbui T, Barosi G, Brossi A et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89 : 215–232.
3. Castro-Malaspina H, Gay RE, Jhanwar SC et al. Characteristics of bonemarrow fibroblast colony-forming cells (CFU-F) and their progeny in patients with myeloproliferative disorders. Blood 1982; 59 : 1046–1054.
4. Cortelazzo S, Finazzi G, Ruggeri M et al. Hydoxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995; 332 : 113.
5. Doubek M, Brychtová Y, Doubek R et al. Anagrelide in pregnancy: report of a case of essential thrombocythaemia. Ann Hematol 2004; 83 : 726–727.
6. Elliott MA, Tefferi A Interferon-α therapy in polycythemia pera and essential phrombocythemia. Semin Thromb Hemost 1997; 23 : 463.
7. Folman CC, Ooms M, Kuenen B et al. The role of thrombopoetin in post–operative thrombocytosis. Br J Haematol 2001; 114 : 126–133.
8. Fruchtmann SM Treatment paradigms in the management of myeloproliferative disorders. Semin Hematol 2004; suppl. 3 : 18–42.
9. Georgii A, Buhr T, Buesche G et al. Classification and staging of Ph–negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leukemia Lymphoma 1996; 22( supl 1): 15–29.
10. Green A, Campbell P, Buck G et al. The Medical Research Council PT1 trial in essentials thrombocythemia. Blood 2004; 104 : 5a–6a.
11. Harrison CN, Gale RE, Machin SK et al. A large proportion of patients with a diagnosis of Essential Thrombocythemia do not have a clonal disorder and may be at lower risk of thombotic complications. Blood 1999; 93 : 417–424.
12. Horikawa Y, Matsumura I, Hashimoto K et al. Markedly reduced expression of platelet c-mpl receptor in essential thrombocythemia. Blood 1997; 90 : 4031–4038.
13. Jaffe ES, Harris NL, Stein H et al. (Eds.): World health organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press 2001.
14. Kralovics R, Buser AS, Teo SS et al. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood 2003; 102 : 1869–1871.
15. Lengfelder E, Hochhaus A, Kronawitter U et al. Should a platelet limit of 600 x 109/l be used as a diagnostic criterion in essential thrombocythaemia? An analysis of the natural course including early stages. Br J Haematol 1998; 100 : 15–23.
16. Lofvenberg E, Wahlin A Management of Polycythemia vera, Essential Thrombocythemia an myelofibrosis with hydroxyurea. Eur J Haematol 1998; 41 : 375.
17. Liu E, Jelinek J, Pastore YD et al. Discrimation of polycythemias and thrombocytoses by novel, simple, accurate clonality assays and comparison with PRV-1 expresion and BFU-E response to erythrocytosis. Blood 2003; 101 : 3294–3301.
18. Martyre MC, Le Bousse–Kerdiles MC, Romquin N et al. Elevated levels of basic fibroblast growth factor in megakaryocytes and platelets from patients with idiopathic myelofibrosis. Br J Haematol 1997; 1997 : 441–448.
19. Mazur EM, Rosmarin AG, Sohl PA et al. Analysis of the mechanism of anagrelid-induced thrombocytopenia in humans. Blood 1992; 79 : 1931.
20. Messa RA, Tefferi A, Jacobsen SJ et al. Population-based incidence and survival figures in Essential Thrombocythemia and agnogenicmyeloid meta-plasia: an Olmsted County study. Am J Hematol 1999; 61 : 10–15.
21. Michiels JJ Diagnostic criteria of the myeloproliferative disorders (MPD): essential thrombocythaemia, polycythaemia vera and chronic megakaryocytic granulocytic metaplasia. Netherlands J Med 1997; 51 : 57–64.
22. Michiels JJ, Juvonen E Proposal for revised diagnostic criteria of essential thrombocythemia and polycythemia vera by the Thrombocythemia Vera Study Group. Semin Thromb Hemost 1997; 23 : 339–347.
23. Michiels JJ, Kutti J, Stark P et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic
metaplasia and myelofibrosis. Neth J Med 1999; 54 : 46–62.
24. Michiels JJ, Barbui T, Finazzi G et al. Diagnosis and treatment of Polycythemia Verand possible future study designsof the PVSG. Leuk Lymphoma 2000, 36 : 239–253.
25. Michiels JJ, Thiele J Clinical and pathological criteria for thed diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol 2002; 76 : 133–145.
26. Murphy S, Peterson P, Iland H et al. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol 1997; 34 : 29–39.
27. Oehler L, Jaeger E, Eser A et al. Imatinib mesylate inhibits autonomous erythropoesis in patients with polycythemia vera in vitro. Blood 2003; 102 : 2240–2242.
28. Penka M, Buliková A, Matýšková M et al. Hematologie I. Neonkologická hematologie. Praha: Grada 2001.
29. Petrides PE Anagrelide: decade of clinical experinces with its use for the treatment of primary thrombocythemia. Expert Opin Pharmacother 2004; 5 : 1781–1798.
30. Petitt RM, Silverstein MN, Petrone ME Anagrelide for controle of thrombocythemia in polycythemia and other myeloproliferative disorders. Semin Hematol 1997; 34 : 51.
31. Prchal JF, Axelrad AA Letter: bone marrow responses in polycythemia vera. N Engl J Med 1974; 290 : 1382.
32. Pytlík R, Cmunt E, Kleibl Z et al. Úloha anagrelidu v léčbě esenciální trombocytémie. Trans Hematol dnes 2004; 10 : 154–160.
33. Rameshwar P, Denny TN, Stein D et al. Monocytes adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-β. J Immunol 1994; 153 : 2819–2830.
34. Reeder TL, Bailey RJ, Dewald GW et al. Both B and T lymphocytes may be clonally involved in myelofibrosis with myeloid metaplasia. Blood 2003; 101 : 1981–1983.
35. Roder S, Steimle C, Meinhardt G et al. STAT3 is constitutively active in some patients with polycythemia rubra vera. Exp Hematol 2001; 29 : 694–702.
36. Sacchi S, Vinci G, Gugliotta L et al. Diagnosis of essential thrombocythemia at platelet counts between 400 and 600 × 109/l. Haematologica 2000; 85 : 492–495.
37. Schafer AI Thrombocytosis. N Engl J Med 2004; 350 : 1211–1219.
38. Schwarz J, Penka M Trombocytózy a trombocytemie. Vnitř Lék 2005; 51: v tisku.
39. Skoda RC. Chronic myeloproliferative disorders: molecular markers and pathogenesis. Hematol J 2004; 5: S122–S125.
40. Spinelli O, Rota B, Finazzi G et al. Quantitative analysis of PRV-l gene expression in chronic myeloproliferative disorders: positive correlation with polycytemia vera diagnosis and leukocyte
alkaline phospahtase expression. Blood 2002; 100 : 3145a.
41. Spivak JL Diagnosis of the myeloproliferative disorders: resolving phenotypic mimicry. Semin Hematol 2003; 40 : 1–5.
42. Thiele J, Kvasnicka HM, Diehl V et al. Clinickopathological diagnosis and differential criteria of thrombocythemias in various myeloproliferative disorders by histopathology, histochemistry and immunostaining from bone marrow biopsies. Leukemia Lymphoma 1999; 33 : 207–218.
43. Thiele J, Kvasnicka HM, Graeff AS et al. Follow-up examinations including sequential bone marrow biopsies in essential thrombocythemia (ET): A retrospective clinicopathological study of 120 patients. Am J Hematol 2002; 70 : 283–291.
44. Thiele J, Kvasnicka HM, Werden C et al. Idiopathic primary osteo-myelofibrosis: a clinico-pathological study on 208 patients with special emphasis on evolution of disease features, differentiation from essential thrombocythemia and variables of prognostic impact. Leukemia Lymphoma 1996; 22 : 303–317.
45. Thiele J, Kvasnicka HM, Zankovich R et al. Clinical and morphological criteria for the diagnosis of prefibrotic idiopathic (primary) myelofibrosis. Ann Hematol 2001; 80 : 160–165.
46. Tefferi A, Solberg LA, Silverstein MN A clinical update on Polycythemia Vera and Essential Thrombocythemia. Am J Med 2000; 109 : 141–149.
47. Weinfeld A, Swolin B, Westin J Acute leukaemia after hydroxyurea therapy in Polycythaemia Vera and allied disorders: prospective study of efficacy and leukemogenicity with therapeutic implications. Eur J Haematol 1994; 52 : 134–139.
48. Wright CA, Tefferi A A single institutional experience with 43 pregnancies in Essential Thrombocythemia. Eur J Haematol 2001; 66 : 152.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 6
Most read in this issue
- Esophageal achalasia
- B-type natriuretic peptide (BNP) – application in differential diagnosis of dyspnoea
- Practice guidelines for diagnosis and therapy of essential thrombocytaemia and thrombocytaemia associated with other myeloproliferative diseases
- Disorders of iron metabolism I. Regulation of iron homeostasis