
Antifosfolipidový syndrom - popisy dvou případů
Antiphospholipid syndrome – the description of two cases
Antiphospholipid syndrome (APS) often occurs in young people, it is defined by the presence of venous or arterial thromboses, repeated miscarriages, thrombocytopenias and increased levels of antiphospholipid antibodies. Clinical symptoms are different, there is often experienced the phlebothrombosis of lower limbs, miscarriages or neurological symptoms characterized by transient ischemic attacks (TIA). If APS is associated with other system disease, most often with systemic lupus erythematosus (SLE), it is called secondary APS. We present two cases of secondary APS in the work. In first case we describe synchronous occurrence of SLE with secondary APS, which was clinically manifested by phlebothrombosis of veins of crus. At another elder patient there was stated the diagnosis of non - differentiated disease of bonding agent with secondary APS with cardial, pneumonic and neurological clinical symptoms.
Key words:
primary and secondary antiphospholipid syndromes – antiphospholipid antibodies – cerebral MR
:
L. Podrazilová 1; V. Peterová 2; M. Olejárová 1; Z. Seidl 2,3; C. Dostál 1
:
Revmatologický ústav, 1. lékařská fakulta UK, Praha, ředitel prof. MUDr. Karel Pavelka, DrSc.
1; MR oddělení Radiodiagnostické kliniky 1. lékařské fakulty UK a VFN, Praha, přednosta doc. MUDr. Jan Daneš, CSc.
2; Neurologická klinika 1. lékařské fakulty UK a VFN, Praha, přednosta prof. MUDr. Soňa Nevšímalová, DrSc.
3
:
Vnitř Lék 2006; 52(1): 89-94
:
Case Report
Antifosfolipidový syndrom (APS) se často vyskytuje u mladých osob, je definován přítomností žilních či tepenných trombóz, opakovanými samovolnými ztrátami plodu, trombocytopenií a zvýšenými hladinami antifosfolipidových protilátek. Klinické projevy jsou rozdílné, často bývá prodělaná flebotrombóza dolních končetin, ztráty plodu nebo neurologické projevy charakterizované přechodnými ischemickými atakami (TIA). Pokud je APS asociován s jiným systémovým onemocněním, nejčastěji systémovým lupus erytematodes (SLE), nazývá se sekundární APS. V práci předkládáme dva případy sekundárního APS. V prvním případě popisujeme současný výskyt SLE se sekundárním APS, který se klinicky projevil flebotrombózou žil bérce. U druhé starší nemocné byla stanovena diagnóza nediferencovaného onemocnění pojiva se sekundárním APS s klinickými projevy kardiálními, plicními i neurologickými.
Klíčová slova:
antifosfolipidový syndrom primární i sekundární - antifosfolipidové protilátky - MR mozku
Antifosfolipidový syndrom (APS) - Hughesův syndrom - je definován jako arteriální i žilní trombóza ve spojení s mnohočetnými spontánními potraty či cévními mozkovými příhodami a s antifosfolipidovými protilátkami v séru nemocných [1,2,3,4,5]. Vyskytuje-li se samostatně, je označován jako primární [3]. Je-li přítomen současně s jiným systémovým onemocněním pojiva, např. systémovým lupus erythematodes (SLE) či léky indukovaným lupusem, nazýváme jej sekundárním APS 6,7). V rámci APS mohou být postiženy jakékoliv cévy, takže dochází k rozsáhlé klinické symptomatologii (tab. 1). Antifosfolipidové protilátky (aPL) byly poprvé zjištěny Wassermanem na začátku 20. století jako falešně pozitivní reakce na syfilis [8]. Teprve v 50. letech 20. století byly detekovány precipitačními nebo komplement fixačními standardními testy na syfilis, které však nebyly specifické [9]. Později bylo zjištěno, že antigenem, který váže reagin, byl kyselý fosfolipid získaný alkoholovou extrakcí z hovězího myokardu a byl nazván kardiolipinem. Antikardiolipinové protilátky (aCL) byly nejprve vyšetřovány metodou radioimunoanalýzy (RIA), kde byl jako antigen použit negativně nabitý fosfolipid kardiolipin [10]. Později se rozšířil jednodušší průkaz pomocí enzymatické metody ELISA [11]. Lupusový antikoagulans (LA) je nespecifický inhibitor vedoucí v in vitro koagulačních testech k jejich prodloužení, které však není provázeno krvácivými, nýbrž trombotickými komplikacemi. V současné době se používají k průkazu nespecifického inhibitoru koagulace LA vhodnější testy, zejména hemokoagulační. Označení LA vyplynulo z časté asociace se SLE a poprvé bylo použito v roce 1972 [12]. V práci Fojtíka byla zjištěna asociace LA u 28 % nemocných se SLE [13]. Teprve před několika lety bylo zjištěno, že vazba autoprotilátek na negativně nabité fosfolipidy je závislá na přítomnosti plazmatického proteinového kofaktoru, kterým je β2-glykoproten I (β2-GPI), původně známý jako apolipoprotein H [14,15]. Molekula β2-GPI prodělá díky vazbě s fosfolipidem konformační změny, jenž vedou k odhalení antigenního epitopu. Protilátky proti tomuto epitopu jsou jednak tzv.„pravé“ autoimunitní antifosfolipidové protilátky a jednak parainfekční, reagující se samotným fosfolipidem. Druhé z nich jsou nezávislé na kofaktoru a nepůsobí klinické projevy a po vyléčení infekce zmizí. Stanovení anti β2-GPI protilátek ELISA testem doplňuje diagnostiku APS, umožňuje rozlišit trombofilní antifosfolipidové protilátky a včas odhalit případné riziko klinických komplikací tohoto syndromu. Stejnou roli mohou však hrát další sérové proteiny, jako protein S, C, anexin V, trombomodulin, vysoko i nízkomolekulární kininogeny a protrombin [16]. Interakcí mezi aPL a přirozenými inhibitory koagulace se zvyšuje riziko tromboembolických příhod. Za opakované potraty a ztráty plodu je předpokládáno, že jsou zodpovědné protilátky proti Anetinu [5,6].

Klasifikační kritéria pro tento syndrom byla vypracována Harrisem (tab. 2) a ke stanovení diagnózy by měli mít nemocní pozitivní alespoň jeden klinický příznak a opakovaně, tj. alespoň dvakrát s časovým odstupem 2 - 3 měsíců, i jeden laboratorní příznak ve středním až vysokém titru. O tzv. pravděpodobném APS hovoříme tehdy, pokud je u nemocného přítomen jeden klinický příznak a vysoká koncentrace aCL nebo jsou přítomny dva klinické příznaky při nízké hladině aCL protilátek [10]. V roce 1998 byla v Sapporu revidována starší Harrisova klasifikační kritéria pro APS (tab. 3) [16].
![Kritéria pro APS, dle [10].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ebfb8d479eaf3ecc885f5e80c4b17b32.png)

Kazuistiky
Případ 1
22letá nemocná s roční anamnézou systémového lupus erythematodes (SLE) a sekundárního antifosfolipidového syndromu (APS) byla vyšetřena v Revmatologickém ústavu (RÚ). V rodinné anamnéze je sledována matčina sestra pro SLE, osobní anamnéza je nevýznamná. Operovaná byla poprvé v lednu roku 1999, ve svých 21 letech, pro apendicitidu s následným rozvojem pravostranné pleuropneumonie středního a dolního laloku s pleuritickým syndromem, pro kterou byla přeléčena antibiotiky s přetrvávajícími pachypleuritickými změnami na kontrolním CT snímku. V květnu stejného roku se objevily kloubní potíže ve formě migrujících artralgií s artritidami (zápěstí a drobné ruční klouby) a ranní ztuhlostí přes 2 hodiny, provázané celkovými projevy onemocnění, subfebriliemi a necíleným váhovým úbytkem (4 kg/3 měsíce) s nočním pocením. O devět měsíců po výskytu pleuropneumonie s pleuritidou, tedy v září 1999, prodělala nemocná flebotrombózu bérce pravé dolní končetiny a proto byla zahájena antikoagulační léčba.
Pro splnění kritérií pro SLE, na podkladě motýlovitého erytému, artritid, serozitidy, laboratorního průkazu leukopenie a antinukleárních protilátek (ANA) s APS byla v prosinci roku 1999 zahájena léčba antimalariky s kortikosteroidy (Medrol 8 mg denně) a ponechána byla stávající antikoagulační léčba.
Během hospitalizace v RÚ u nemocné dominovaly kloubní potíže s objektivně prokazatelnou polyartritidou drobných ručních kloubů a náznakem motýlovitého erytému na kůži obličeje. Z laboratorních hodnot byly středně zvýšené reaktanty akutní fáze a v krevním obraze se potvrdila leukopenie s průkazem protilátek proti leukocytům a trombocytům. Při imunologickém vyšetření byla konstatována středně vysoká protilátková aktivita onemocnění, s přítomností ANA protilátek v titru 1 : 1280 a anti ds DNA protilátek v titru 1 : 80. V imunoblottingu byly prokázány protilátky proti Ro a La a antihistonové protilátky. Antikardiolipinové (ACLA) protilátky byly zvýšené v obou třídách IgG i IgM, rovněž byla hematologickými testy prokázána přítomnost LA, spolu s prodlouženým APTT testem. Genetickým vyšetřením nebyla prokázána přítomnost leidenské mutace v genu pro koagulační faktor V ani mutace v protrombinovém genu.
V rámci systémového přešetření při SLE sekundárním APS nebylo zjištěno orgánové postižení, provedeno bylo i vyšetření mozku pomocí MR s normálním nálezem. Při očním vyšetření Schirmerův test ukázal ještě normální hodnoty. Při hospitalizaci nebyl prokázán sekundární Sjögrenův syndrom při laboratorní pozitivitě Ro a La protilátek. Pro přetrvávající klinickou i autoprotilátkovou aktivitu byla nemocné terapeuticky navýšena dávka kortikosteroidů na 16 mg Medrolu denně, po úpravě medikace došlo k mírnému klinickému i laboratornímu zlepšení. Nemocná je i nadále sledována v naší ambulanci, při plánování mateřství bude pod odborným lékařským dohledem po celou dobu gravidity.
Případ 2
71letá nemocná s 15letou anamnézou ischemické choroby srdeční a hypertenze (od 22 let) byla přeložena z interního oddělení s podezřením na APS. Rodinná i gynekologická anamnéza nemocné je negativní. Již 15 let je nemocná léčena pro algickou formu ischemické choroby srdeční s recidivujícími atakami anginózních bolestí. V 70 letech nemocné, tedy rok před přijetím, prodělala diafragmatický infarkt myokardu, při provedené srdeční katetrizaci byla pro 50% stenózu na ramus circumflexus a významnou stenózu ramus marginalis sinister indikována k chirurgickému řešení. Stav nemocné se však komplikoval nejprve rozvojem sukcesivní plicní embolizace v lednu roku 2001, v únoru roku 2002 prodělala ischemickou cévní mozkovou příhodu s pravostrannou lokalizací a následovalo několik tranzitorních ischemických atak (TIA), poslední z nich byla v březnu roku 2003, tj. 2 měsíce před přijetím do RÚ. Od výskytu neurologických projevů byla nemocná trvale na antikoagulační léčbě. Provedené CT vyšetření mozku (únor roku 2002) prokázalo nález periventrikulární mozkové atrofie s hypodenzními zónami paraventrikulárně odpovídající encefalopatii bez známek hemoragie. Sonografické vyšetření karotid (únor roku 2002) ozřejmilo sklerotické pláty v bifurkacích bilaterálně bez stenotického zrychlení toku. Pro laboratorní pozitivitu antinukleárních, Ro, La a antikardiolipinových protilátek byla doporučena k diagnostickému pobytu. Subjektivně byla nemocná zcela bez obtíží, objektivně na kůži byly patrné četné hematomy, orientačně neurologický nález byl zcela v normě.
Nemocná byla během hospitalizace vyšetřena v rámci orgánového přešetření u SLE, pro tuto diagnózu však nebyla splněna kritéria. V imunologii byla potvrzena pozitivita ANA protilátek v titru 1 : 320 s jemně zrnitou a homogenní fluorescencí, dále anti ds DNA protilátek v titru 1 : 40. V testu extrahovatelných nukleárních antigenů (ENA) byla potvrzena pozitivita Ro a La. Splněna byla klinická kritéria s anamnézou infarktu myokardu, sukcesivní plicní embolizace, CMP a opakovaných TIA i laboratorní kritéria pro APS: tedy pozitivita antikardiolipinových protilátek (ACLA) obou izotypů IgG i M s prokázanou přítomností inhibitorů typu LA i anti β2-GPI. V krevním obraze byla potvrzena sekundární normochromní, normocytární anémie s hemoglobinem 101 g/l kombinované etiologie při základním systémovém onemocnění a chronické renální insuficienci.
Chronická renální insuficience nejasné etiologie s významnou proteinurií (přes 1 g denně) a retencí dusíkatých látek, bez nálezu erytrocyturie a válců byla v souladu s nefrologickým konziliem hodnocena spíše jako projev intersticiální nefritidy než glomerulonefritidy a renální biopsie nebyla indikována vzhledem ke klinickému stavu a warfarinizaci nemocné.
Podrobné neurologické vyšetření nemocné po prodělané CMP s lateralizací a opakovaných TIA prokázalo pouze frustní známky po proběhlé pravostranné cévní mozkové příhodě. MR mozku i krční míchy prokázala přítomnost splývavých ložiskových hyperintenzit v T2 vážených obrazech supra - i infratentoriálně (obr. 1). Tento nález je však nespecifický vzhledem k věku, odpovídá nejspíše postischemickým změnám staršího data v kombinaci s hypertenzní encefalopatií, bez poruchy hematoencefalické bariéry (HEB).
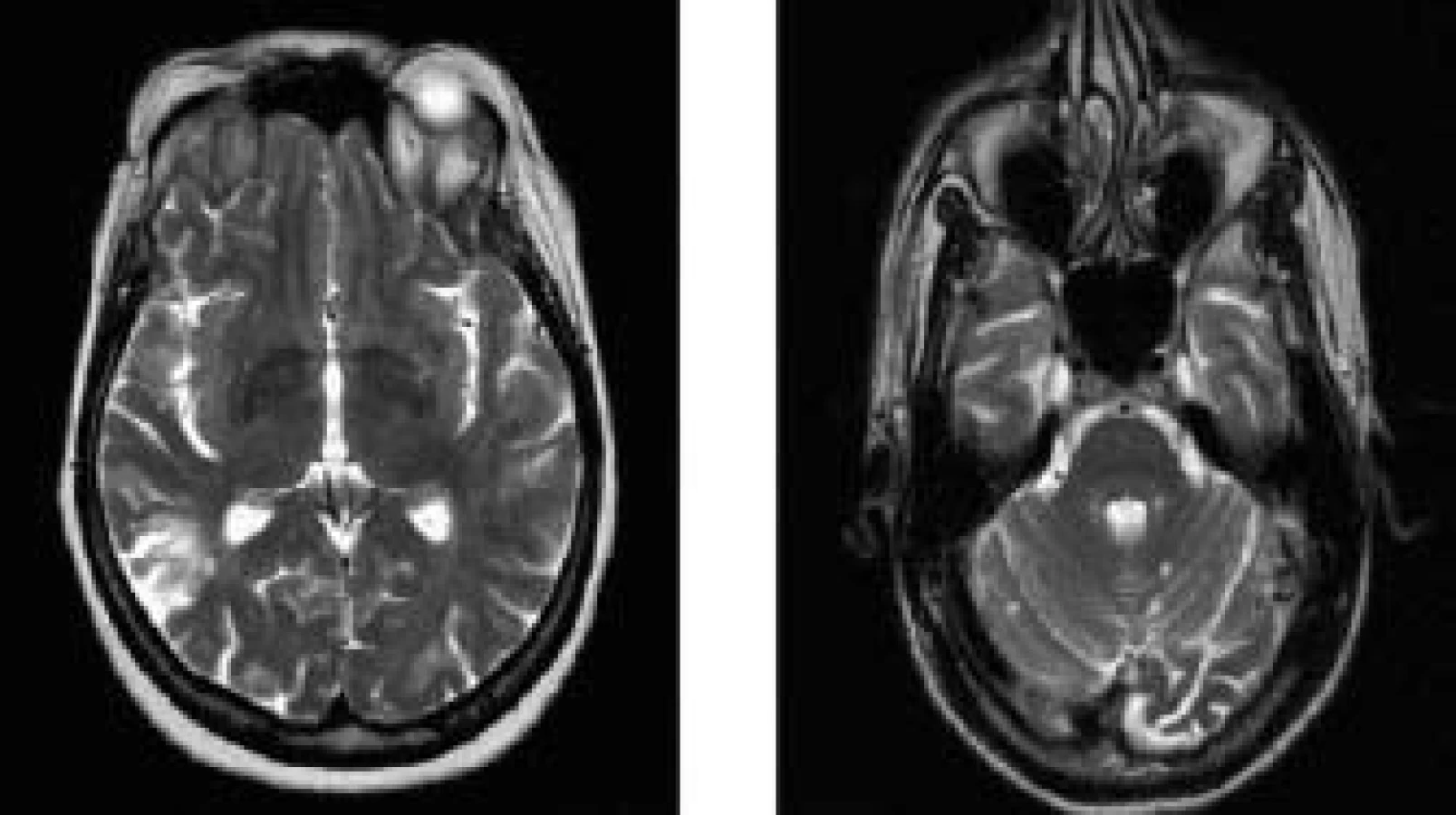
Zhodnocením veškerých klinických i laboratorních výsledků jsme stav nemocné uzavřeli jako nediferencované onemocnění pojiva se sekundárním APS a ponechána byla trvalá warfarinizace s pečlivým monitorováním INR. Další léčba systémového onemocnění vzhledem k věku nemocné a chybění jiných orgánových projevů nebyla indikována.
Diskuze
Předložené případy ilustrují, jak rozdílné mohou být klinické projevy antifosfolipidového syndromu, a zejména pak věk nemocných v době manifestace postižení. V prvním případě se jedná o typický výskyt u mladé ženy se SLE, která je po prodělané flebotrombóze DK trvale warfarinizována. Při plánované graviditě je nutná její trvalá dispenzarizace s léčebnou prevencí, neboť tato nemocná je ohrožena opakovanými ztrátami plodu. U gravidních žen s APS se podává dle tíže klinických projevů kyselina acetylsalicylová, heparin či nízkomolekulární hepariny, případně kortikosteroidy. V těžších případech je popisována i aplikace intravenózních imunoglobulinů či plazmaferéza [16].
V druhém případě starší ženy se první projevy nemoci vyskytují v pozdním věku, tedy v 7. dekádě života a během jednoho roku jsou postupně postiženy srdce, plíce i mozek nemocné. Zde nejsou splněna kritéria pro SLE a stav je hodnocen jako nediferencované onemocnění pojiva se sekundárním APS opět s nutností trvalé antikoagulační léčby. V obou případech je potvrzena asociace sekundárního APS s pozitivitou ANA a v druhém případě i s anémií [17,18].
Při neurologických symptomech u nemocných s APS i systémovými onemocněním jiné geneze je nejspecifičtější vyšetření mozku pomocí magnetické rezonance [19,20]. Na MR snímcích jsou nejčastěji patrná drobná (do 3 mm) difuzní, hyperintenzní ložiska odpovídající mikroinfarktům v bílé hmotě mozku v několika arteriálních teritoriích zasahujících až do subkortikální oblasti, způsobené vaskulárními mikrolézemi. Větší (> 6 mm) klínovité ložisko v bílé hmotě mozku odpovídá klinicky významnému infarktu. Glióza ložiska tvořená astrocyty a demyelinizací má v MR izosignální obraz. Porušení HEB se projeví enhancementem po podání kontrastní látky, chelátově vázaného gadolinia [19]. V rámci diferenciální diagnostiky magneticko-rezonančího nálezu přicházejí v úvahu všechna ložisková postižení bílé hmoty: roztroušená skleróza (RS) [21,22,23,24], neuroinfekce, jiná systémová onemocnění s postižením mozku [25], vaskulitidy [26], prosté postischemické změny cévní a migrény. Neuropsychiatrické projevy v rámci SLE, tzv. neuropsychiatrický lupus erythematodes (NPSLE) [27,28,29,30,31], se vyskytují u více než 50 % pacientů se SLE. Neurologické projevy postihují nejčastěji centrální nervový systém, dále periferní i vegetativní nervový systém. Rovněž se vyskytují i psychiatrické projevy a jednotlivé příznaky se mohou vyskytovat kdykoliv v průběhu SLE a mohou se vzájemně kombinovat. Neuropsychiatrické projevy mohou být vyvolány působením autoprotilátek nebo postižením cév zánětlivou vaskulitidou či častěji nezánětlivou vaskulopatií s mikroinfarzací drobných cév. Při výskytu CMP je nezbytné vyloučit možnost embolizace z endokardu pomocí jícnové echokardiografie. S rostoucím věkem může činit problémy odlišení postischemických ložisek v bílé i šedé hmotě, zároveň se zvyšuje počet lézí při arteriální hypertenzi [32], jako u námi prezentované druhé nemocné.
Přítomnost antifosfolipidových protilátek je většinou prokazována u nemocných s postižením mozku při SLE, velice často je asociována s cévní mozkovou příhodou [33,34,35]. Dokladem toho je studie, která prokázala přítomnost antikardiolipinových protilátek třídy IgG v autopsiích u více než 90 % pacientů zemřelých na SLE s prokázanými vaskulárními histopatologickými změnami. Kromě abnormální koagulace za přítomnosti aCL se pravděpodobně uplatňuje i přímé poškození endoteliálních buněk [17].
Závěr
Antifosfolipidový syndrom primární či sekundární se může objevit v různém věku a je nutno na něj v rámci diferenciální diagnostiky systémových onemocnění myslet, zejména pokud jsou přítomny klinické známky. Ke klinickým projevům mladších žen patří i riziko samovolných potratů a ztrát plodu, a proto je vhodné tyto ženy dispenzarizovat a léčit po celou dobu gravidity ve specializované poradně. V pozdějším věku jsou pozorovány neurologické projevy jako jsou ikty a TIA, při vyšetření MR neodlišíme změny postichemické a hypertenzní od mikroinfarktů při APS. Neurologické klinické projevy jsou různé: iktus v úvodu s očními příznaky mohou imitovat jak RS, tak i NPSLE [23]. MR vyšetření se jeví jako nejsenzitivnější metoda, avšak nález u APS není specifický, diferenciálně diagnosticky odlišujeme od jiných ložiskových postiženích bílé hmoty mozku. Podle dosavadních literárních údajů mají někteří nemocní s APS v nálezech na MR mozku ložiskové hyperintenzity v T2 vážených obrazech [19,20], které mohou jevit určitou podobnost nálezů u nemocných s RS [21,22,23,24] nebo se SLE s postižením nervového systému (NPSLE) [27,28,29,30,31]. Proto doporučujeme vyšetřit hladiny antifosfolipidových protilátek u všech nemocných nejenom žen mladšího věku s akutní symptomatologií a aborty, ale i u ostatních, u kterých je podezření na antifosfolipidový syndrom. Dosud však nebyla prokázána souvislost mezi titrem antifosfolipidových protilátek, klinickou manifestací a obrazem mozku v MR.
Práce vychází z grantu IGA MZČR NR 8459-3.
MUDr. Lucie Podrazilová roz. Linková
www.revma.cz
e-mail: link@revma.cz
Doručeno do redakce: 3. 10. 2005
Přijato po recenzi: 7. 11. 2005
Sources
1. Khamasta MA, Hughes GR. The antifosfolipid syndrome. Rheumatology in Europe 1994; 23 : 23-25.
2. Asherson RA, Khamashta MA, Ordi-Ros J et al. The primary antiphospholipid syndrome: major clinical and serological features. Medicine 1989; 68 : 366-374.
3. Vencovský J. Antifosfolipidové protilátky, Antifosfolipidový syndrom. In: Autoimunitní systémová onemocnění. Praha: Triton 1998; 27, 107-110.
4. Scott TF, Hess D, Brillmanm J. Antiphospholipid antibody syndrome mimicking multiple sclerosis clinically and by magnetic resonance imaging. Arch Intern Med 1994; 154 : 917-920.
5. Dostál C. Autoprotilátky u systémového lupus erytematodes. In: Dostál C., Vencovský J (eds). Systémový lupus erythematodes. Praha: Medprint 1997; 38-56.
6. Dostál C, Vencovský J et al. Antifosfolipidové protilátky, Antifosfolipidový syndrom. In: Dostál C., Vencovský J (eds). Systémový lupus erytematodes. Praha: Medprint 1997; 57-64, 136-147.
7. Cervera R, Piette JC, Font J et al. Antiphospholipid syndrome. Arthritis Rheumatism 2002; 46 : 1019-1027.
8. Wasserman A, Neisser A, Bruck C. Eine serodiagnostiche Reaktion bei Syphylis. Deutch Med Wochenschr 1906; 32 : 475-489.
9. Caterall RD. Biological false positive reactions and systemic disease. In: Walker G (ed). Ninth Symposium in Advanced Medicine. London: Pitman Medical: 1973; 97-111.
10. Harris EN, Gharavi AE, Boey ML et al. Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 1983; 1211-1214.
11. Loizou S, Mc Crea JD, Rudge AC et al. Measurement of anti-cardiolipin antibodies by an enzyme-linked immunosorbent assay (ELISA): Standartization and quantitation of results. Clin Exp Immunol 1985; 62 : 3, 738-745.
12. Feinstein DI, Rapaport SI. Acquired inhibitors of Blood Coagulation. Prog Hemos Thromb 1972; 1 : 75-95.
13. Fojtík Z, Beránek M, Klabusay M et al. Výskyt vybraných antifosfolipidových protilátek u skupiny nemocných se systémovým lupus erythematodes. Vnitř Lék 2004; 50 : 267-273.
14. Galli M, Comfurius P, Maasem C et al. Anticardiolipin antibodies (ACA) are directed not to cardiolipin but to a plasma cofactor. Lancet 1990; 335 : 1544-1547.
15. Mc Neil HP, Simpson RJ, Chesterman CN et al. Antiphospholipid antibodies are directed against a complex antigen that includes a lipid binding inhibitor of coagulation: beta2-glykoprotein I (apolipoprotein H). Proc Natl Acad Sci USA 1990; 87 : 4120-4124.
16. Dostál C. Antifosfolipidový syndrom. In: Pavelka K, Rovenský J. Klinická revmatologie. Praha: Galén 2003; 265-272.
17. Sipek-Dolnicar A, Hojnik M, Bozic B et al. Clinical presentations and vascular histopatology in autopsied patients with systemic lupus erythematosus and anticardiolipin antibodies. Clin Exp Rheumatol 2002; 20 : 335-342.
18. Marai I, Levi Y, Godard G et al. Following 90 patients with antiphospholipid syndrome with antibody titers and correlations with clinical manifestations: symptoms of the disease, a new antibody and correlations with clinical manifestations in the Israeli population. Harefuah 2001; 140 : 495-500.
19. Molad Y, Sidi Y, Gornish M et al. Lupus anticoagulant: correlation with magnetic resonance imagingof brain lesions. J Rheumatol 1992; 19 : 556-561.
20. Appenzeller S, Zeller CB, Annichino-Bizzachi JM et al. Cerebral venous thrombosis: influence of risk factors and imaging findings on prognosis. Clin Neurol Neurosurg. 2005; 107 : 371-378.
21. Kelly BJ, Cronin M, Curran JJ. Anticardiolipin syndrome resembling multiple sclerosis. Arthritis Rheum 1989; 32: S71.
22. Isaac C, Li DK, Genton M et al. Multiple sclerosis: a serial study using MRI in relapsing patients. Neurology 1988; 38 : 1511-1515.
23. Ferreira S, Cruz DPD, Hughes GRV. Multiple sclerosis, neuropsychiatric lupus and antiphospholipid syndrome: where do we stand? Rheumatology 2005; 44 : 434-442.
24. Paran D, Chapman J, Korczyn AD et al. Evoked potential studies in the antiphospholipid syndrome: differential diagnosis from multiple sclerosis. Ann Rheum Dis 2005; 17, abstrakt.
25. Totolian NA. Magnetic - resonance tomography in differential diagnosis of brain lesions in demyelinating and systemic autoimmune diseases. Zh Nevrol Psikhiatr Im S S Korsakova 2005; 105 : 42-46.
26. Ingram SB, Goodnight SH, Bennett RM. An unusual syndrome of a devastating noninflammatory vasculopathy associated with anticardiolipin antibodies. Ann Rheum Dis 1988; 47 : 681-683.
27. Asherson RA, Derksen RHWM, Harris EN et al. Chorea in systemic lupus erythematosus and „lupus-like“ disease: association with antiphospholipid antibodies. Semin Arthritis Rheum 1987; 16 : 253-259.
28. Cauli A, Montaldo C, Peltz MT et al. Abnormalities of magnetic resonance imaging of the central nervous system in patients with systemic lupus erythematosus correlate with disease severity. Clin Exp Rheumatol 1994; 12 : 389-394.
29. Sibbitt WL jr, Sibbitt RR, Griffey RH et al. Magnetic resonance and computed tomographic imaging in the evaluation of acute neuropsychiatric disease in systemic lupus erythematosus. Ann Rheum Dis 1989; 48 : 1014-1022.
30. Peterová V, Dostál C, Linková L et al. The distribution of MR lesions in neuropsychiatric lupus erythematosus and multiple sclerosis patients. J Neuroradiol 2002, Suppl: S40.
31. Linková L, Peterová V, Olejárová M et al. Neuropsychiatrické postižení u pacientů se systémovým lupus erythematodes. Čes Revm 2003; 2, 78-82.
32. Jensen MC, Brant-Zawadski MN, Jacobs BC. Ischemia. In: Magnetic Resonance Imaging. Ed. Mosby 1999, USA, 1255-1274.
33. Harris EN, Gharavi AE, Asherson RA. Cerebral infarction in systemic lupus: association with anticardiolipin antibodies. Clin Exp Rheumatol 1984; 2 : 47-51.
34. Toubi E, Khamashta MA, Panarra A et al. Association of antiphospholipid antibodies with central nervous system disease in systemic lupus erythematosus. Amer J Med 1995; 99 : 397-401.
35. Levine SR, Deegan MJ, Futrell A et al. Cerebrovascular and neurologic disease associated with antiphospholipid antibodies: 48 cases. Neurology 1990; 40 : 1181-1189.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2006 Issue 1
Most read in this issue
- AIP – Atherogenic index of plasma like significant predictor of cardiovascular risk: from research to practice
- The volumes of the thyroid gland in adults aged 18-65 years in Czech republic - determination of the norms
-
Diagnostika a léčba jaterní encefalopatie
Doporučený postup vypracovaný skupinou pro portální hypertenzi při České hepatologické společnosti České lékařské společnosti J. E. Purkyně a schválený výborem České hepatologické společnosti České lékařské společnosti J. E. Purkyně - Antiphospholipid syndrome – the description of two cases