
Nádory štítné žlázy a Hirschsprungova choroba: desetileté zkušenosti s molekulárně genetickou diagnostikou RET proto-onkogenu
Thyroid carcinomas and Hirschsprung’s disease – 10-year experience with molecular genetic testing of the RET proto-oncogene
Research of the last ten years approved the role of the RET proto–oncogene in the pathogenesis of thyroid cancer such as medullary thyroid carcinoma (MTC) and papillary thyroid carcinoma (PTC), multiple endocrine neoplasia type 2 syndromes (MEN 2) and Hirschsprung’s disease that could be associated with MTC or MEN 2. Thanks to the molecular genetic testing, which enables to detect the gene mutations, the course of the disease could be predicted and the mutation carriers among the at-risk persons could be cured at the very early, clinically asymptomatic stage of the disease. Then the prophylactic total thyreoidectomy is recommended. Recently, the physiological role of the RET proto-oncogene in normal cell proliferation, differentiation and survival has been intensively studied. Thanks to this new knowledge the possibility of the gene therapy at the RET signaling cascade level is supposed in the treatment of these patients in near future.
Key words:
medullary thyroid carcinoma – papillary thyroid carcinoma – Hirschsprung’s disease – molecular genetic testing – RET proto-oncogene – gene therapy
Authors:
B. Bendlová 1
![]() ; Š. Dvořáková 1; E. Václavíková 1; V. Sýkorová 1; P. Vlček 2; R. Škába 3
; Š. Dvořáková 1; E. Václavíková 1; V. Sýkorová 1; P. Vlček 2; R. Škába 3
Authors‘ workplace:
Endokrinologický ústav, Praha, ředitel doc. MUDr. Vojtěch Hainer, CSc.
1; Klinika nukleární medicíny a endokrinologie 2. lékařské fakulty UK a FN Motol, Praha, přednosta doc. MUDr. Petr Vlček, CSc.
2; Klinika dětské chirurgie 2. lékařské fakulty UK a FN Motol, Praha, přednosta prof. MUDr. Jiří Šnajdauf, DrSc.
3
Published in:
Vnitř Lék 2006; 52(10): 926-934
Category:
Review
Overview
Výzkumy posledních 10 let prokázaly patogenetickou roli RET proto-onkogenu u nádorových onemocnění štítné žlázy – medulárního karcinomu štítné žlázy (MTC), papilárního karcinomu štítné žlázy (PTC), u syndromů MEN 2 a u Hirschsprungovy choroby, která může být spojena s MTC či MEN 2. Molekulárně genetická diagnostika umožňuje nalézt mutace genu, na základě nichž lze předvídat průběh onemocnění, ale zejména je přínosná u rizikových osob, v jejichž případech je možno při pozitivním nálezu mutace terapeuticky zasáhnout již v presymptomatickém stadiu onemocnění. Nositelům zárodečné mutace je doporučena profylaktická totální tyreoidektomie. Nyní se intenzivně zkoumá fyziologická role RET proto-onkogenu v normální proliferaci, diferenciaci a přežívání buněk. Díky novým poznatkům se rýsuje i možnost terapeutického využití genové terapie na úrovni RET-signalizační kaskády.
Klíčová slova:
medulární karcinom štítné žlázy – papilární karcinom štítné žlázy – Hirschsprungova choroba – molekulárně genetická diagnostika – RET proto–onkogen – genová terapie
Úvod
Již téměř 10 let studujeme molekulárně genetické příčiny medulárního karcinomu štítné žlázy, nověji i papilárního karcinomu štítné žlázy a Hirschsprungovy choroby. Tato klinicky zdánlivě vzdálená onemocnění mají společnou patogenetickou příčinu - mutace v RET proto-onkogenu. Studium RET proto-onkogenu má význam nejen vědecký, ale detekce genetických alterací tohoto genu slouží dnes již k rutinní molekulárně genetické diagnostice, která významně přispívá k predikci onemocnění a umožňuje včasný profylaktický zákrok snižující morbiditu a mortalitu spojenou s těmito onemocněními.
RET proto-onkogen - normální funkce
Proteinkinázy jsou zapojeny do většiny signálních cest. Velkou skupinu (90 z 518 známých proteinkináz) představují receptorové či nereceptorové tyrozinkinázy, v jejichž aktivaci hrají roli specifické tyrozinové zbytky [39]. Defekty tyrozinkinázových receptorů jsou příčinou řady onemocnění a tyrozinkinázové receptory jsou tak slibnými terapeutickými cíli [24]. Jsou to transmembránové receptory s vnitřní kinázovou aktivitou, která je stimulována ligandy. Navázání extracelulárního ligandu vede k dimerizaci receptoru, kdy dojde ke spojení dvou katalytických domén, což umožní transfosforylaci tyrozinových zbytků. Fosfotyroziny šíří dále signál prostřednictvím intracelulárních proteinů, které nesou SH2 (src-homology 2) a fosfotyrozinové vazebné domény. Aktivace tyrozinkináz tak iniciuje intracelulární signalizační kaskádu, která nakonec vede k modulaci genové exprese a biologické odpovědi [50].
RET proto-onkogen je transmembránovou receptorovou tyrozinkinázou s typickými strukturními a funkčními oblastmi (obr. 1) [34]. Jeho extracelulární část obsahuje 4 adherinu podobné domény, doménu vážící vápník a oblast bohatou na cysteiny. Tato oblast obsahuje 28 cysteinových zbytků, z nichž 27 je konzervováno a vyskytuje se u mnoha živočišných druhů. Mezi cysteiny se tvoří disulfidické vazby, které jsou rozhodující při formování terciární struktury RET proteinu. Intracelulární část obsahuje typickou tyrozinkinázovou doménu. V důsledku alternativního sestřihu vznikají izoformy RETu. RET9 a RET51 jsou dvě hlavní proteinové izoformy o 1072 a 1114 aminokyselinách [15].
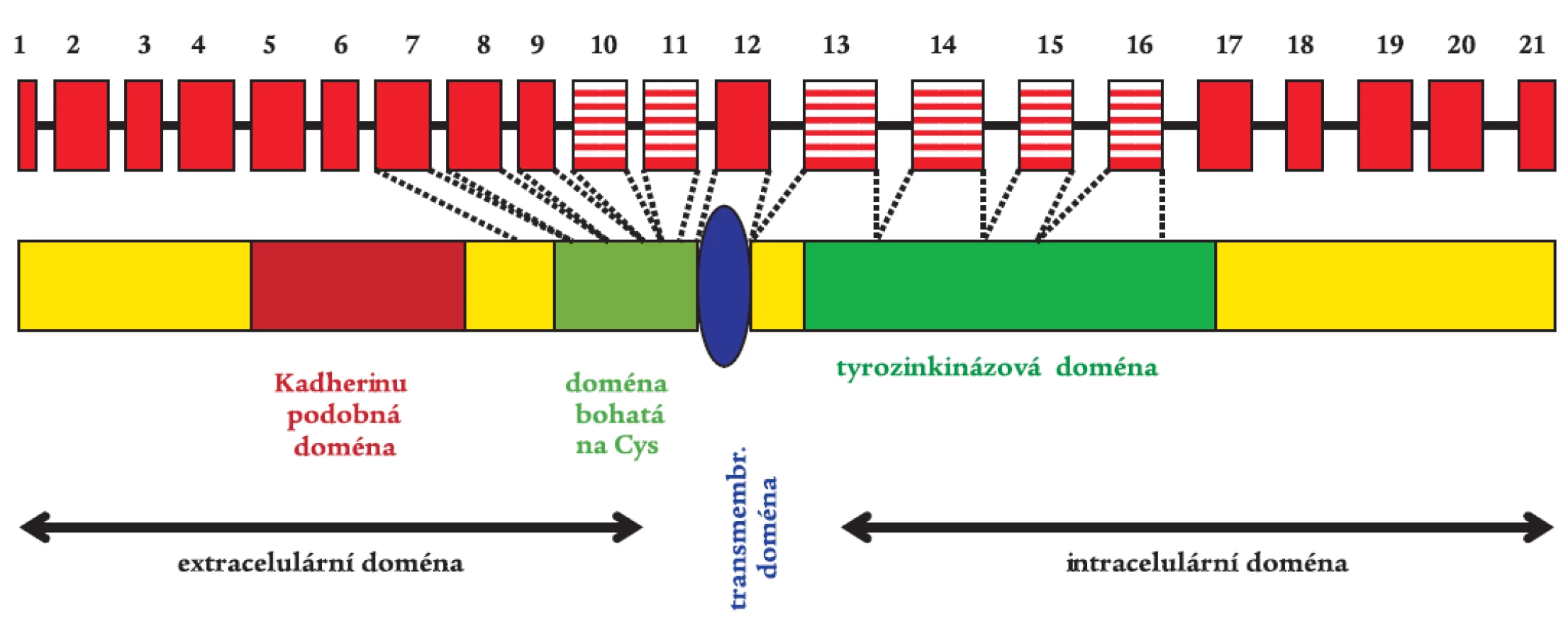
Aktivace RETu je velmi přísně regulovaný, tkáňově specifický proces. Za normálních okolností je RET protein exprimován v raných vývojových fázích v buňkách odvozených od neuroektodermu, tedy z neurální lišty. RET protein je při aktivaci součástí multikomponentního komplexu, který váže růstové faktory z rodiny GDNF (glial derived neurotrophic factor). Byly izolovány 4 ligandy aktivující RET, jejichž specifita se vzájemně částečně překrývá. Jsou to GDNF, neurturin, artemin a persefin. Ligandy se nejprve musí navázat na své specifické koreceptory (GFRα1-4), které jsou ukotveny v membráně prostřednictvím glykosylfosfatidylinositolu. Ligandy se poté spolu se svými koreceptory vážou na dvě molekuly RETu, dojde k dimerizaci receptoru, která spustí autofosforylaci klíčových tyrozinových zbytků a následnou intracelulární signalizaci (obr. 2) [1,38,49]. Vazbou GDNF na koreceptor se zároveň mění lipidové okolí RETu v plazmatické membráně. Intracelulární doména RETu obsahuje nejméně 12 autofosforylačních míst. Tyrozinové zbytky (Tyr) Tyr1090 a Tyr1096 jsou přítomny pouze u izoformy RET51. Fosforylované tyroziny RETu slouží jako místa vazebné interakce s intracelulárními signalizačními proteiny. Tyr905 je vazebným místem pro adaptory Grb7/10, Tyr1015 pro fosfolipázu Cγ, Tyr981 pro c-Src a Tyr1096, specifický jen pro RET51, pro Grb2. Tyr1062 je vazebným místem pro takové proteiny jako jsou Shc, ShcC, IRS1/2, FRS2, DOK1/4/5 a Enigma. Tvorba těchto proteinových komplexů s Tyr1062 vede ke stimulaci Ras/ERK a fosfatidylinositol-3-kináza/AKT kaskády. Dle výsledků studií prováděných na buněčných kulturách a transgenních zvířecích modelech je přítomnost tyrozinu v pozici 1062 nezbytnou podmínkou pro transformační schopnost RET proto-onkogenu [14,26,33,36,46]. Ligandy ovlivňují aktivaci RET proteinu nejspíše autokrinním, možná i parakrinním mechanizmem. RET je nepostradatelný pro vývoj sympatického, parasympatického a enterického nervového systému a ledvin [43,51].
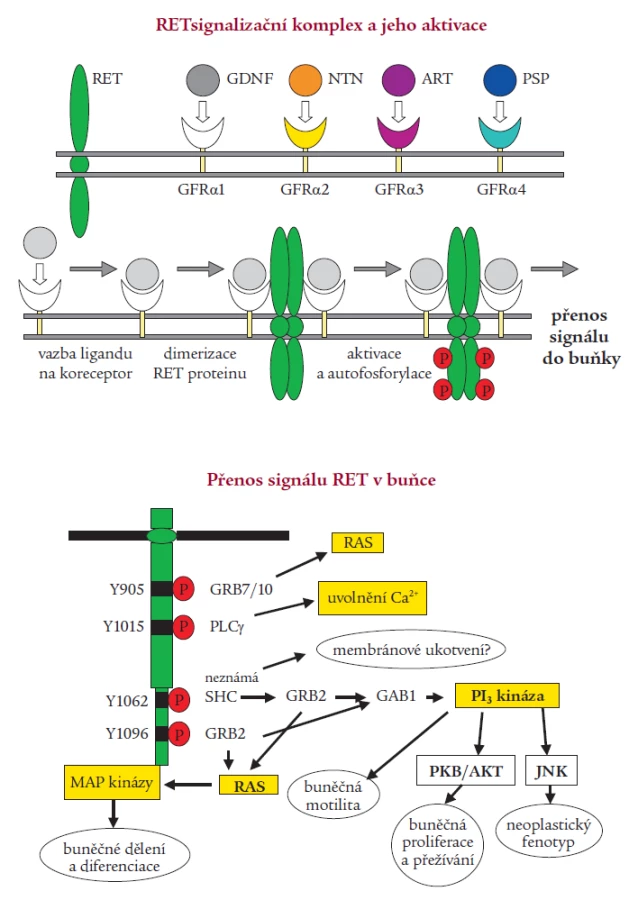
Poruchy RET signalizace
Za normálních okolností je aktivita receptorových tyrozinkináz přísně regulována. Pokud dojde k poruše této regulace, stávají se účinnými onkogeny [23,49]. K onkogenní konverzi tyrozinkinázových receptorů dochází v důsledku různých mechanizmů, zejména retrovirovou transdukcí, v důsledku genomového přeskupení, bodovými mutacemi, případně zvýšenou expresí [6]. RET je jediným příkladem tyrozinkinázového receptorového genu, jehož alterace způsobují různé typy lidských nádorů. Dysfunkce RET signalizační kaskády je příčinou nádorových onemocnění štítné žlázy - medulárního karcinomu a papilárního karcinomu, kdy dochází k nepatřičné aktivaci této kaskády. U Hirschsprungovy choroby je naopak RET signalizace inhibována [22].
1. Medulární karcinom štítné žlázy, syndromy MEN 2
Zárodečné bodové mutace v RETu jsou příčinou 3 nádorových syndromů s autozomálně dominantním typem dědičnosti: mnohočetné endokrinní neoplazie typu 2A (MEN 2A), typu 2B (MEN 2B) a familiárního medulárního karcinomu štítné žlázy (FMTC) (Multiple Endocrine Neoplasia Type 2: www.genetests.org) [41]. Pro tyto hereditární nádorové syndromy je příznačný familiární výskyt medulárního karcinomu štítné žlázy (MTC), což je maligní tumor vycházející z parafolikulárních buněk (C-buněk) štítné žlázy secernujících kalcitonin. Feochromocytom je přítomen u přibližně 50 % postižených v rodinách s MEN 2A, hyperparatyreóza pak asi u 15-30 % pacientů. Pacienti se syndromem MEN 2B mívají typický astenický marfanoidní habitus, u více než poloviny pacientů je přítomen feochromocytom, častá je ganglioneuromatóza gastrointestinálního traktu, mukózní neurinomy na rtech a jazyku, případně další léze. V rodinách s FMTC se vyskytuje pouze MTC bez dalších lézí. U pacientů s MEN 2B se MTC manifestuje již v raném dětství, u pacientů s MEN 2A obvykle v rané dospělosti a u FMTC ve středním věku. Přes 95 % pacientů se syndromy MEN 2 a až 88 % pacientů s FMTC nese zárodečné mutace v RET proto-onkogenu. Tento nález vedl k zavedení genetického screeningu, který umožňuje včasnou diagnózu hereditárního MTC u příbuzných v riziku onemocnění, u pacientů pak predikci vývoje onemocnění. Většina mutací nalézaných u pacientů s MEN 2A a s FMTC postihuje extracelulární oblast RETu bohatou na cysteiny. MEN 2A fenotyp je spojován nejčastěji s mutacemi v kodonu 634 (85 %), hlavně Cys634Arg, zatímco mutace spojené s FMTC fenotypem postihují rovnoměrně různé cysteiny extracelulární domény. FMTC může být také spojen s bodovými mutacemi v kinázové doméně RETu (Glu768Asp, Leu790Phe, Tyr791Phe, Val804Leu, Val804Met, Ser891Ala). Většina pacientů s MEN 2B nese mutaci Met918Thr v tyrozinkinázové doméně, jen u malé frakce těchto pacientů byla nalezena mutace Ala883Phe. Většina MTC (75 % případů) však vzniká sporadicky. Zhruba u poloviny nádorových tkání jsou pak detekovány somatické mutace pouze v nádorové tkáni, nejčastěji Met918Thr. Vzhledem k tomu, že až u 5-20 % prvotních záchytů MTC je detekována zárodečná, tedy na potomstvo přenosná mutace, doporučuje se geneticky testovat i tyto rodiny [8,9,21,31,37]. Mutace v RET proto-onkogenu jsou vzácně detekovány i u sporadických feochromocytomů [5]. Mechanizmus vedoucí k onkogenní konverzi RET proteinu závisí na typu a místě aminokyselinové záměny. RET s mutací v extracelulární doméně v kodonu pro některý z klíčových cysteinů tvoří kovalentní dimery, které vykazují konstitutivní kinázovou aktivitu nezávislou na ligandu. Záměna cysteinu za jinou aminokyselinu totiž zabrání tvorbě intramolekulárních disulfidických vazeb a umožní tvorbu intermolekulárních disulfidických vazeb mezi nepárovými cysteiny [48]. Mutace spojené s FMTC mívají nižší transformační aktivitu než mutace asociované s MEN 2A [55]. Mutace Met918Thr, typická pro syndrom MEN 2B, je spojena se změnou substrátové specifity. Dochází k aktivaci RET signalizační kaskády nezávisle jak na ligandu, tak na dimerizaci. V tomto ohledu jsou mutace spojené s MEN 2B rozdílné od mutací spojených s MEN 2A, liší se ve stechiometrii fosforylace tyrozinů RET proteinu a dalších intracelulárních proteinů. Navíc nádory typu MEN 2B mají odlišný genový expresní profil oproti nádorům typu MEN 2A [27]. Všechny mutace RET proto-onkogenu asociované se syndromy MEN 2 či s FMTC zvyšují aktivaci kinázy a onkogenní konverzi (gain-of-function). Nádorový fenotyp může ještě zhoršovat nerovnováha mezi mutantní a zdravou alelou, např. nález zárodečných mutací na jedné alele a somatických mutací na druhé, či ztráta heterozygozity a/nebo amplifikace mutantního RET proteinu [25].
2. Papilární karcinom štítné žlázy
Papilární karcinom štítné žlázy (PTC) představuje 80-90 % všech karcinomů štítné žlázy [53]. Častou genetickou příčinou PTC jsou somatické chromozomální inverze nebo translokace, které způsobují rekombinaci intracelulární kinázové domény RET proto-onkogenu s heterologními geny za vzniku chimérických onkogenů RET/PTC (tab. 1) [28]. Dnes již není pochyb o tom, že nejčastější příčinou vzniku RET/PTC přeskupení je radioaktivní ozáření. Různé typy RET/PTC přeskupení byly nalezeny u více než 60 % „postčernobylských“ nádorových tkání PTC [57]. Indukce tvorby RET/PTC onkogenů vlivem radiace byla potvrzena i experimentálně, jak in vivo, tak in vitro [40]. Jak ionizující záření může indukovat vznik RET/PTC přeskupení vysvětluje zajímavý model, který dokládá, že ač jsou fúzní geny, např. v případě RET/PTC1 geny RET a H4, od sebe daleko vzdálené na lineární mapě 10. chromozomu, v jádrech buněk štítné žlázy jsou vlivem terciární a kvartérní struktury často blízko sebe, což umožňuje jejich rekombinaci [42]. Vlivem fúze RET/PTC dojde k zahájení exprese RET fúzního proteinu, který ovšem může být exprimován pouze v cytoplazmě, transmembránová a extracelulární část proteinu totiž chybí. Část partnerského genu umožní dimerizaci tyrozikinázových domén a aktivaci RETu.
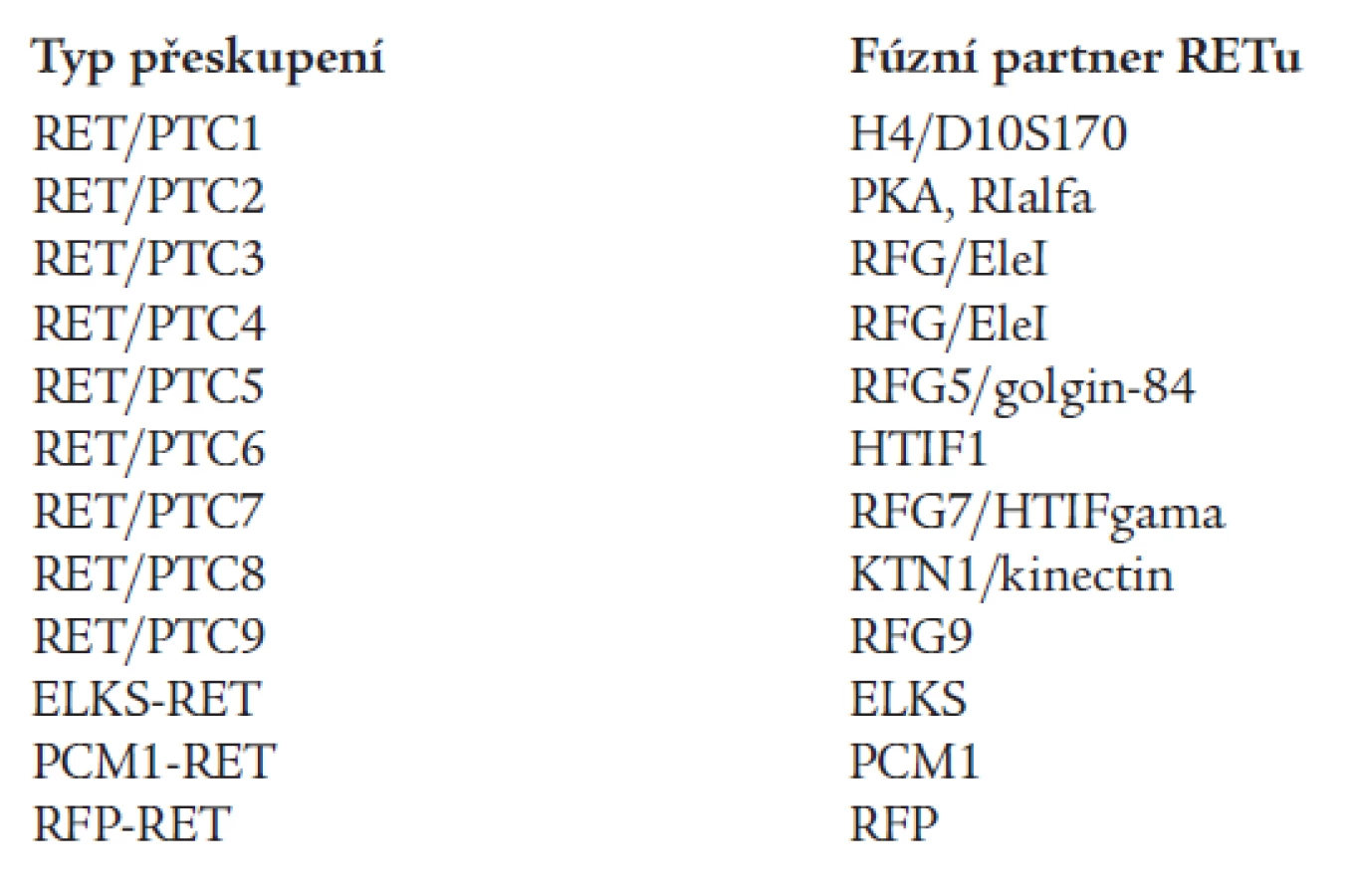
3. Hirschsprungova choroba
Hirschsprungova choroba (HSCR) je vrozené onemocnění enterického nervového systému, při kterém chybí gangliové buňky v myenterickém a submukózním plexu. Nad aganglionárním střevním úsekem, který je v 75-80 % případů HCSR lokalizován v rektu a colon sigmoideum, vzniká typické megakolon. Aganglionární úsek může ve 3-12 % postihovat celé tlusté střevo a přecházet na ileum. Vzácně se může vyskytnout i excesivní střevní aganglionóza postihující nejen tlusté, ale i téměř celé tenké střevo. HSCR se zřídka vyskytuje jako jeden ze symptomů chromozomálních abnormalit či některých monogenních syndromů, častěji pak samostatně bez jiných anomálií. Fenotyp HSCR je tedy poměrně rozmanitý. Familiární i sporadické formy jsou asociovány s mutacemi nejméně 6 genů (tab. 2), jejichž penetrace a exprese je velmi variabilní (Hirschsprung Disease Overview: www.genetests.org) [44]. Nejčastěji jsou nalézány zárodečné inaktivující mutace RET proto-onkogenu, jejichž dědičnost je autozomálně dominantní [2]. Fenotypická variabilita zde může být ovlivněna i některými polymorfizmy či haplotypy RET proto-onkogenu [7]. Vztah genotypu a fenotypu je stále předmětem výzkumu. Kromě řady inaktivujících mutací RET proto-onkogenu, které jsou typické pouze pro HSCR, byly nalezeny i mutace aktivující (v 10. exonu - v kodonech 611, 618, 620 a 634), které jsou typické pro MTC - v těchto případech se HSCR sdružuje s nádorovým postižením štítné žlázy, případně s feochromocytomem [16,45]. Jedním z možných vysvětlení koexistence MEN 2 a HSCR fenotypů může být nastartování apoptózy v buňkách embryonálního enterického ganglia jako odpovědi na nepatřičný mitogenní signál způsobený mutovanou alelou RET proto-onkogenu. Jiným vysvětlením může být hypotéza, že proteinové izoformy obsahující tyto specifické mutace pronikají na buněčný povrch s velmi malou účinností - HSCR vzniká v důsledku nedostatku RET proteinu ve vyvíjející se střevní inervaci, zatímco zároveň dochází k aktivaci signalizační kaskády RETu ve štítné žláze a v nadledvině, která postačuje ke vzniku hyperplazie a k tvorbě nádoru. Při léčbě pacientů s HSCR je důležité si uvědomit, že nejméně u 5 % pacientů s mutacemi RET proto-onkogenu je riziko vzniku MTC, proto je doporučován screening mutací RET proto-onkogenu, zejména 10. exonu, i u pacientů s HSCR.

Naše zkušenosti
Na našem pracovišti provádíme rutinně u pacientů s MTC a nověji i u pacientů s HSCR molekulárně genetický screening 6 rizikových exonů (exon 10, 11, 13, 14, 15 a 16) RET proto-onkogenu pomocí sekvenace. Při pozitivním záchytu mutace vyšetřujeme cíleně i příbuzné v riziku. Tab. 3a a 3b udávají typy zachycených mutací a záchyt mutací u jednotlivých fenotypů. Na základě molekulárně genetického stanovení byla retrospektivně u řady pacientů potvrzena diagnóza a dle typu mutace predikován další průběh onemocnění, u mnoha prediktivních jedinců byla na základě detekované mutace doporučena profylaktická totální tyreoidektomie, řada příbuzných mohla být na základě negativního výsledku genetického screeningu vyřazena z pravidelného biochemického testování [29,30]. Díky výzkumným projektům můžeme pátrat i po výskytu minoritních mutací a polymorfizmů ve zbývajících 15 exonech a v přilehlých intronických oblastech. U jedné rodiny s FMTC jsme nalezli dosud nepopsanou mutaci v 5. exonu, která je zřejmě zodpovědná za onemocnění [18]. Objevili jsme také zajímavé kazuistiky, podle kterých jsou pacienti nositeli dvojitých či trojitých mutací, kdy se zdá, že výsledný fenotyp determinuje vždy ta nejagresivnější z mutací [19,20]. Snažíme se prosadit optimální protokol genetického testování RET proto-onkogenu u pacientů s MTC (obr. 3). Abychom mohli jednoznačně vyloučit familiaritu klinicky sporadického MTC (prvotní záchyt v rodině), je třeba mít k dispozici i odoperovanou nádorovou tkáň.


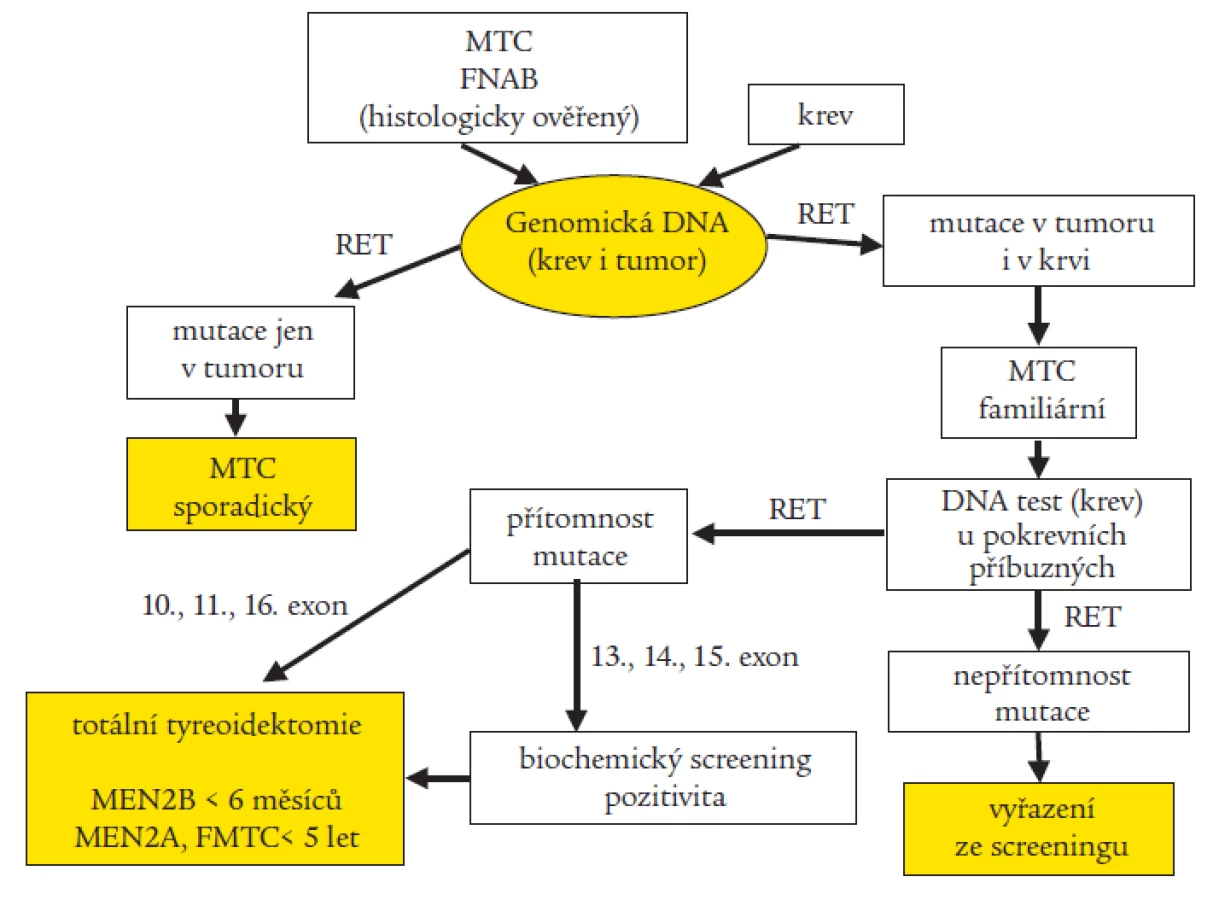
U 5 rodin s HSCR (celkem testováno zatím 57 rodin) jsme zachytili mutace v 10. exonu spojené s MTC, nebo atypické mutace v rizikovém 13. exonu (tab. 4) [17]. Tři z nich dosud nebyly popsány, bez provedení funkční studie lze jen těžko spekulovat, jaký dopad budou mít na fenotyp. Problémem je i neúplná penetrace v rodině. To dokládá, že ani molekulárně genetická diagnostika neposkytne vždy jednoznačné doporučení. Největší přínos genetického testování vidíme tedy v možnosti včasné predikce, kdy lze dosud klinicky asymptomatickým nositelům mutace nabídnout profylaktickou totální tyreoidektomii (TTE), případně s disekcí krčních lymfatických uzlin, kam nádor často již ve velmi raných fázích karcinogeneze metastazuje [12]. Otázkou je, jak tento chirurgický výkon načasovat. O časnosti a radikálnosti zákroku rozhoduje typ nalezené zárodečné mutace. Doporučení se s přibývajícím počtem diagnostikovaných rodin, kdy se pečlivě vyhodnocuje korelace mezi genotypem a fenotypem, mění a obecně zpřísňují [32,35,37,54,58]. Dle konsenzu [8] byly mutace podle agresivity rozděleny do 3 skupin, od toho se odvíjí i doporučení, v jakém věku provést TTE, abychom skutečně předešli rozvoji onemocnění:

Skupina 3 - nejagresivnější mutace v kodonech 883, 918, 922, bývají příčinou MEN 2B (až v 50 % případů vznikají de novo) - TTE provést během prvních 6 měsíců života, nejlépe do 1 měsíce po narození.
Skupina 2 - mutace v kodonech 609, 611, 618, 620, 630, 634, bývají příčinou MEN 2A, případně FMTC, první čtyři se vyskytují u pacientů s HSCR spojeným s MTC - TTE provést do 5 let věku.
Skupina 1 - nejméně agresivní mutace v kodonech 768, 790, 791, 804, 891 - TTE provést mezi 5. a 10. rokem života.
Pacienti by i po provedené TTE měli být nadále sledováni pro možnost rozvoje reziduálního či metastatického MTC, feochromocytomu a sledována by měla být i případná hypoparatyreóza u odoperovaných pacientů. Pacienti odoperovaní pro mutaci v kodonu 634 by měli být sledováni pro možnost případného rozvoje adenomu či hyperplazie příštítných tělísek. V některých zahraničních laboratořích se provádí i prenatální diagnostika, u nás jsme zatím toto vyšetření prenatálně neprováděli.
Studujeme i genetické alterace u papilárních karcinomů štítné žlázy. Zavedena byla detekce RET/PTC forem ze zamražených nádorových tkání i z biopsií, problematičtější je zatím stanovení těchto forem u nádorových buněk izolovaných ze starších parafinových bločků, kde je genetický materiál značně narušen. Přesto věříme, že se nám podaří zhodnotit vliv černobylské havárie na patogenezi PTC v České republice.
Moderní strategie léčby karcinomů štítné žlázy založená na inhibici RET kinázové aktivity.
Tyrozinkinázové geny jsou velmi slibnými terapeutickými cíli [13]. Pro blokaci tyrozinkinázové funkce RETu je k dispozici několik strategií, zahrnujících RNA interferenci k zastavení exprese RETu, genovou terapii s dominantně negativní mutantou nebo využití malých molekul, které slouží jako inhibitory tyrozinkinázy [24]. Genová terapie je pro léčbu nádorů štítné žlázy zvláště atraktivní, protože je zde možnost selektivního zacílení terapeutických genů do nádorových buněk aplikací tkáňově specifických promotorů, jako je např. kalcitoninový promotor u MTC, čímž dochází ke snížení mimonádorové toxicity. Možnosti terapie karcinomů štítné žlázy, které jsou testovány zatím zejména in vitro na tkáňových kulturách nebo na zvířecích modelech, jsou následující [3,4,52]:
- Korektivní genová terapie: cílem je obnovení normální funkce mutovaného genu nebo alespoň zamezení jeho účinku, což představuje inhibici onkogenní RET signalizace expresí dominantně-negativní RET mutanty. Gen nesoucí její sekvenci je zabudován do adenovirového vektoru a exprimován pod kontrolou promotoru pro kalcitonin nebo tyreoglobulin. Dominantně-negativní mutanty dimerizují s onkogenním RET proteinem v endoplazmatickém retikulu, a tak brání expresi jak dominantně negativního, tak onkogenního RET proteinu na buněčném povrchu. Výsledkem je silná inhibice přežívání buněk způsobená indukcí apoptózy.
- Cytoreduktivní genová terapie: principem je vpravení exogenního „sebevražedného“ genu, který způsobuje zastavení syntézy DNA a buněčnou smrt nebo umožňuje aplikaci cytotoxické látky do nádorové tkáně. V praxi došlo u laboratorních zvířat k destrukci nebo stabilizaci jen malých tumorů, zatím bez terapeutického efektu na větší nádory.
- Imunomodulační genová terapie: smyslem této metody je indukce genové exprese, která zvyšuje imunitní odpověď proti nádorové tkáni. Mnoho tumorů exprimuje nádorově asociované antigeny, které mohou být rozeznatelné imunitním systémem. Tyto antigeny jsou uvolňovány z nádorových buněk fyziologicky nebo po cytotoxické léčbě. Antigeny jsou pak vychytávány fagocytózou pomocí antigen prezentujících buněk. Nádory často vyvolávají slabou T-buněčnou odpověď, a tak unikají imunitnímu systému. Lokální expresí určitých cytokinů je tak možno vyvolat reakci imunitního systému proti nádoru. Cytokiny s protinádorovou aktivitou zahrnují interferon γ, tumor nekrotický faktor α (TNF-α), interleukiny 2 a 12 (IL-2, IL-12). Výsledkem pokusů prováděných na zvířatech byla regrese malých nádorů a stabilizace velkých nádorů, což ukazuje na vývoj dlouhotrvající protinádorové imunity.
- Monoklonální protilátky: příprava několika lidských monoklonálních protilátek proti RETu byla již publikována, ale zatím nebyly použity v léčbě [47].
- Inhibitory tyrozinkinázy: další skupinou nadějných protinádorových léčiv jsou malé molekuly tyrozinkinázových inhibitorů, které kompetují s adenozintrifosfátem (ATP), a tím brání autofosforylaci, blokují kinázovou aktivitu a signální transdukci [10,24]. Inhibice RETu může být efektivní strategií pro léčbu MTC, ovšem selektivní tyrozinkinázový inhibitor využitelný pro léčbu MTC nebyl zatím nalezen. Nejslibnějším kandidátem z testovaných potenciálních léčiv se zatím jeví anilinquinazolin ZD6474, protože efektivně blokuje fosforylaci a signalizaci RETu in vivo, inhibuje nádorový růst a působí zároveň jako antiangiogenní agens, přitom má nízkou toxicitu pro okolní tkáně [11].
Vzhledem k tomu, že se počet objevených malých organických sloučenin, které blokují RET tyrozinovou fosforylaci, stále zvyšuje, doufáme, že se už brzy použijí pro léčbu RET-pozitivních nádorů a možná se zapojí do preventivní strategie léčby pacientů se zárodečnými mutacemi RET proto-onkogenu.
Závěr
Nádory štítné žlázy představují nejpočetnější skupinu onkologických onemocnění v endokrinologii. Jejich incidence, zejména incidence papilárních karcinomů, bohužel rok od roku stoupá. V patogenezi medulárního a papilárního karcinomu, ale i zdánlivě klinicky vzdálené Hirschsprungovy choroby hrají roli genetické aberace RET proto-onkogenu. Identifikace jednotlivých genetických změn v RET proto-onkogenu může u pacientů pomoci odhadnout prognózu onemocnění, popř. zpřesnit diagnózu, ale hlavně indikuje včasný terapeutický zásah u rizikových osob ještě v presymptomatickém stadiu onemocnění.
Tímto článkem jsme chtěli upozornit naši lékařskou veřejnost a pacienty na dostupnost tohoto stanovení v České republice.
Podpořeno granty IGA MZ ČR NR/7806-3 a GAČR 301/06/P425.
RNDr. Běla Bendlová, CSc.
www.endo.cz
e-mail: bbendlova@endo.cz
Doručeno do redakce: 20. 6. 2006
Sources
1. Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002; 3 : 383-394.
2. Attie T, Pelet A, Edery P et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995; 4 : 1381-1386.
3. Barzon L, Bonaguro R, Palu G et al. New perspectives for gene therapy in endocrinology. Eur J Endocrinol 2000; 143 : 447-466.
4. Barzon L, Boscaro M, Palu G. Endocrine aspects of cancer gene therapy. Endocr Rev 2004; 25 : 1-44.
5. Bender BU, Gutsche M, Glasker S et al. Differential genetic alterations in von Hippel-Lindau syndrome-associated and sporadic pheochromocytomas. J Clin Endocrinol Metab 2000; 85 : 4568-4574.
6. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001; 411 : 355-365.
7. Borrego S, Ruiz A, Saez ME et al. RET genotypes comprising specific haplotypes of polymorphic variants predispose to isolated Hirschsprung disease. J Med Genet 2000; 37 : 572-578.
8. Brandi ML, Gagel RF, Angeli A et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86 : 5658-5671.
9. Bugalho MJ, Domingues R, Sobrinho L. Molecular diagnosis of multiple endocrine neoplasia Type 2. Expert Rev Mol Diagn 2003; 3 : 769-779.
10. Carlomagno F, Santoro M. Identification of RET kinase inhibitors as potential new treatment for sporadic and inherited thyroid cancer. J Chemother 2004; 16(Suppl 4): 49-51.
11. Ciardiello F, Caputo R, Damiano V et al. Antitumor effects of ZD6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin Cancer Res 2003; 9 : 1546-1556.
12. Cohen MS, Moley JF. Surgical treatment of medullary thyroid carcinoma. J Intern Med 2003; 253 : 616-626.
13. Cote GJ, Gagel RF. Lessons learned from the management of a rare genetic cancer. N Engl J Med 2003; 349 : 1566-1568.
14. Coulpier M, Anders J, Ibanez CF. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J Biol Chem 2002; 277 : 1991-1999.
15. de Graaff E, Srinivas S, Kilkenny C et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 2001; 15 : 2433-2444.
16. Decker RA, Peacock ML, Watson P. Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet 1998; 7 : 129-134.
17. Dvorakova S, Dvorakova K, Malikova M et al. A novel Czech kindred with familial medullary thyroid carcinoma and Hirschsprung's disease. J Pediatr Surg 2005; 40(Suppl e): e1-e6.
18. Dvorakova S, Vaclavikova E, Duskova J et al. Exon 5 of the RET proto-oncogene: a newly detected risk exon for familial medullary thyroid carcinoma, a novel germ-line mutation Gly321Arg. J Endocrinol Invest 2005; 28 : 905-909.
19. Dvorakova S, Vaclavikova E, Ryska A et al. Double Germline Mutations in the RET Proto-oncogene in MEN 2A and MEN 2B Kindreds. Exp Clin Endocrinol Diabetes 2006; 114 : 192-196.
20. Dvorakova S, Vaclavikova E, Sykorova V et al. New multiple somatic mutations in the RET proto-oncogene associated with a sporadic medullary thyroid carcinoma. Thyroid 2006; 16 : 311-316.
21. Eng C, Clayton D, Schuffenecker I et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996; 276 : 1575-1579.
22. Eng C, Mulligan LM. Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumours, and Hirschsprung disease. Hum Mutat 1997; 9 : 97-109.
23. Futreal PA, Coin L, Marshall M et al. A census of human cancer genes. Nat Rev Cancer 2004; 4 : 177-183.
24. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 2004; 4 : 361-370.
25. Huang SC, Torres-Cruz J, Pack SD et al. Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J Clin Endocrinol Metab 2003; 88 : 459-463.
26. Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett 2004; 204 : 197-211.
27. Jain S, Watson MA, DeBenedetti MK et al. Expression profiles provide insights into early malignant potential and skeletal abnormalities in multiple endocrine neoplasia type 2B syndrome tumors. Cancer Res 2004; 64 : 3907-3913.
28. Jhiang SM. The RET proto-oncogene in human cancers. Oncogene 2000; 19 : 5590-5597.
29. Jindrichova S, Kodet R, Krskova L et al. The newly detected mutations in the RET proto-oncogene in exon 16 as a cause of sporadic medullary thyroid carcinoma. J Mol Med 2003; 81 : 819-823.
30. Jindrichova S, Vcelak J, Vlcek P et al. Screening of six risk exons of the RET proto-oncogene in families with medullary thyroid carcinoma in the Czech Republic. J Endocrinol 2004; 183 : 257-265.
31. Jindrichova S, Vlcek P, Bendlova B. Genetic causes of the thyroid carcinomas. Cas Lek Cesk 2004; 143 : 664-668.
32. Kahraman T, de Groot JW, Rouwe C et al. Acceptable age for prophylactic surgery in children with multiple endocrine neoplasia type 2a. Eur J Surg Oncol 2003; 29 : 331-335.
33. Kawamoto Y, Takeda K, Okuno Y et al. Identification of RET autophosphorylation sites by mass spectrometry. J Biol Chem 2004; 279 : 14213-14224.
34. Kwok JB, Gardner E, Warner JP et al. Structural analysis of the human ret proto-oncogene using exon trapping. Oncogene 1993; 8 : 2575-2582.
35. Lips CJ, Hoppener JW, Van Nesselrooij BP et al. Counselling in multiple endocrine neoplasia syndromes: from individual experience to general guidelines. J Intern Med 2005; 257 : 69-77.
36. Liu X, Vega QC, Decker RA et al. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J Biol Chem 1996; 271 : 5309-5312.
37. Machens A, Ukkat J, Brauckhoff M et al. Advances in the management of hereditary medullary thyroid cancer. J Intern Med 2005; 257 : 50-59.
38. Manie S, Santoro M, Fusco A et al. The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet 2001; 17 : 580-589.
39. Manning G, Whyte DB, Martinez R et al. The protein kinase complement of the human genome. Science 2002; 298 : 1912-1934.
40. Mizuno T, Iwamoto KS, Kyoizumi S et al. Preferential induction of RET/PTC1 rearrangement by X-ray irradiation. Oncogene 2000; 19 : 438-443.
41. Mulligan LM, Eng C, Healey CS et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 1994; 6 : 70-74.
42. Nikiforova MN, Stringer JR, Blough R et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 2000; 290 : 138-141.
43. Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 1993; 119 : 1005-1017.
44. Parisi MA, Kapur RP. Genetics of Hirschsprung disease. Curr Opin Pediatr 2000; 12 : 610-617.
45. Romeo G, Ceccherini I, Celli J et al. Association of multiple endocrine neoplasia type 2 and Hirschsprung disease. J Intern Med 1998; 243 : 515-520.
46. Salvatore D, Barone MV, Salvatore G et al. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in ret and ret-derived oncoproteins. J Clin Endocrinol Metab 2000; 85 : 3898-3907.
47. Salvatore G, Nagata S, Billaud M et al. Generation and characterization of novel monoclonal antibodies to the Ret receptor tyrosine kinase. Biochem Biophys Res Commun 2002; 294 : 813-817.
48. Santoro M, Carlomagno F, Romano A et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 1995; 267 : 381-383.
49. Santoro M, Melillo RM, Carlomagno F et al. Minireview: RET: normal and abnormal functions. Endocrinology 2004; 145 : 5448-5451.
50. Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Sci STKE 2003; 2003: RE12.
51. Schuchardt A, D'Agati V, Larsson-Blomberg L et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994; 367 : 380-383.
52. Shelling A, Schweder, PM, During, MJ. Principles of gene therapy. In: Baxter JD (ed). Genetics in endocrinology. Philadelphia: Lippincott, Williams and Wilkins 2002 : 491-520.
53. Sherman SI. Thyroid carcinoma. Lancet 2003; 361 : 501-511.
54. Szinnai G, Meier C, Komminoth P et al. Review of multiple endocrine neoplasia type 2A in children: therapeutic results of early thyroidectomy and prognostic value of codon analysis. Pediatrics 2003; 111(Suppl E): E132-E139.
55. Takahashi M, Iwashita T, Santoro M et al. Co-segregation of MEN2 and Hirschsprung's disease: the same mutation of RET with both gain and loss-of-function? Hum Mutat 1999; 13 : 331-336.
56. Vitagliano D, Carlomagno F, Motti ML et al. Regulation of p27Kip1 protein levels contributes to mitogenic effects of the RET/PTC kinase in thyroid carcinoma cells. Cancer Res 2004; 64 : 3823-3829.
57. Williams D. Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat Rev Cancer 2002; 2 : 543-549.
58. Yip L, Cote GJ, Shapiro SE et al. Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 2003; 138 : 409-416.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2006 Issue 10
Most read in this issue
- Hashimotova encefalopatie
- Radionuklidové zobrazovací metody používané v endokrinologii
- Kongenitální adrenální hyperplazie na podkladě deficitu 3-β-hydroxysteroidní dehydrogenázy
- Autoimunitní tyreoiditida: vybrané etiopatogenetické mechanizmy