
Histiocytóza z Langerhansových buněk u osob dospělého věku - zkušenosti jednoho pracoviště a přehled léčebných možností
Histiocytosis from Langerhans cell in adults – experience of one department and review of treatment alternatives
Thirteen patients with Langerhans cell histiocytosis (LCH) have been treated in hospital Brno Masaryk University during the last 15 years. In 4 cases of this total amount, the diagnosis was made in childhood and these young adults were referred to our department from Pediatric cancer center. In 9 cases, the diagnosis has been made in people elder than 18 years. The disease recidived in 3 patients with LCH diagnosed in childhood, in one case with neurodegenerative impairment of brain. In 4 patients from the total of 9 with LCH diagnosed in age over 18 years, the disease had aggressive course with several recidives. In one case it was pulmonal and multifocal osseal manifestation, in two cases multifocal osseal disease. Isolated pulmonal form of LCH was diagnosed only in one patient. By the first patient, high dose melphalan and etoposide with peripheral blood stem cell transplantation was performed after failure of vincristin and prednison therapy and failure of etoposide therapy. The first remission after this high dose therapy lasted only 2.5 years. For relapse second cycle of the same high dose therapy was administered, but the next remission was much shorter. By the two patients with multifocal recidives in bones 2-chlordeoxyadenosine was administered as initial therapy. These patients are followed up for more then 24 months and they are without relapse of this disease. The 2-chlordeoxyadenosine is very efficient in multifocal bone form of LCH and has potential to reach long remission. Therefore in a case of aggressive multifocal disease we would prefer 2-chlordeoxyadenosine therapy as therapy of the first choice.
Key words:
Langerhans cell histiocytosis - 2-chlordeoxyadenosine
Authors:
Z. Adam 1; J. Vaníček 2; P. Šlampa 3
![]() ; P. Čoupek 3; I. Mareschova 1; J. Neubauer 4; L. Babičková 5; Z. Adamová 6; M. Tomíška 1; M. Navrátil 1
; P. Čoupek 3; I. Mareschova 1; J. Neubauer 4; L. Babičková 5; Z. Adamová 6; M. Tomíška 1; M. Navrátil 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
1; Klinika zobrazovacích metod Lékařské fakulty MU a FN u sv. Anny, Brno, přednosta doc. MUDr. Petr Krupa, CSc.
2; Radioterapeutické oddělení Masarykova onkologického ústavu, Brno, přednosta doc. MUDr. Pavel Šlapa, CSc.
3; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil Válek, CSc.
4; Klinika tuberkulózy a respiračních nemocní Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednostka doc. MUDr. Jana Skřičková, CSc.
5; Zdavotní středisko pro děti a dorost, Obilní trh, Brno, vedoucí MUDr. Zdenka Adamová
6
Published in:
Vnitř Lék 2006; 52(4): 355-370
Category:
Original Contributions
Overview
V průběhu 15 let bylo na Interní hematoonkologické klinice LF MU a FN Brno, pracoviště Bohunice (IHOK) léčeno a sledováno 12 pacientů s prokázanou histiocytózou z Langerhansových buněk (LCH) a u jednoho pacienta byla tato nemoc diagnostikována na Klinice tuberkulózy a respiračních nemocí LF MU a FN Brno, pracoviště Bohunice (KTRN). Celkem 4 pacienti k nám byli předáni s LCH diagnostikovanou a léčenou v raném dětském věku a u 9 pacientů byla choroba diagnostikována až ve věku dospělém. U 3 ze 4 pacientů s diagnózou stanovenou v dětství došlo k recidivě nemoci, v jednom případě z nich k mnohočetným recidivám s následným neurodegenerativním poškozením CNS. Z 9 pacientů, u nichž byla nemoc diagnostikována v dospělosti, měla u 4 velmi agresivní průběh. U jednoho pacienta se kombinovalo poškození plic a skeletu, u 2 šlo o recidivující mnohočetné postižení skeletu a pouze u jednoho pacienta byla diagnostikována izolovaná plicní forma LCH. V prvním případě byla po selhání klasické léčby (vinca-alkaloidy a prednison a posléze etoposid s lokální radioterapií) použita opakovaná vysokodávkovaná chemoterapie (etoposid a melfalan) s autologní transplantací krvetvorných buněk. Po první vysokodávkované chemoterapii trvala remise 2,5 roku, po opakované vysokodávkované chemoterapii byla remise podstatně kratší. U dalších 2 pacientů s mnohočetným kostním postižením (v jednom případě s expanzí do CNS) byl podán v rámci iniciální léčby 2-chlordeoxyadenozin. Oba pacienti, léčení 2-chlordeoxyadenozinem, jsou sledováni déle než 2 roky a jsou v kompletní remisi. Naše zkušenosti potvrzují, že 2-chlordeoxyadenozin je velmi účinným lékem pro dospělé pacienty s agresivním multiložiskovým postižením. V rámci diskuse je podán přehled léčebných možností a jsou diskutovány naše zkušenosti s nimi.
Klíčová slova:
Langerhansova histiocytóza - 2-chlordeoxyadenozin
Úvod
Histiocytóza z Langerhansových buněk (LCH) je vzácné onemocnění, jehož incidence se udává 0,5/100 000 obyvatel. Jiné prameny uvádějí incidenci v rozmezí 0,1-1,0/100 000 dětí do 15 let [42,67]. Většina případů je diagnostikována v dětském věku, ve věku nad 18 let je LCH velmi vzácným onemocněním.
Pro nízkou incidenci není ani velký počet publikací o této nemoci. Neexistují žádné prospektivní randomizované studie u dospělých osob, které by vyhodnocovaly jednotlivé léčebné postupy. U dětí je tomu o něco lépe, ale ani zde není počet randomizovaných klinických studií vysoký. Informace o této nemoci v dospělém věku lze získat dominantně z popisů případů či nerandomizovaných studií, či zobecněním informací, získaných v pediatrických studiích, i na dospělé. Z tohoto důvodu jsme se rozhodli popsat zkušenosti Interní hematoonkologické kliniky LF MU a FN Brno, pracoviště Bohunice, s léčbou této nemoci a doufáme, že střípky informací zde předložené mohou pomoci jiným lékařům, když se s touto výjimečnou diagnózou setkají. Úvodem několik slov ke klasifikaci.
Klasifikace histiocytárních chorob
Termín histiocytóza X byl poprvé použitý Lichtensteinem v roce 1953. Vzhledem k nejasné etiologii a rozpakům, zdali toto onemocnění řadit k nádorovým či infekčním onemocněním, nebo k lipidovým tezaurismózám, byla tato klinická jednotka původně nazvána histiocytóza X. V posledních letech se vedla dlouho diskuse, zda jde o atypický reaktivní granulomatózní proces na neznámé vyvolávající agens, nebo zde jde o klonální proliferaci maligního onemocnění. Teorie atypického zánětlivého onemocnění byla podkladem klinického testování imusupresiv, která se v této indikaci neosvědčila. Dnes již byl prokázán klonální původ Langerhansových buněk, takže v současnosti se tato choroba spíše považuje za maligní nemoc než za reaktivní zánětlivé onemocnění, a tak je i uvedena v platné WHO klasifikaci maligních chorob. Reaktivní původ se stále zvažuje u hemofagocytující lymfohistiocytózy [99].
Proto byl původní název, histiocytóza X, přeměněn na termín histiocytóza z Langerhansových buněk (Langerhans cell histiocytosis - LCH). Tato nemoc je řazena WHO klasifikací krevních chorob do skupiny maligních histiocytárních chorob [34].
Klasifikaci histiocytárních chorob shrnujeme v tab. 1.

LCH se manifestuje širokou škálou příznaků, počínaje náhodným RTG-nálezem jednoložiskového lytického procesu v kosti (označovaného také termínem eozinofilní granulom), až po generalizované systémové postižení. To, jakou formou se choroba projeví, souvisí zatím z neznámého důvodu s věkem první manifestace.
LCH je tedy dominantně chorobou dětského věku a v dětském věku má také podstatně agresivnější průběh, obvykle multisystémový, zatímco LCH, která je poprvé diagnostikována v dospělosti, má průběh méně agresivní a v této věkové skupině dominuje poškození kostí, takže vzdáleně může LCH připomínat svým chováním mnohočetný myelom.
V dětském věku popisují pediatrické učebnice dvě formy nemoci, byť mezi nimi může být kontinuální přechod, v dospělosti dominuje eozinofilní granulom. Charakteristiku těchto jednotek uvádíme v přehledné tab. 2.

V dospělosti LCH postihuje dominantě kosti formou osteolytických ložisek (eozinofilní kostní granulom) a s menší frekvencí potom kůži, plíce nebo lymfatické uzliny. V životě však vždy existují výjimky, a tak i v dospělosti byly popsány případy s dominujícím kožním postižením, následujícím diabetes insipidus, pleurálním výpotkem a osteolýzou či izolovaným postižením jater [35,51]. Centrální diabetes insipidus může být tedy v každém věku příznakem LCH, proto považujeme za velmi účelné se v tom případě řídit diferenciálně diagnostickým schématem dle Prosche (2004) [72].
Z hlediska biologického chování můžeme tedy LCH rozdělovat na chorobu multisystémovou (převažující v dětství) a monosystémovou (převažující v dospělosti). V případě monosystémového postižení rozlišovat formu unifokální či multifokální.
Pokud se hovoří souhrnně o manifestaci jak u dětí, tak u dospělých, tak u 80 % bývají postiženy kosti, u 60 % kůže, u 33 % játra, slezina a uzliny, u 30 % kostní dřeň, u 25 % plíce, u 25 % orbita a u 20 % orodentální a otologická oblast. Diabetes insipidus bývá vyjádřen u méně než 15 % pacientů. K prognosticky nepříznivým faktorům patří první manifestace do dvou let, orgánová dysfunkce, multiorgánové postižení nebo kombinace orgánové dysfunkce s infiltrací jater. V literatuře zatím nebylo popsáno postižení srdce, svalů, ledvin, gonád a periferních nervů [39,82-84]; další informace www.histio.org.
Stanovení diagnózy
Pro nepatologa je vhodné vědět, že v době akutní choroby jsou v ložisku přítomny četné Langerhansovy buňky, exprimující proliferační markery Ki-5l a Ki-67 a eozinofily, a v té době je nemoc dobře rozpoznatelná.
Později v ložisku ubývá Langerhansových buněk, přibývá makrofágů a fibrocytů a nakonec obraz odpovídá postnekrotické fibróze. Je nutné si uvědomit, že čím později se přistupuje k excizi z ložiska a histologickému vyšetření, tím méně je v ložisku patologických Langerhansových buněk, a tím hůře je proces diagnostikovatelný. Někdy může být obtížné histologické rozlišení osteomyelitidy od starých ložisek LCH. Také diferenciace plicní formy LCH od jiných granulomatózních procesů může být ze stejného důvodu obtížná.
Mikroskopické změny v ložisku v průběhu času, k nimž nutno přihlížet při interpretaci histologického nálezu a morfologické znaky LCH, uvádí tab. 3.

Průběh nemoci u dospělých
Pokud se nemoc projeví až v dospělosti, dominuje kostní osteolytická manifestace, provázená často zduřením měkkých tkání nad lytickým ložiskem.
Méně časté je izolované plicní postižení či plicní postižení v kombinaci s osteolýzou. Plicní postižení lze detekovat nejlépe pomocí high resolution computer tomography - HRCT. Za počáteční formu lze považovat nodulární postižení o rozměrech 1-3 mm, někdy ale až 15 mm, což koresponduje s tvorbou intersticiálních granulomů. Pokročilejší forma je pak charakterizována ubýváním nodularit a narůstáním počtu cyst. Kombinace nodulárního a cystického plicního postižení je pro LCH typická. Počínající forma plicního postižení může být zcela asymptomatická, proto je na zvážení u pacienta s LCH provedení screeningového HRCT plic. Později mohou způsobovat cysty spontánní pneumotoraxy [55]. Frekvence plicního postižení je udávána kolem 24 % [10,13,19,77,82-84,92].
Třetím nejčastějším projevem v dospělosti je prvotní kožní manifestace, nebo souběžná s kostním postižením.
Podle analýzy provedené Baumgartnerem je postižení hypofýzy, lymfatických uzlin a jater v dospělém věku celkem vzácné. U dospělých pacientů má onemocnění velmi různorodý průběh. U některých osob vznikne pouze jedno ložisko a po léčbě se již neobjeví, u jiných má LCH recidivující charakter, vznikají stále další nová ložiska a choroba může být příčinou omezené hybnosti či může dokonce přivodit smrt [6].
Periferní krevní obraz je buď zcela normální nebo je přítomna mírná anémie chronického onemocnění. Monocytóza je vzácná a spíše reaktivní. Ačkoliv prekurzory Langerhansových buněk mají svůj původ v kostní dřeni, nebyla jejich přítomnost v periferním krevním obraze zachycena. U většiny pacientů je necílená biopsie kostní dřeně normální, a proto biopsie kostní dřeně nepatří do standardního vyšetření, pokud jsou normální hodnoty krevního obrazu [52].
Osteolytické defekty se objevují hlavně na kalvě a osovém skeletu, méně v periferních kostech. U některých pacientů lze detekovat ložiska různého stáří, jak nově vznikající, tak i hojící se ložiska, která mívají sklerotický lem. Ne všechna musí bolet. Zduření tkání přiléhajících ke kosti signalizuje, že choroba prorůstá do okolí. Ložiska v kostech lze detekovat jak radioizotopy, tak RTG či pomocí MR. RTG-snímky a scintigrafie skeletu technecium-difosfonátem se doplňují, jak vyplývá ze studie srovnávající scintigrafické a RTG-vyšetření. U 42 pacientů bylo nalezeno celkem 191 kostních ložisek. Z tohoto počtu 36 (19 %) nebylo patrných na scintigrafii a 55 (29 %) nebylo patrných na RTG-snímcích. RTG-vyšetření často neznázornilo změny na žebrech, pánvi a páteři, radionuklidové vyšetření nezobrazilo někdy defekty v lebce. Proto se doporučuje kombinovat obě vyšetření, nebo alespoň provést při prvním vyšetření jak scintigrafické, tak i RTG-vyšetření a další monitorování provádět hlavně scintigraficky [26,44]. Nejcitlivější metodou zobrazení patologického děje v kosti je však magnetická rezonance (MR). Informaci o tom, zda v ložisku zůstává aktivní nádorová tkáň, lze získat také pomocí pozitronové emisní tomografie (PET) [14].
Popis případů s LCH diagnostikovanou v dětském věku
Pacientka nar. 1978. Diagnóza LCH byla stanovena v 10 měsících (nemoc Lettererova a Siweho) s dominující kožní formou. Pacientka byla léčena v letech 1979-1981 prednisonem a vinkristinem. Poslední kontrola u nás byla v roce 2004 - bez recidivy. Je to jediná pacientka předaná k nám z pediatrického pracoviště, která zůstává stále bez recidivy nemoci.
U druhého pacienta, nar. 1975, se nemoc projevila také kožní formou v 1. roce života a od počátku nemoci byl zjištěn diabetes insipidus. K druhé atace nemoci zřejmě došlo v roce 1993, kdy náhle vznikly neurologické kmenové příznaky, centrální vestibulární syndrom, neúplný neocerebelární syndrom vpravo a naznačená pravostranná pyramidová symptomatologie. Tyto problémy spontánně téměř ustoupily a mladý muž je dále u nás sledován bez recidivy nemoci, od dětství však je nutná trvalá substituce adiuretinu. V době příznaků bylo sice provedeno zobrazení CNS metodou MR, ale žádná patologie není v popisu MR uvedena. V literatuře jsou popisovány podobné ataky LCH s uvedenými neurologickými příznaky [45].
Také třetí pacientka, nar. 1974, měla diagnózu LCH stanovenu ve dvou letech (1976), kdy se nemoc manifestovala kožními příznaky a diabetes insipidus. Po léčbě vinkristinem a prednisonem byla dlouhodobě bez známek recidivy, ale s nutností trvalé substituce adiuretinu. Na rozdíl od předchozích však v roce 1996 (po 20 letech) byla zjištěna indurace v okcipitální oblasti s defektem kosti, zřetelným na RTG-snímku. Pacientka byla ozářena ložiskovou dávkou 20 Gy. K další recidivě do roku 2005 nedošlo.
Čtvrtý pacient, nar.1976, s LCH diagnostikovanou v dětském věku, měl téměř chronický průběh této nemoci. Prvním příznakem nemoci byl diabetes insipidus (1981) zjištěný v 5. roce života. V roce 1985 bylo zjištěno zpomalení růstu. Diagnóza LCH byla stanovena až v roce 1988. V té době měl exoftalmus, diabetes insipidus, kostní defekt v oblasti levé orbity, čili klasický obraz Handovy-Schüllerovy-Christianovy nemoci.
V roce 1993 byla zjištěna infiltrace kůže perianální krajiny - později histologicky ověřena LCH této kožní oblasti. Při předání z pediatrického pracoviště do péče Interní hematoonkologické kliniky LF MU a FN Brno, pracoviště Bohunice, v roce 1996 došlo k recidivě v mandibule, což se projevilo bolestivým zduřením. Histologicky byla potvrzena další ataka LCH. Lokální recidiva v mandibule byla řešena radioterapií a na ni navázala léčba vinblastinem 10 mg i.v. v týdenních intervalech, celkem 8krát, ale od 4. aplikace byla redukována dávka na 5 mg pro neutropenii. Další progrese byla zjištěna v roce 1998, na scintigrafii skeletu i RTG-snímku bylo nové ložisko v os parietale a pouze na scintigrafii ložisko v pravém akromiu. Následovala chemoterapie etoposidem a dexametazonem a lokální ozáření kalvy cíleně na kostní ložiska.
V září roku 2001 došlo k výraznému zhoršení, které se ohlásilo zduřením nad levým okem. Zduření a infiltrace v této oblasti někdy ohlašuje současné postižení CNS. Zduření nad levým okem se zřetelným ložiskem na kalvě bylo léčeno lokální radioterapií a pulzy dexametazonu.
Po měsíci se objevila diplopie, ataxie a další neurologické problémy. O několik týdnů později prudce vzestoupily veškeré standardně vyšetřované jaterní enzymy, aniž byla prokázána infekční hepatitida. Pro neurologické příznaky jsme provedli MR-vyšetření CNS. Interpretace MR-nálezu nebyla jednoznačná, a protože jde o komplikaci, která je velmi, ale opravu velmi výjimečná a běžný rentgenolog se s ní nesetká, konzultovali jsme MR-obraz s prof. Gadnerem z AKH Vídeň. Naši lékaři z oddělení radiologické kliniky i prof. Gadner se shodli na závěru: neurodegenerativní změny, odpovídající pozdní komplikaci Langerhansovy histiocytózy.
Přesný popis MR zněl: v oblasti bazálních ganglií a dále v bílé a šedé hmotě mozkové byly nalezeny změny odpovídající neurodegenerativním změnám typickým pro LCH. Difuzní hyperintenzivní ložiska v oblasti mostu odpovídala spíše leukoencefalopatii než LCH. Infundibulum bylo zesílené. Vzhledem k nejasnosti, zda se jedná o nádorovou infiltraci, či degeneraci, bylo provedeno také PET-vyšetření v Praze, kde byla popsána zvýšená kumulace v oblasti mozkového kmene. Při kontrole PET po roce již v této oblasti nebyla zvýšená kumulace značené glukózy, byl zřetelný pouze lehce asymetricky vyšší metabolizmus glukózy bez jednoznačných ložiskových změn. Ovšem ani tento zprvu pozitivní, posléze negativní PET-nález neodpovídá na otázku, zda šlo o infiltraci Langerhansovými histiocyty, nebo zda šlo o zánětlivé a degenerativní změny, neboť v obou případech může být zvýšeně vychytávána 18fluorodeoxyglukóza.
Vzhledem k rychlému zhoršování stavu jsme usoudili na další, tentokráte výrazně agresivnější ataku této nemoci s postižením jater a CNS. V říjnu roku 2001 byla proto zahájena léčba 2-chlordeoxyadenozinem.
Po první aplikaci 2-chlordeoxyadenozinu rychle poklesly hodnoty jaterních enzymů, takže jsme předpokládali postižení jater touto chorobou i bez histologického ověření.
Po 6 cyklech chemoterapie byl v únoru roku 2002 PET-nález negativní, neurologické změny zůstávaly však beze změn, MR-obraz CNS se nezměnil. Vyjma normalizace hodnot jaterních enzymů měla tato léčba ještě jeden pozitivní dopad: kožní exantém (histologicky prokázaná kožní histiocytóza) v anální krajině dlouhodobě vymizel a při poslední kontrole v prosinci roku 2005 je pacient bez recidivy kožního postižení. To se předtím při lokální kortikoidní léčbě nepodařilo.
Neurologický stav nemocného se postupně zhoršuje, takže dále již připadá v úvahu pouze symptomatická léčba.
Poslední laboratorní hodnoty v lednu roku 2006: krevní obraz v normě, základní biochemie (urea, kreatinin, ionty, bilirubin, všechny jaterní enzymy, LD v normě, celková bílkovina 66 g/l, albumin 42 g/l, CRP 0, β2-mikroglobulin 1,31 mg/l). Žádná z uvedených laboratorních hodnot nekorelovala s aktivitou nemoci.
RTG skeletu: vyjma ložisek v oblasti kalvy nebyla na snímcích zřetelná osteolytická ložiska. Kostní hustota Z skóre, SD od -4 do -5 SD. Těžká osteoporóza souvisí zřejmě s insuficientní funkcí hypofýzy. Přetrvávající neurodegenerativní poškození CNS nelze hodnotit jako aktivitu nemoci, takže stav hodnotíme jako remisi nemoci trvající více než 4 roky od ukončení léčby 2-chlordeoxyadenozinem.
Popis případů s jednoložiskovou formou Langerhansovy histiocytózy a první manifestací v dospělém věku bez dalších recidiv
Muž, nar. 1973. Diagnóza byla stanovena ve 25 letech (1998), kdy byla zjištěna izolovaná osteolýza postihující obratle C4-C5. Následovala operační léčba s odstraněním měkkých nádorových hmot a fúzí obratlů C3-C6 kovovou dlahou, fixovanou 6 šrouby, s následnou radioterapií. Poslední kontrola pacienta byla v květnu roku 2005 v rozsahu scintigrafie skeletu, RTG-snímku a MR-páteře. Žádné vyšetření neprokázalo recidivu nemoci. Doufáme, že se v tomto případě jednalo o izolované jednoložiskové postižení.
Stejně tak se u slečny, nar. 1980, jednalo o jednoložiskové postižení. Diagnóza byla stanovena v 18 letech (1998), kdy bylo pro zvýšenou citlivost v oblasti temporální kosti provedeno RTG-vyšetření s nálezem osteolýzy. Histologie ložiska prokázala eozinofilní granulom jako jednu z forem LCH. Následovala lokální radioterapie (1998) a slečna je od té doby bez recidivy.
V jednom případě jsme se setkali s uzlinovou formou LCH vzniklou v dospělosti. U pacienta, nar. 1962, byla diagnóza stanovena v roce 2001 v jeho 39 letech, z izolované zvětšení uzliny na krku. Následovalo CT mediastinálních a břišních uzlin, RTG skeletu doplněný magnetickou rezonancí zaměřenou na páteř a dále na femory, kde byl RTG-nález nejasný. Žádné další kostní ložisko ani lymfadenopatie nebyla zjištěna. Vzhledem k tomu, že šlo o totálně exstirpovanou uzlinu s nálezem histiocytózy, nebylo ani ozářeno místo po exstirpaci. Poslední kontrola u nás byla v roce 2005, kdy byl pacient bez známek relapsu nemoci.
Popis případů Langerhansovy histiocytózy s první manifestací v dospělém věku a s recidivami nemoci
V případě pacienta, nar. 1981 se první příznak objevil v 18. roce života (1999): bolest a rezistence v oblasti 8. žebra. Žebro bylo totálně resekováno a histologicky stanovena diagnóza unifokální LCH. Další ložisko nebylo v té době patrné. Po 5 letech, v roce 2004, přišel s bolestmi v oblasti temene, kde si nahmatal rezistenci. Na RTG-snímku kalvy měl neostré mapovité projasnění o velikosti 3x1 cm, odpovídající kostní formě nemoci. Následovala radioterapie cílená na kostní ložisko. Poslední kontrola proběhla v únoru roku 2005, mladý muž byl bez známek recidivy.
S primárně kožním projevem LCH přišel mladý muž, nar. 1986. Tento muž dříve neprodělal žádné vážnější onemocnění. V roce 2004 (18 let) se objevilo nejasné kožní ložisko v oblasti metakarpofalangeálních kloubů. Morfa byla excidována a histolog morfu popsal jako dermatofibrom s příměsí Langerhansových buněk. Elektronová mikroskopie potvrdila Birbeckova granula, takže diagnóza z tohoto ložiska byla nakonec uzavřena jako kožní forma LCH. Následovalo vyšetření s cílem potvrdit či vyloučit generalizaci nemoci (zobrazení skeletu, CT-břicha). Místo původního kožního defektu bylo i s bezpečnostním lemem ozářeno dávkou 30 Gy. V roce 2005 se objevily bolesti bederní páteře. RTG-snímek neprokázal patologii, zato MR-vyšetření bederní páteře prokázalo difuzní změny kostní dřeně, které jsou jednoznačně abnormálním nálezem. Pro podezření na nepoznanou kostní formu histiocytózy provedli ortopedové cílenou biopsii kosti s odběrem materiálu na histologii. Histologie však byla nekonkluzivní, LCH nebyla potvrzena. PET byla před biopsií negativní. Asi nelze stoprocentně vyloučit starší ložisko LCH v páteři. Další sledování ukáže, zda opravdu šlo o jedno kožní ložisko, nebo zda časem vzniknou další nová kostní ložiska.
Velmi komplikovaný průběh měla LCH u pacienta, nar. 1964. Tento mladý muž neměl do svých 37 let závažnější onemocnění. V roce 1991 (ve 37 letech věku) bylo nalezeno osteolytické ložisko ve femoru - řešeno ortopedicky, histologicky ověřena LCH. Po operaci následovala radioterapie. Za dva roky, v roce 1993, byla RTG zjištěna další ložiska v oblasti pánve a obou femorů. Scintigrafie kostí popsala ještě ložiska v mandibule a pravé lopatce. Ložiska v pánvi a v L5 byla ověřena CT-zobrazením.
U této víceložiskové formy jsme se rozhodli pro podání 7 cyklů chemoterapie CHOP (adriamycin, vinkristin, cyklofosfamid a prednison). První remise trvala tři roky.
V březnu roku 1996 byla zjištěna nová ložiska v oblasti žeber a Th-páteře - řešeno zářením a podáváním vinblastinu 1krát týdně po dobu 6 týdnů.
V červenci roku 1997 byla opět nová ložiska na paži v hlavici humeru a pravděpodobně i další ložiska v páteři. Obraz na HRCT plic byl kompatibilní s plicní formou histiocytózy. Následovala chemoterapie (etoposid a prednison) a radioterapie na oblast humeru. Vzhledem k častým recidivám a neuspokojivému výsledku běžné chemoterapie byl v září roku 1997 proveden sběr kmenových krvetvorných buněk a následně podána první vysokodávkovaná chemoterapie (melfalan a etoposid) s podporou transplantace autologních krvetvorných buněk. Remise po této léčbě trvala jenom 2,5 roku, do června roku 2001, kdy byla scintigraficky zjištěna nová ložiska. Následovaly tedy opět 3 cykly chemoterapie (etoposid a dexametazon) a ozáření těchto ložisek. Po krátkém klidovém období se v červenci roku 2002 objevila ložiska na žebrech a páteři, zřetelná nejen na scintigrafii, ale i na RTG-snímku. Pro tento relaps byla opět provedena stimulace s opakovaným sběrem kmenových krvetvorných buněk z periferní krve a následně podána druhá vysokodávkovaná chemoterapie.
Po této druhé vysokodávkované chemoterapii měla remise jen krátké trvání, několik měsíců, a opět se objevovala další kostní ložiska, takže dále pokračovala jen paliativní perorální chemoterapie melfalanem. V únoru roku 2002 byl pacient s aktivním onemocněním na naší ambulanci naposledy, jeho další osud je neznámý, předpokládáme, že zemřel.
V roce 1993 (21 let) vznikla u pacienta, nar. 1972, první ataka LCH, která se projevila bolestí v pravé stehenní kosti a dále v obratli Th12. Osteolytická ložiska byla operována, čímž byla stanovena diagnóza a choroba byla léčena operačně alogenními štěpy. Pak byl pacient pouze sledován bez další léčby.
V roce 2003, po 10 letech, se nově objevila bolest žeber, páteře a kalvy. Na RTG-snímcích bylo osteolytické ložisko v kalvě, které bylo potvrzeno CT-zobrazením, průměr ložiska byl 21 mm. Na pravé straně hrudníku bylo velké osteolytické ložisko v 7. žebru a velmi suspektní nález byl také na 6., 5. a 4. žebru. Na páteři byla nápadná nerovná dolní krycí ploténka Th8 a byla zřetelná přestavba krčku pravého femoru. V těle L4 CT prokázalo 3 osteolytická ložiska, největší 15x14x9 mm, a další ložiska byla i v těle obratle L5. Zobrazení páteře pomocí MR potvrdilo ložiska také v obratlích Th11, L2, L4, L5. Vzhledem k 10letému klidovému intervalu bylo resekováno 7. žebro. Histologické vyšetření prokázalo recidivu LCH.
Protože zobrazovací metody (RTG-snímky, scintigrafie skeletu, CT a MR) prokázaly mnohočetné osteolytické postižení skeletu, přistoupili jsme k systémové chemoterapii. Pacient dostal 5 cyklů chemoterapie (2-chlordeoxyadenozin 5 mg/m2 i.v. 1. až 5. den). Léčba byla ukončena v listopadu roku 2003.
Poslední kontrola proběhla v prosinci roku 2005. Nález PET-vyšetření byl negativní a MR páteře neprokázalo žádnou progresi, jsou však stále zřetelná původní ložiska, remise trvá déle než 2 roky.
Stejně tak mladý muž, nar. 1978, neměl v předchozím životě žádné vážné onemocnění, až v letech 2002/2003 (ve věku 24 let) bylo nalezeno osteolytické postižení femoru vlevo. V lednu roku 2003 byla provedena v nemocnici Na Bulovce v Praze exkochleace a zpevnění hřebem. Histologie ložiska: eozinofilní granulom. Již asi od roku 2002 si povšiml rezistencí (bulek) na hlavě průměru 2-3 cm. Největší byla v okcipitální krajině a zvětšovala se. Způsobovala bolest hlavy a posléze i poruchy zraku. Z chirurgicky odstraněných hmot opět histologicky vyšel eozinofilní granulom (leden roku 2003). Rozsah kostního postižení: na snímku lebky osteolytické ložisko okcipitálně o průměru 2 cm. V levé lopatě kyčelní bylo ložisko 8x4 cm. Celotělová MIBI-scintigrafie: vyšší aktivita v oblasti pánve, další ložisko v oblasti levé orbity, pokračující do os temporale. Závěry scintigrafie skeletu: ložiska v kalvě, fronto-parietálně a okcipitálně, další ložisko v kosti spánkové vpravo, velké ložisko v lopatě kosti kyčelní vlevo. Dále ložiska v pravém femoru. Vyšší aktivita také v oblasti kolenního a talokrurálního kloubu vpravo. CT kalvy a mozku: defekty v okcipitální oblasti oboustranně. MR mozku: extraaxiální expanzivní proces oboustranně parasagitálně okcipitálně, šířící se defektem kalvy epidurálně až do oblasti zadní jámy lební, bez infiltrace dury. Suspektní další kostní ložiska frontoparietálně vpravo a parietálně vlevo.
Vzhledem k mnohočetným kostním ložiskům, která však u tohoto nemocného měla již i mimokostní šíření, byl proveden sběr periferních kmenových buněk po předchozí stimulační chemoterapii (etoposid a cyklofosfamid). Pak následovaly 4 cykly chemoterapie - 2-chlordeoxyadenozin 5 mg/m2 i.v. inf, 1. až 5. den v měsíčních intervalech. Léčba byla ukončena v srpnu roku 2003.
Po ukončení léčby při kontrolním vyšetření bylo nalezeno: MR: jen velmi drobné reziduum měkkotkáňové expanze. V rozsahu původního defektu kalvy okcipitálně nyní jen pruh středního signálu v rozsahu diploe, bez známek sycení či expanzivních projevů, mající charakter původního infiltrátu, nebo regresivních změn v místě původního infiltrátu, což nelze na MR odlišit. Kontrolní MIBI skeletu: je patrná difuzní kumulace aktivity v páteři, náznakem v pánvi. Nejsou však patrná původní ložiska v kalvě, zřetelná při prvním MIBI-vyšetření. PET po léčbě: bez ložisek aktivity nemoci, PET-vyšetření před léčbou bohužel nebylo provedeno.
Domníváme se, že i u tohoto pacienta je nemoc v remisi a že agresivní léčba 2-chlordeoxyadenozinem navodila dlouhodobou kompletní remisi. Pacient má v zásobě periferní kmenové buňky pro případ recidivy. Stav nemoci u tohoto pacienta před a po léčbě znázorňují MR a RTG obr. 1-7.








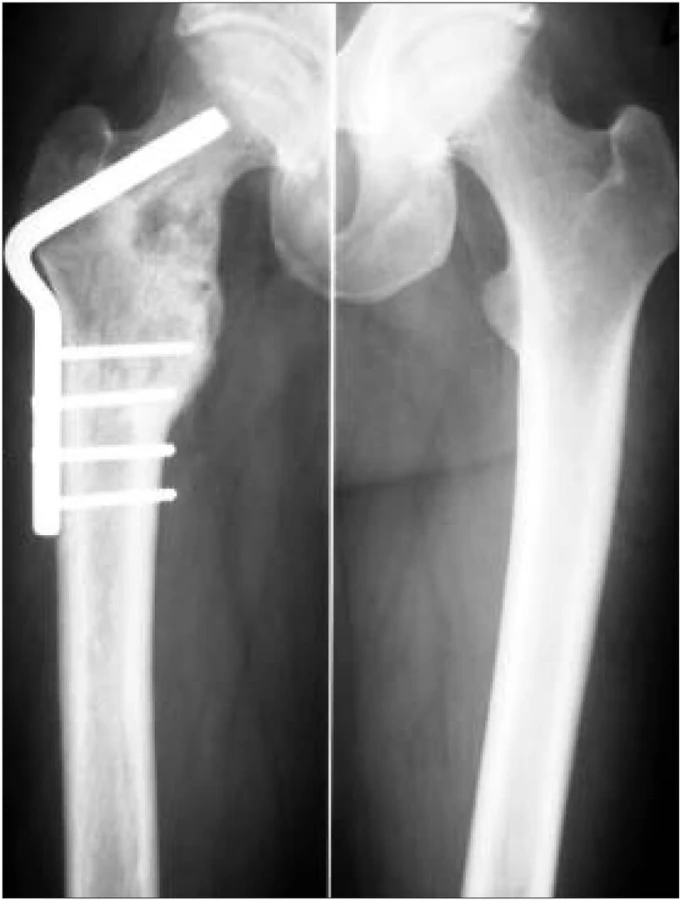
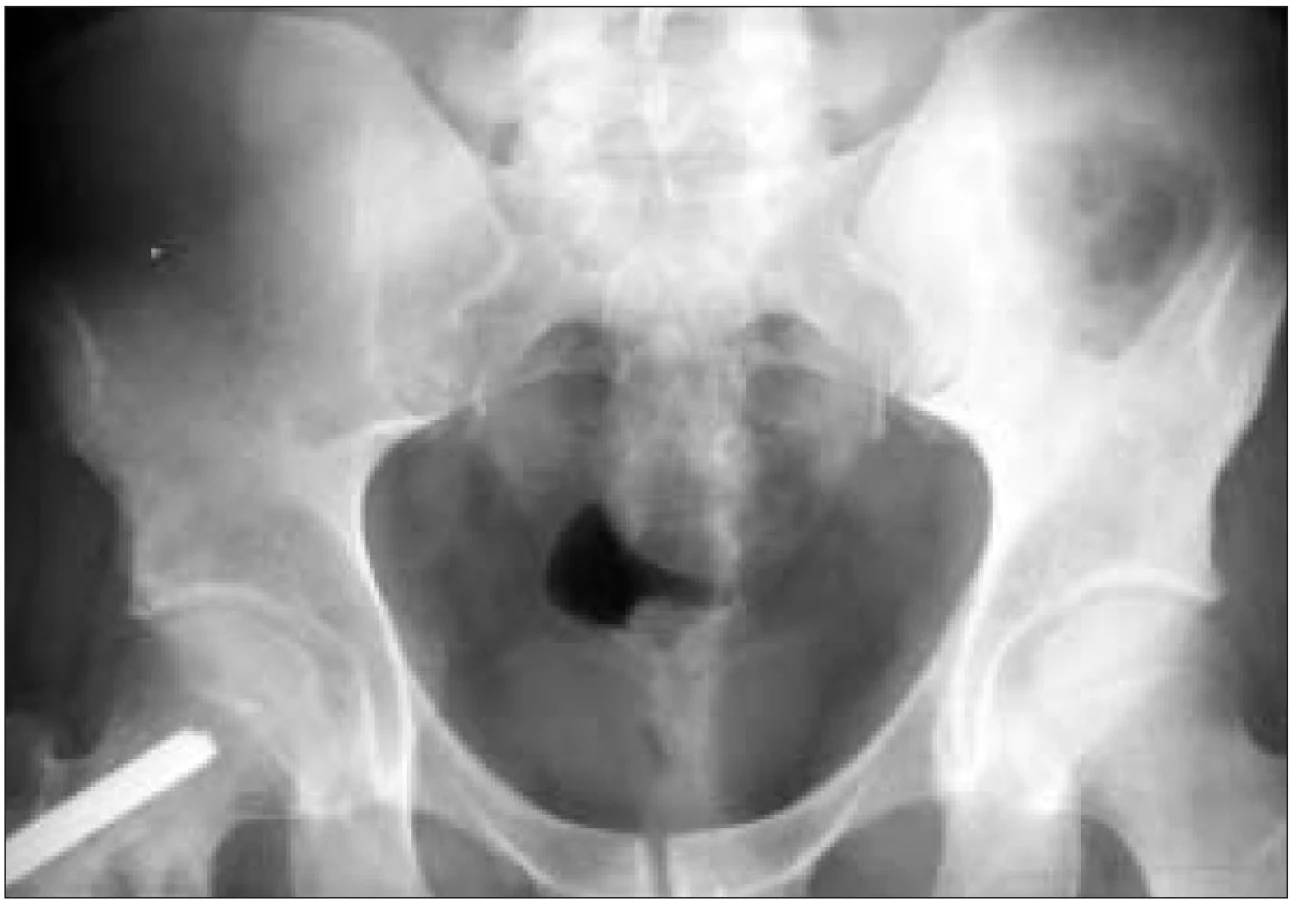
Poslední pozorování, které uvádíme, byl diagnostikován na Klinice tuberkulózy a respiračních nemocní LF MU a FN Brno, pracoviště Bohunice (KTRN).
Pacient, nar. 1944, kuřák, poprvé vyšetřen na ambulanci KTRN Brno-Bohunice v dubnu roku 1999 pro chřipkové příznaky, febrilie, dráždivý suchý kašel, bez váhového úbytku. Opakovaná kultivace sputa na Mycobacterium tuberculosis byla negativní. Na snímku hrudníku byla patrna drobněložisková zastínění difuzně ve všech plicních polích, ale bronchoskopie neodhalila patologii, stejně jako spirometrické vyšetření. Na high resolution CT (HRCT) plic byl však obraz chomáčkovitých opacit a nodularit. Vyšetření pacienta v té době žádné maligní onemocnění neprokázalo. V květnu roku 1999 byla provedena torakoskopie s resekcí dolního laloku levé plíce. Histologickým vyšetřením byla zjištěna ložiskově zvýšeně vzdušná plicní tkáň s obrazem dystelektatického emfyzému s překrvenými cévami a ložiskově ztluštělou pleurou. Z rozpaků bylo nakonec přistoupeno k léčbě antituberkulotiky jako terapeutický test, tato léčba byla ukončena v lednu roku 2000. Klinický stav ani kontrolní skiagram hrudníku při antituberkuloticích nebyl zlepšen. V lednu roku 2000 pacient podstoupil druhou torakoskopii, při které mu byla provedena resekce středního laloku vpravo. Zde již byla histologicky verifikována infiltrace plic Langerhansovou histiocytózou. Na základě této diagnózy byly podány glukokortikosteroidy, které však nepřinesly zlepšení plicního nálezu. Pro dyspeptické potíže byla v červnu roku 2000 provedena gastroskopie s nálezem mukoepidermoidního karcinomu jícnu a kardie pT2 pN1. Po operaci v době od září do listopadu roku 2000 dostával nemocný chemoterapii 5-fluorouracilem. Po této chemoterapii, indikované jako adjuvantní léčba karcinomu jícnu, byla v listopadu roku 2000 zjištěna regrese oboustranného plicního nálezu. Po další dva roky, kdy byl sledován na KTRN, byl nález na plicích v pořádku. Nicméně koncem roku 2002 došlo k recidivě nádoru jícnu, která byla příčinou smrti pacienta. Uvedeným příkladem ilustrujeme obtížnost diagnostiky plicní formy Langerhansovy histiocytózy a vhodnost provést při takovémto nálezu vyšetření bronchoalveolární tekutiny elektronovou mikroskopií na Birbeckova granula a dále imunohistochemické barvení buněk na CD1a antigen. Tato vyšetření nebyla při první nekonkluzivní bronchoskopii provedena.
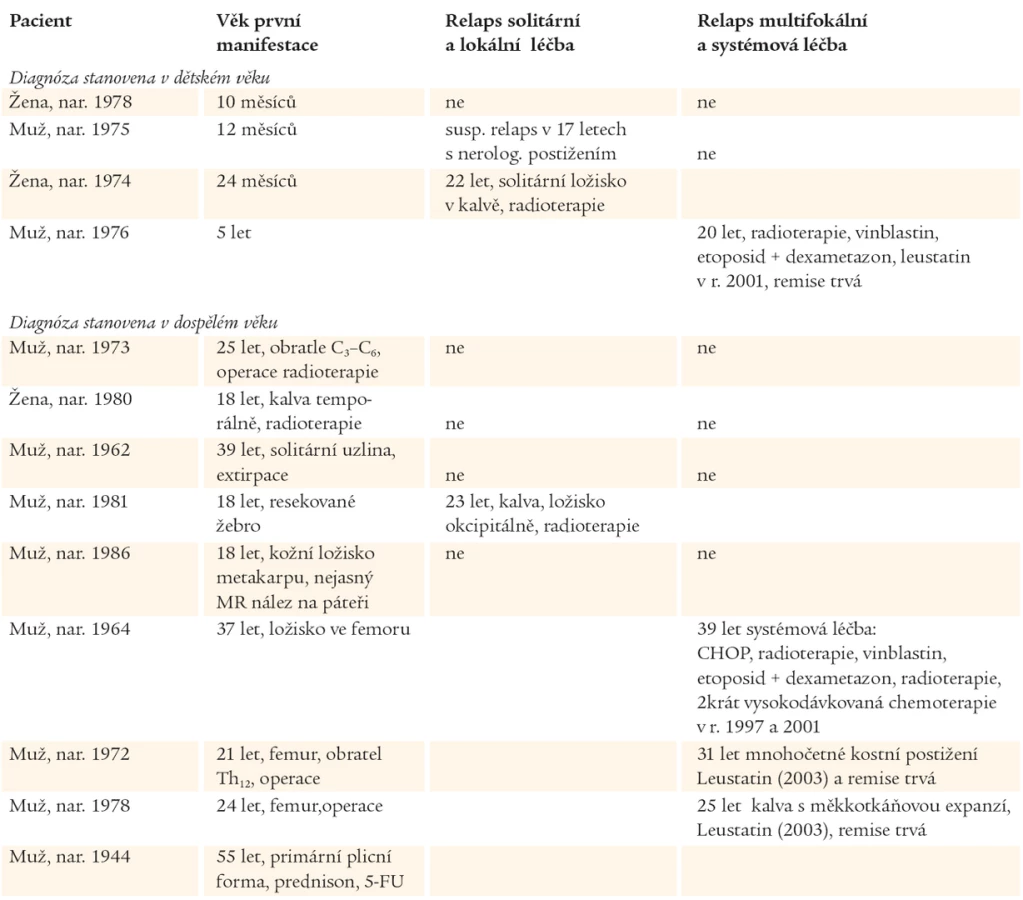
Přehledně je osud popisovaných nemocných znázorněn v tab. 4.
Diskuse
Náš soubor obsahuje celkem 13 osob postižených LCH, ve 4 případech se jedná o pacienty předané do našeho sledování pediatry. Pouze u jedné z těchto 4 osob jsme nepozorovali žádnou recidivu. V jednom případě (pacient, nar. 1975) byla v anamnéze ataka neurologických potíží, netypických pro mladého muže. Tyto potíže výrazně ustoupily, byť ne zcela ad integrum. Kontrolní MR mozku bylo dle lékařských zpráv negativní, nicméně vzhledem k obtížnosti interpretací změn na MR při neurodegenerativních změnách v důsledku LCH se domníváme, že pravděpodobně šlo o nerozpoznanou mozkovou ataku LCH.
V případě muže, nar. 1976, je evidentní, jak původně dětská forma LCH přešla v dospělosti do závažného chronického onemocnění. Zpočátku jsme se domnívali, že recidivy budou omezené pouze na skelet. Výjimečné u něj bylo, že mnohočetné kostní recidivy byly prokázány vždy pouze v oblasti kalvy. Po poslední recidivě v oblasti oka (rok 2001) následovalo za několik měsíců těžké zhoršení stavu s diplopií, skandovanou řečí a příznaky z postižení cerebella. MR obraz i další průběh odpovídal neurodegenerativnímu poškození CNS, pro nějž neexistuje žádná účinná léčba [45].
Trvalé postižení CNS je v dospělosti největším problémem. Vzhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi, musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií. Dle posledních informací o formě CNS postižení je možné diferencovat tři typy postižení:
- Ohraničené granulomy při MR zobrazení v oblasti mozkové pojivové tkáně, odpovídající tumorózním ložiskům v meningách nebo v chorioidním plexu. Jejich histologie odpovídá Langerhansovým granulomům v pojivové tkáni.
- Granulomy v oblasti pojivové mozkové tkáně s částečnou infiltrací okolního nervového parenchymu jak CD1a+ tak i reaktivními histiocyty. Tyto infiltráty byly provázeny výraznou T-buněčnou infiltrací a neurodegenerací, ztrátou neuronů, axonů a gliózou.
- Neurodegenerativní ložiska postrádající CD1a+ buňky, nejčastěji postihující cerebellum a mozkový kmen s výraznou zánětlivou infiltrací, obsahující CD8 lymfocyty. Tento proces vede k degeneraci a glióze nervové tkáně. Dle této studie [40] se neurodegenerace v případě LCH objevuje na základě dominantně T-buněčného zánětlivého procesu a je provázena destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu. V případě tohoto pacienta, nar. 1976, se jednalo o tento typ poškození, pro nějž není známa účinná léčba [40,86].
Tento pacient, nar. 1976, byl prvním pacientem, u něhož jsme se přesvědčili o výborném účinku 2-chlordeoxyadenozinu. Bohužel však tato léčba byla podána až v počátku projevů neurodegenerativního poškození CNS. Zpětně můžeme spekulovat, zda by k tomuto neurodegenerativnímu poškození došlo, kdyby byl 2-chlordeoxyadenozin použit dříve.
Za 15 let existence Interní hematoonkologické kliniky (IHOK) LF MU a FN Brno-Bohunice zde bylo registrováno a léčeno celkem 8 pacientů s LCH, zjištěnou v době dospělosti, u dalšího, devátého pacienta byla rozpoznána plicní forma LCH na pracovišti KTRN. Z těchto 9 pacientů se u 3 jednalo pouze o jednu ataku nemoci, která byla léčena chirurgicky s případným ozářením. Ve 4. případě šlo o kožní formu LCH v dospělém věku s následnými bolestmi v bederní páteři s abnormálním MR-nálezem a nekonkluzívním závěrem histologického hodnocení punktátu obratle. V tomto případně nevylučujeme souběžné kostní postižení, které však již bylo zastiženo ve fázi jizvení, kdy se histologická diagnóza stanovuje obtížněji než v iniciálním stadiu, bohatém na Langerhansovy buňky. V 5. případě jsme izolované ložiskové kostní recidivy řešili pouze ozářením.
Ve 3 případech však šlo o pacienty s recidivující multiložiskovou formou, v jednom případě postihující plíce i skelet, v dalších 2 případech se jednalo pouze o mnohočetná kostní ložiska s expanzí mimo skelet, která vyžadovala komplexní léčbu. Pouze v jednom případě se jednalo izolovanou plicní formu LCH, která nereagovala dostatečně na glukokortikoidní léčbu, k regresi plicního nálezu došlo až při aplikaci 5-fluorouracilu, který byl podán v rámci adjuvantní léčby karcinomu jícnu.
V rámci diskuse zmíníme existující léčebné alternativy a naše zkušenosti s nimi.
Léčba unifokálního postižení
V případě jednoložiskového kostního postižení můžeme zvolit buď pouhé operační ošetření, exkochleaci, a dále jen pacienta sledovat, nebo na operaci navázat další lokální terapií. První možností je lokální aplikace glukokortikosteroidů (50-100 mg hydrokortisonu nebo 75-150 mg metylprednisolonu) [9,30,66,73]. S touto intralezionální aplikací glukokortikosteroidů do kosti nemáme vlastní zkušenosti.
Lokální aplikace glukokortikosteroidních mastí byla používána u pacienta, nar. 1976, do oblasti konečníku, kde měl urputně svědící kožní formu LCH, ale efekt těchto mastí byl pouze dočasný, zevní aplikace glukokortikosteroidů neměla dlouhodobější léčebný efekt.
Druhou alternativou pro ošetření jednoložiskového procesu je radioterapie, následující po chirurgickém výkonu. V literatuře jsou uváděny poměrně nízké ložiskové dávky záření, podstatně menší, než jsou používány pro kurativní léčbu plazmocytomu. Baumgartner použil nejčastěji dávky od 22 do 25 Gy (s rozptylem od 16 do 45 Gy). Dornfeld a Cassady doporučují pro dospělé dávky 10-20 Gy, zatímco pro děti, které mají obvykle kombinovanou léčbu, kolem 10 Gy. Závislost dávky a účinku nebyla stanovena pro vzácnost této nemoci a individuálně odlišný průběh [6,20,28].
Cílem radioterapie je zastavit progresi. S úpravou kosti ad integrum nelze po radioterapii počítat, byť by byly v ložisku zničeny všechny patologické buňky. Naši pacienti byli ozařováni v Masarykově onkologickém ústavu s průměrnou ložiskovou dávkou 25 Gy.
Klasická léčba multifokálního postižení
V případě víceložiskového postižení je vždy na místě systémová léčba. Langerhansovy buňky obsahují receptory pro glukokortikosteroidy, které v nich, podobně jako v lymfatické tkáni, mají potenciál indukovat apoptózu. Proto jsou glukokortikoidy součástí léčebných režimů. Jako iniciální léčba se nejčastěji podává prednison 2 mg/kg po 4 týdny. Tato dávka se pak pomalu v průběhu dalších 4 až 6 týdnů snižuje do vysazení.
Vzhledem k tomu, že cílem léčby u dětí je minimalizovat pozdní následky, byly testovány a osvědčily se v této indikaci deriváty vinka-alkaloidů, vinkristin a vinblastin (6 mg/m2 1krát týdně). Podávají se obvykle v kombinaci s glukokortikosteroidy. Jsou úspěšné u 50 až 60 % nemocných [15,16].
Vinblastin jsme použili u jednoho pacienta s velmi dobrou tolerancí. Vikristinem nebo vinblastinem v kombinaci s glukokortikosteroidy byli léčeni všichni 4 pacienti se vznikem nemoci v dětském věku a u všech tato léčba navodila dlouhodobou remisi, a v jednom případě vyléčení.
Alternativou při neúspěchu vinca-alkaloidů je etoposid (150 mg/m2 i.v. nebo 300 mg/m2 p.o. 1., 2. a 3. den s pauzou do 21. dne). Oba režimy dosahují 60 až 70 % léčebných odpovědí [15,16,36,50,61,62,69,95,96].
U pacientů léčených etoposidem vzniká méně často pozdější funkční následek - diabetes insipidus. Na druhé straně má etoposid větší mutagenní potenciál ve srovnání s vinca-alkaloidy. Léčbu etoposidem jsme použili u dvou pacientů s recidivující nemocí. V obou případech došlo po čase k recidivě nemoci. Perorální forma etoposidu umožňuje ambulantní léčbu, což je výhodné, neboť žilní systém po předchozí léčbě vinca-alkaloidy bývá vždy poškozen a najít vhodnou žílu pro aplikaci cytostatika není jednoduché.
V literatuře lze nalézt informace o léčebném využití dalších léků ze skupiny cytostatik, ty se však používají až jako léky druhé volby: cyklofosfamid, chlorambucil, cytosin-arabinosid, daunorubicin, 6-merkaptopurin, metotrexát, mechloretamin, prokarbazin. Monoterapie chlorambucilem dosáhla 56 % léčebných odpovědí a přidání prednisonu zvýšilo jejich počet na 64 %. Léčba metotrexátem a prednisonem byla úspěšná v 53 %, prednison a vinkristin však byly účinnější (64 % léčebných odpovědí) [3,29,46,54]. S těmito léčebnými režimy nemáme vlastní zkušenosti.
2-chlordeoxyadenozin
2-chlordeoxyadenozin má do jisté míry selektivní účinek, namířený proti monocytům a lymfocytům v kterékoliv fázi buněčného cyklu. Tento účinek léku na buňky monocytární řady, kam patří i buňky Langerhansovy histiocytózy, činí z 2-chlordeoxyadenozinu kandidáta na lék prvé volby u všech histiocytárních chorob. Je používán jak u dětí, tak u dospělých. V literatuře lze nalézt popisy izolovaných případů či skupin několika nemocných, u nichž konvenční chemoterapie již nebyla účinná, a přesto 2-chlordeoxyadenozin navodil dlouhodobou kompletní remisi. V jednom případě, u dítěte s postižením hypotalamu, se po léčbě obnovila sekrece antidiuretického hormonu. Velmi příznivý účinek se popisuje dokonce v případě familiární hemofagocytující lymfohistiocytózy, což je jinak velmi špatně léčitelné onemocnění. Standardní dávka je 0,7 mg/kg/léčebný cyklus, rozdělená do 5 až 7 dnů. Lék se podává v kontinuálních infuzích, nebo formou dvouhodinových infuzí po dobu 5 dnů. Zda je mezi oběma způsoby aplikace rozdíl v účinnosti, není jasné [8,21,24,37,68,70,75,80,81,87,88,100].
My jsme tuto léčbu použili ve třech případech. První, pacient, který tuto léčbu dostal, byl pacient, nar. 1976. U něj jsme však tuto léčbu použili relativně pozdě, v době nástupu neurologických příznaků po poměrně bohaté předchozí léčbě. Efekt 2-chlordeoxyadenozinu se u tohoto pacienta dostavil velmi rychle, jaterní enzymy se po prvním cyklu dostaly do fyziologických hodnot a konečně při léčbě 2-chlordeoxyadenozinem vymizela infiltrace kůže anální oblasti (rok 2002). V průběhu dalších kontrol v letech 2001-2005 nebyla nalezena nová kostní ložiska ani recidiva infiltrace kůže kolem konečníku. Tento případ byl poučný i v dalších směrech. Ověřili jsme si, že vznik měkkotkáňového infiltrátu v oblasti oka může také u dospělého signalizovat hrozící poškození CNS, což je jinak zkušenost pediatrů vídeňského centra pro léčbu této nemoci, ale i jiných [2]. Zpětnou analýzou léčebných účinků jsme dále zjistili, že 2-chlordeoxyadenozin byl u tohoto muže účinnější než léčba vinca-alkaloidy a než léčba etoposidem v kombinaci s glukokortikoidy. Nicméně, bohužel, tento pacient dostal 2-chlordeoxyadenozin relativně pozdě, v době nástupu neurodegenerativního poškození CNS, které nelze zatím žádnou známou léčbou ovlivnit.
Tato naše první zkušenost s 2-chlordeoxyadenozinem potvrdila literární údaje o vysoké účinnosti této léčby i u rezistentních forem.
Pod tímto dojmem jsme v roce 2003 u dalších dvou mladých mužů s mnohočetnou recidivou nemoci v kostech použili 2-chlordeoxyadenozin jako první léčbu s vynikajícím efektem. Oba pacienti jsou v kompletní remisi více než 2 roky od ukončení léčby. U mladého muže, nar. 1978, jsme pro jistotu ještě provedli sběr kmenových krvetvorných buněk po stimulačním režimu obsahujícím etoposid. Jeho choroba s expanzí nádorových hmot z okcipitální kosti do CNS se nám jevila velmi agresivní a měli jsme obavy z časné progrese a případného poškození CNS.
Vysokodávkovaná chemoterapie s auto - či alogenní transplantací kostní dřeně
V případě recidivujících forem nemoci je v pediatrii i v dospělém věku používána vysokodávkovaná chemoterapie s autologní nebo alogenní transplantací. Alogenní přístup je komplikovanější než autologní, a přesto byl použit častěji, a to jak po klasickém, tak po redukovaném přípravném režimu. Z uvedených zpráv lze pouze usoudit, že alogenní transplantace představuje alternativu pro jinak nezvladatelnou chorobu.
Vysokodávkovaná chemoterapie s autologní transplantací je postup, který umožní podání vyšší dávky alkylačních cytostatik a etoposidu, než je obvyklé při běžné chemoterapii. Z publikací vyplývá, že lze tímto způsobem dosáhnout výraznějšího léčebného efektu než při běžné léčbě etoposidem. V literatuře jsou však pouze jednotlivé popisy případů a otázka, zda je účinnější při progresivní chorobě vysokodávkovaná chemoterapie, nebo léčba 2-chlordeoxyadenozinem, není řešena žádnou prospektivní studií [1,11,22,41,48,56,64,85,89,91].
V našem případě jsme použili 2krát vysokodávkovanou chemoterapii (melfalan a etoposid) pouze u jednoho pacienta. První remise po této léčba trvala 2,5 roku, druhá byla podstatně kratší. Je škoda, že u tohoto pacienta jsme nepoužili 2-chlordeoxyadenozin, abychom mohli srovnat délku remisí. Z popisů uvedených případů by se mohlo jevit, že 2-chlordeoxyadenozin může mít větší účinek než vysokodávkovaná chemoterapie (melfalan a etoposid), ale z tak malého počtu pacientů nelze dělat směrodatné závěry, neboť efekt léčby vždy záleží na míře agresivity nemoci.
Nezodpovězenou otázkou tedy zůstává, zda by aplikace 2-chlordeoxyadenozinu nebyla účinnější než vysokodávkovaný melfalan a etoposid, či zda by se v případě autologní transplantace neměl do předtransplantačního režimu dostat i 2-chlordeoxyadenozin. Problémem jsou zde však nehematologické nežádoucí účinky 2-chlordeoxyadenozinu, které znemožňují jeho podání ve vyšších dávkách v rámci předtransplantačního režimu. Netransplantuje se pouze kostní dřeň, jsou publikovány případy s provedenou transplantací plic u rezistentní LCH [90].
Bisfosfonáty
Bisfosfonáty mají svoji roli v léčbě LCH nejen proto, že mají potenciál inhibovat osteoklasty zprostředkovanou osteolýzu. Bisfosfonáty se však kumulují v retikuloendoteliálním (monocytomakrofágovém) systému, tedy i v LCH buňkách a mají potenciál inhibovat aktivitu těchto buněk, a tedy snižovat aktivitu nemoci. Publikované zprávy hovoří o tom, že bisfosfonáty nejen mírní kostní projevy a odstraňují bolesti, ale při léčbě bisfosfonáty regredovaly i mimokostní projevy nemoci [5,18,31-33,38]. Proto bisfosfonáty užívají všichni naši pacienti s kostním poškozením.
Další, námi zatím nepoužité léčebné alternativy
V době, kdy se zvažovala možnost, že LCH je atypickým zánětlivým onemocněním na neznámý vyvolávající podnět, bylo logické testovat imunosupresivní monoterapii cyklosporinem A, nebo kombinovanou imunosupresivní terapii, obsahující antitymocytární globulin a prednison. Tato léčebná alternativa se neukázala přínosnou [4,57-59].
Interferon α byl u Langerhansovy histiocytózy poprvé úspěšně použit v roce 1987 u dvou homozygotních bratrů (1,3 milionů jednotek denně s.c.). V literatuře lze nalézt popisy případů, kdy iniciální monoterapie interferonem α u dětí navodila kompletní remisi. U dospělých nastal léčebný efekt po vysokých dávkách interferonu α, 12 milionů jednotek 3krát týdně, což je ale velmi špatně tolerovatelná dávka. Z publikovaných dat nelze přesně usoudit, jak velkým přínosem je interferon α pro dospělé pacienty s LCH, uvedené publikace však mohou být podkladem k testování této léčby u pacientů nereagujících na běžnější postupy. Nicméně skutečnost, že všechny práce s interferonem α v této indikaci jsou staršího data, asi signalizuje, že nepřinesl zásadní převrat [7,23,79,93].
Účinek interferonu α se u LCH vysvětluje downregulací proteinové kinázy C-α (PKC-α), jejíž zvýšená exprese souvisí s proliferací LCH, podobný účinek na PKC-α má také alterace mevalonátové cesty bisfosfonáty [17]. Účinek interferonu se také vysvětluje jeho antiangiogenním působením [25]. S podáváním interferonu α v této indikaci nemáme vlastní zkušenosti.
U kožní formy se popisuje příznivý účinek retinoidů, což také nemůžeme z vlastní zkušenosti potvrdit [97]. Etanercept - monoklonální protilátky proti TNF (tumor necrosis factor) navodil u rezistentní formy vymizení příznaků, které se však po přerušení léčby obnovily [43]. Tento lék pro jeho nákladnost těžko budeme testovat.
V roce 2005 byl pro mnohočetný myelom dostupný talidomid (preparát Myrin) v rámci specifického léčebného programu na schválení revizním lékařem. Proto by pozitivní zkušenosti s tímto lékem u LCH teoreticky mohly být i přínosem pro naše nemocné, byť zatím by to muselo být na náklady nemocného, pokud by pojišťovna neudělala výjimku, specifický léčebný program se týkal pouze mnohočetného myelomu, v případě jiného onemocnění, které je také léčitelné talidomidem, by musel revizní lékař schválit výjimku nebo by si lék kupoval pacient na vlastní náklady. Myrin (talidomid) se podává se v dávce 100 mg denně po dobu 1 až 2 let [12,49,53,78,94]. U kožní formy byl popsán příznivý efekt léčby PUVA [76].
Mezi nové alternativy patří kombinovaná protizánětlivá a angiostatická léčba. V ložiscích LCH je jednak exprimována COX-2 a dále také je možné histiocytární buňky ovlivnit ligandem takzvaného peroxysome proliferator - activated receptor γ - PPARγ. U systémové LCH byly také popsány signifikantně zvýšené hladiny vaskulárního endoteliálního růstového faktoru (VEGF). Autoři této zprávy zacílili léčbu na tyto tři overexprimované signální cesty. Použili kombinovanou léčbu sestávající z COX-2 inhibitoru rofekoxibu 25, později 12,5 mg denně, dále z analogu PPARγ ligandu a preparátu pioglitazone, 45 mg denně a pravidelně podávanou dávku angiostaticky účinného trofosfamidu 50 mg 3krát denně. Pozitivní výsledek této léčby u dvou chemorezistentních případů považují za signál k jejímu dalšímu testování [25,74]. Bez zajímavosti není ani zpráva o expresi CD52 antigenu, což by signalizovalo možnost léčit tyto pacienty preparátem Campath [47]. Testován byl s nejasným výsledkem také imatinib-mesylat [60].
Monitorování nemoci
Monitorování aktivity Langerhansovy histiocytózy je možné pouze zobrazovacími metodami, což pro dospělého pacienta znamená zobrazení skeletu metodou scintigrafie, RTG a při nejasnostech nákladnějšími metodami, CT a MR. Žádný z běžných výsledků laboratorních vyšetření nekoreluje s aktivitou nemoci a nesignalizuje remisi či recidivu. Jediným nově popsaným vyšetřením, které může korelovat s aktivitou nemoci, je S-100-β sérový protein. Zvýšená koncentrace tohoto proteinu signalizuje také akutní poškození mozku a je výborným prognostickým ukazatelem léčebného efektu u pacientů s maligním melanomem [98]. Toto vyšetření není však u nás prováděno a základem sledování zůstávají zobrazovací metody.
Závěr
Z našich zkušeností a publikovaných zkušeností vyplývá nutnost pečlivě monitorovat aktivitu nemoci. V případě jednoho izolovaného či dvou ložisek se lze spokojit s dominantně lokálními způsoby léčby. V případě opakovaných recidiv nemoci, které mohou signalizovat nepříznivou prognózu, je vhodné ihned přistoupit k léčbě 2-chlordexyadenozinem. Vysokodávkovaná chemoterapie je u této diagnózy proveditelná, vliv vysokodávkované chemoterapie s autologní transplantací kostní dřeně na morbitidu a délku přežití zatím není definován.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 3. 1. 2006
Přijato po recenzi: 31. 1. 2006
Sources
1. Akkari V, Donadieu J, Piguet C et al. Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group. Bone Marrow Transplant 2003; 31 : 1097-1103.
2. Anton M, Holoušova M, Řehůřek J et al. Histiocytoza X a dětská očnice. Čs Ophthal 1992; 48 : 176-180.
3. Arceci RJ, Brenner MK, Pritchard J et al. Contraversis and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998; 12 : 339-357.
4. Arico M, Colella R, Conter V et al. Cyclosporine therapy for refractory Langerhans cell histiocytosis. Med Pediatr Oncol 1995; 25 : 12-16.
5. Arzoo K, Sadeghi S, Pullarkar V. Pamidronate for bone pain from osteolytic lesions in Langerhans cell histiocytosis. N Engl J Med 2001; 345 : 225-226.
6. Baumgartner I, Hochstetter A, Baumert B et al. Langerhans cell histiocytosis in adults. Med Pediatric Oncol 1997; 28 : 9-14.
7. Bellmunt J, Albanell J, Salud A et al. Interferon and disseminated Langerhans cell histiocytosis. Med Pediatr Oncol 1992; 20 : 336-337.
8. Bernard F, Thomas C, Bertrand Y et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer 2005; 41 : 2682-2689.
9. Bernstrand C, Björk O, Ahnström L et al. Intralesional steroids in Langerhans cell histiocytosis of bone. Acta Paediatr 1996; 85 : 502-504.
10. Bernstrand C, Cederlund K, Sandstedt B et al. Pulmonary abnormalities at long term follow up of patients with Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 459-468.
11. Berry J, Russel JA. Salvage of relapsed malignant histiocytosis by autologous bone marrow transplant. Bone Marrow Transplant 1989; 4 : 123-124.
12. Bertolini F, Mingrone W, Alietti A et al. Thalidomide in multiple myeloma, myelodysplastic syndromes and histiocytosis. Ann Oncol 2001; 12 : 987-990.
13. Bittenglova R, Pešek M, Mukenšnabl P et al. Granulomatóza z Langerhansových buněk. Stud Pneumol Phtiseol 2002; 62 : 196-202.
14. Blum R, Seymour JF, Hicks RJ. Role of FDG-positron emission tomography scannin in the management of histiocytosis. Leukem Lymphoma 2002; 43 : 2155-2157.
15. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998; 12 : 327-336.
16. Broadbent V, Pritchard J, Yeomans E. Etoposide in the treatment of multisystem Langerhans cell hitocytosis. Med Pediatr Oncol 1989; 17 : 97-100.
17. Brown RE. Interpheron alpha therapy protein kinse C alpha and Langerhans cell histiocytosis. Med Pediatr Oncol 2003; 41 : 63-64.
18. Brown RE. Bisphosphonates as antialveolar macrophage therapy in pulmonary Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 641-643.
19. Bunanska E, Stančokova T, Dluholucky S. Histiocytóza z Langerhansových buněk. Čes Slov Pediat 1998; 53 : 18-19.
20. Cassady JR. Current role of radiation therapy in the management of histiocytosis X. Hematol Oncol N Am 1987; 1 : 123-125.
21. Choi SW, Bangaru BS, Wu CD et al. Gastrointestinal involvement in disseminated Langerhans cell histiocytosis (LCH) with durable complete response to 2-chlorodeoxyadenosine and high-dose cytarabine. J Pediatr Hematol Oncol 2003; 25 : 503-506.
22. Conter V, Reciputo A, Arrigo C et al. Bone marrow transplantation for refractory Langerhans' cell histiocytosis. Haematologica 1996; 81 : 468-471.
23. Culic S, Jakobson A, Culic V. Etoposide a the basis and interferon alfa as the maitenance therapy for Langerhans cell histiocytosis. Pediatr Hematol Oncol 2001; 18 : 291-294.
24. Dallafior S, Pugin P, Cerny T et al. Successful treatment of a case of cutaneous Langerhans cell granulomatosis with 2-chlorodeoxyadenosine and thalidomide. Hautarzt 1995; 46 : 553-560.
25. Dina A, Zahava V, Iness M. The role of vascular endothelial growth factor in Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2005; 27 : 62-66.
26. Dogan S, Conway J, Miller JH et al. Detection of bone lesions in Langerhans cell histiocytosis. Complementary roles of scintigraphy and conventional radiograpy. J Pediatric Hematol/Oncol 1996; 18 : 51-58.
27. Donadieu J, Rolon MA, Pion I et al. Incidence of growth hormone difficienci pediatric onset Langerhans cell histiocytosis: Efficacy and safety of Growth hormone treatment. J Clin Endokrinol Metabolism 2004; 89 : 604-609.
28. Dornfeld S, Winkler C, Dörr W et al. Stralentherapie der Histiocytosis X. Stralenther. Onkologie 1998; 174 : 534-535.
29. Egeler MR, de Kraker J, Voute PA. Cytosine-arabinoside, vincristine and prednisolone in the treatment of children with disseminated Langerhans cell histiocytosis with organs dysfunction. Med Pediatr Oncol 1993; 20 : 265-270.
30. Egeler MR, Thompson RC, Voute PA et al. Intralesional infiltration of corticosteroids in localized Langerhans cell histiocytosis. J Pediatr Orthop 1992; 12 : 811-814.
31. Egeler RM. More on pamidronate in Langerhans cell histiocytosis. N Engl J Med 2001; 345 : 1502-1503.
32. Elomaa I, Blomquist C, Porkka L et al. Experience of clodronate treatment of multifocal eosinophilic granuloma of bone. J Intern Med 1989; 225 : 59-61.
33. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 2001; 23 : 54-56.
34. Favara BE, Feller A and members WHO committee on Histiocyte/Retikulum Cell Proliferations. Contemporary classification of histiocyte disorders. Medical Pediatric Onkol 1997; 29 : 157-166.
35. Ferreli C, Aste N, Pinna LA et al. Langerhans cell histiocytosis in an adult. J Eur Acad Dermatol Venereol 1997; 9 : 253-255.
36. Gadner H, Heitger A, Grois N et al. Treatment strategy for disseminated Langerhans cell histiocytosis. Med Pediatr Oncol 1994; 23 : 72-80.
37. Goh NS, McDonald CE, MacGregor DP et al. Successful treatment of Langerhans cell histiocytosis with 2-chlorodeoxyadenosine. Respirology 2003; 8 : 91-94.
38. Goto H, Inaba M, Kobayashi K et al. Seccessful treatment of multicentric reticulohistiocytosis with alendronate. Arthritism Rheumatism 2003; 48 : 3538-3541.
39. Gotz G, Fichter J. Langerhans'-cell histiocytosis in 58 adults. Eur J Med Res 2004; 9 : 510-514.
40. Grois N, Prayer D, Prosch H et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829-838.
41. Hale GA, Bowman LC, Woodard JP et al. Allogeneic bone marrow transplantation for children with histiocytic disorders: use of TBI and omission of etoposide in the conditioning regimen. Bone Marrow Transplant 2003; 31 : 981-986.
42. Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol. 2004; 50 : 157-174.
43. Henter JI. Succesful treatment of Langerhans cell histiocytosis with etanercept. N Engl J Med 2001; 345 : 1577-1578.
44. Howarth DM, Mullan BP, Wiseman GA et al. Bone scintigraphy evaluated in diagnosis and staging Langerhans´s cell histiocytosis and related disorders. J Nuclear Med 1996; 37 : 1456-1460.
45. Imashuku S, Ishida S, Koike K et al. Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2004; 26 : 735-739.
46. Jones B, Kung F, Chevalier L et al. Chemotherapy of retikuloendotheliosis. Comparison of methotrexate plus prednisone versus vincristine plus prednisone. Cancer 1974; 34 : 1011-1017.
47. Jordan MB, McClain KL, Yan X et al. Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer 2005; 44 : 251-254.
48. Kinugawa N, Imashuku S, Hirota Y et al. Hematopoietic stem cell transplantation (HSCT) for Langerhans cell histiocytosis (LCH) in Japan. Bone Marrow Transplant 1999; 24 : 935-938.
49. Kolde G, Schulze P, Sterry W. Mixed response to thalidomide therapy in adults: two cases of multisystem Langerhans' cell histiocytosis. Acta Derm Venereol 2002; 82 : 384-386.
50. Ladisch S, Gadner H, Arico M et al. LCH-1: A randomized trial of etoposide vs. vinblastine in disseminated Langerhans cell histiocytosis. Medical Pediatric Oncol 1994; 23 : 107-110.
51. Lévy S, Capron D, Joly JP et al. Hepatic nodules as single organ involvement in an adult with Langerhans celle Granulomatosis. J Clin Gastroentrol 1998; 26 : 69-73.
52. Malpas JS. Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Amer 1998; 12 : 259-268.
53. Mauro E, Fraulini C, Rigolin GM et al. A case of disseminated Langerhans' cell histiocytosis treated with thalidomide. Eur J Haematol 2005; 74 : 172-174.
54. McClain KL. Drug therapy for the treatment of Langerhans cell histiocytosis. Expert Opin Pharmacother 2005; 6 : 2435-2441.
55. Mendez JL, Nadrous HF, Vassallo R et al. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125 : 1028-1032.
56. Meyer-Wentrup F, Foell J. Unrelated cord blood transplantation in an infant with severe multisystem Langerhans cell histiocytosis: clinical outcome, engraftment and culture of monocyte-derived dendritic cells. Bone Marrow Transplant 2004; 33 : 875-876.
57. Minkov M, Grois N, Braier J et al. Immunosuppressive treatment for chemotherapy-resistant multisystem Langerhans cell histiocytosis. Med Pediatr Oncol 2003; 40 : 253-256.
58. Minkov M, Grois N, Broadbent V et al. Cyclosporin A therapy for multisystem Langerhans cell histiocytosis. Med Pediatr Oncol 1999; 33 : 482-485.
59. Minkov M, Grois N, Heitger A et al. Treatment of multisystem Langerhans cell histiocytosis. Results of the TAL HX83 and DAL-HX 90 studies. Klinische Pediatrie 2000; 212 : 139-144.
60. Montella L, Insabato L, Palmieri G. Imatinib mesylate for cerebral Langerhans'-cell histiocytosis. N Engl J Med 2004; 351 : 1034-1035.
61. Mottl H, Koutecký J, Ganevová M. Strategie léčby histiocytózy z Langerhansových buněk u dětí. Čes Slov Pediat 1994; 49 : 81.
62. Mottl H, Mracek J, Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530-533.
63. Munn S, Chu AC. Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Amer 1998; 12 : 269-286.
64. Nagarajan R, Neglia J, Ramsay N et al. Successful treatment of refractory Langerhans cell histiocytosis with unrelated cord blood transplantation. J Pediatr Hematol Oncol 2001; 23 : 629-632.
65. Nanduri VR, Lillywhite L, Chapman C et al. Cognitive outcome of long term survivors of multisystem Langerhans cell histiocytosis. J Clin Oncol 2003; 21 : 2961-2967.
66. Nebelung W, Ropke M, Kluba U et al. Treatment concepts in osseous manifestations of Langerhans cell histiocytosis. Z Orthop Ihre Grenzgeb 1999; 137 : 236-243.
67. Nicholson SH, Egeler M, Nesbit ME. The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998; 12 : 379-348.
68. Ottaviano F, Finlay JL. Diabetes insipidus and Langerhans cell histiocytosis: a case report of reversibility with 2-chlorodeoxyadenosine. J Pediatr Hematol Oncol 2003; 25 : 575-577.
69. Pacovska V, Bortlova A, Homolka J et al. Granulomatóza z Langerhansových buněk. Trendy Med 2002; 4 : 59-61.
70. Pardanani A, Phyliky R, Chin-Yang L et al. 2-chlordeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc 2003; 78 : 301-306.
71. Prosch H, Feldges A, Grois N et al. Demonstration of CD1a positive cells in the cerebrospinal fluid--A clue to diagnosis of isolated Langerhans cell histiocytosis of the hypothalamic-pituitary axis? Med Pediatr Oncol 2003; 41 : 474-476.
72. Prosch H, Grois N, Prayer D et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 594-599.
73. Putters TF, deVisscher JG, van Veen A et al. Intralesional infiltration of corticosteroids in the treatment of localised Langerhans´ cell histiocytosis of the mandibule. Int J Oral Maxillofac Surg 2005; 34 : 571-575.
74. Reichle A, Vogt T, Kunz-Schughart L et al. Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans' cell histiocytosis of adults. Br J Haematol 2005; 128 : 730-732.
75. Rodriguez-Galindo C, Kelly P, Jeng M et al. Treatment of children with Langerhans cell histiocytosis with 2-chlorodeoxyadenosine. Am J Hematol 2002; 69 : 179-184.
76. Rosen K, Jontell M, Mobacken H et al. Epidermal Langerhans-cells in chronic eczematous dermatitis of the palms treated with PUVA and UVB. Acta Derm Venereol 1989; 69 : 200-205.
77. Rožánek P, Molnar V, Rešl M. Tři případy plicní granulomatózy z Langerhansových buněk. Lék Zpr Lék Fak Univ Karlovy Hr Králové 1998; 43 : 127-132.
78. Sander CS, Kaatz M, Elsner P. Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology 2004; 208 : 149-152.
79. Sato Y, Ikeda Y, Ito E et al. Histiocytosis X: Successful treatment with recombinant interferon alpha-2a. Acta Paediatr Jpn 1990; 32 : 151-154.
80. Saven A, Burian C. Cladribine activity in Langerhans cell histiocytosis. Blood 1999; 93 : 4125-4130.
81. Saven A, Kenneth A, Foon A. 2-Chlorodeoxyadenosine induced complete remissions in Langerhans cell histiocytosis. Ann Intern Med 1994; 121 : 430-433.
82. Šímová B, Mališ J, Neuwirt J. Klinické projevy histiocytózy z Langerhansových buněk. Zdrav Nov ČR Lék Listy 2003; 52 : 18.
83. Skacel Z, Marel M, Vraštilova P et al. Histiocytóza z Langerhansových buněk. Přehled literatury a vlastní pozorování. Stud Pneumol Phtiseol 2000; 60 : 150-156.
84. Smilek P, Krejčová B, Čada K et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994; 43 : 263-265.
85. Steiner M, Matthes-Martin S, Attarbaschi A Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant 2005; 36 : 215-225.
86. Steiner M, Prayer D, Asenbaum S et al. Modern imaging methods for the assessment of Langerhans' cell histiocytosis-associated neurodegenerative syndrome: case report. J Child Neurol 2005; 20 : 253-257.
87. Stime KC, Saylors RL, Williams LL et al. 2-Chlordeoxyadenosine for the treatment of refractory or recurent Langerhans cell histiocytosis in pediatric patients. Medical Pediatric Oncol 1997; 29 : 288-292.
88. Stine KC, Saylors RL, Saccente S et al. Efficacy of continuous infusion 2-CDA (cladribine) in pediatric patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 81-84.
89. Stoll M, Freund M, Schmid H et al. Allogeneic Bone Marrow transplantation for Langerhans cell histiocytosis. Cancer 1990; 66 : 284-288.
90. Sulica R, Teirstein A, Padilla ML. Lung transplantation in interstitial lung disease. Curr Opin Pulm Med 2001; 7 : 314-322.
91. Suminoe A, Matsuzaki A, Hattori H et al. Unrelated cord blood transplantation for an infant with chemotherapy-resistant progressive Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2001; 23 : 633-636.
92. Sundar KM, Gosselin MV, Chung HL et al. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology and clinical evolution of the disease. Chest 2003; 123 : 1673-1683.
93. Takemori H, Sakata Y, Suzuki H et al. A case of malignant histiocytosis succesfuly treated with combination of interferon alpha and etoposide therapy. Jpn J Clin Oncol 1990; 20 : 431-435.
94. Thomas L, Ducros B, Secchi T et al. Successful treatment of adults Langerhans cell histiocytosis with thalidomide. Arch Dermatol 1993; 129 : 1261-1264.
95. Tichy M jr, Tichy M, Krč I et al. Multicentrická retikulohistiocytóza. Ces Slov Derm 1999; 74 : 168-171.
96. Toušovská K, Slavík Z. Histiocytóza z Langerhansových buněk v dětském věku. Lék Zprav Lék Fak Univ Karlovy Hr Králové 1997; 42 : 127-132.
97. Tsambaos D, Georgiu S, Kapranos N et al. Langerhans cell histiocytosis: complete remission after oral Isotretinoid therapy. Acta Derm Venereol 1995; 75 : 62-64.
98. Ugurel S, Pröhler G, Tilgen W et al. S100-β serum proein - a new marker in the diagnosis and monitoring of Langerhans cell histiocytosis. Brit J Dermatol 2000; 143 : 201-202.
99. Večer J, Charvát J, Kubátová H et al. Sekundární hemofagocytující syndrom při systémovém onemocnění. Čas Lék Čes 2000; 139 : 379-381.
100. Watts J, Files B. Langerhans cell histiocytosis: central nervous system involvement treated successfully with 2-chlorodeoxyadenosine. Pediatr Hematol Oncol 2001; 18 : 199-204.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2006 Issue 4
Most read in this issue
- Videotorakoskopie v lokální anestezii v diagnostice a léčbě pohrudničních výpotků
- Primární prevence ischemické choroby srdeční u mužů středního věku v Praze: výsledky dvacetiletého sledování
- Ubikvitiny, proteazomy, sumoylace a použití dnes a zítra v terapii nádorů i jiných chorob I. Ubikvitin-proteazomový systém a transkripční faktor NF-κB
- Profylaxe žilních tromboembolických komplikací v interních oborech - rozpor mezi teorií a praxí