
Waldenströmova makroglobulinemie - klinické projevy a diferenciální diagnostika a prognóza nemoci
Waldenström macroglobulinemia – clinical manifestations and differential diagnosis and prognosis of the disease
Waldenström macroglobulinemia is defined by the presence of IgM type monoclonal immunoglobulin and histological prove of lymphoplasmocytary lymphoma in the bone marrow. Clinical symptoms of the disease depend on the pressure on the bone marrow by the malignant clone with subsequent cytopenia, extramedullary infiltration and toxic manifestations of monoclonal immunoglobulin. 6q deletion and absence of translocation in the sphere of the heavy immunoglobin chain gene, which is otherwise typical for other lymphoproliferative disorders, is the typical cytogenetic abnormality. The text describes the symptoms of the disease and the issues of its differential diagnosis.
Keywords:
Waldenström macroglobulinemia – IgM type multiple myeloma
:
Z. Adam 1; J. Šmardová 2; V. Ščudla 3
:
Interní hematoonkologické klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
1; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jirka Mačák, CSc.
2; III. interní klinika Lékařské fakulty UP a FN Olomouc, přednosta prof. MUDr. Vlastimil Ščudla, CSc.
3
:
Vnitř Lék 2007; 53(12): 1325-1337
:
Review
Waldenströmova makroglobulinemie je definována přítomností monoklonálního imunoglobulinu typu IgM a histologickým průkazem lymfoplazmocytárního lymfomu v kostní dřeni. Klinické příznaky této nemoci se odvíjejí od útlaku kostní dřeně maligním klonem s následnou cytopenií, infiltrací extramedulárních struktur a toxických projevů monoklonálního imunoglobulinu. Typickou cytogenetickou abnormalitou je delece 6q a dále nepřítomnost translokace v oblasti genu pro těžký řetězec imunoglobulinů, která je jinak typická pro jiné lymfoproliferativní choroby. V textu jsou popsány příznaky nemoci a otázky její diferenciální diagnostiky.
Klíčová slova:
Waldenströmova makroglobulinemie - mnohočetný myelom typu IgM - monoklonální gamapatie nejistého významu
Úvod
Je tomu více než 60 let, co Jan Gosta Waldenström poprvé popsal případ 2 pacientů, trpících opakovaným krvácením z nosu a z úst, kteří měli lymfadenopatii, trombocytopenii, zvýšenou sedimentací erytrocytů, zvýšenou viskozitu séra, zvýšený počet lymfoidních buněk v kostní dřeni a normální nález na snímcích kostí [1].
Waldenströmova makroglobulinemie zůstala vzhledem k malé incidenci po mnoho následujících let definována velmi volně, jako choroba s přítomností zvýšené koncentrace monoklonálního gamaglobulinu typu IgM, bez upřesnění histologického podkladu nemoci. Teprve WHO klasifikace definovala histopatologický základ této nozologické jednotky - lymfoplazmocytární lymfom infiltrující kostní dřeň, který produkuje monoklonální imunoglobulin typu IgM. Tato definice jasně odlišila Waldeströmovu makroglobulinemii od jiných lymfoproliferativních jednotek, které také mohou produkovat monoklonální imunoglobulin typu IgM.
Epidemiologické údaje
Nemáme statistické údaje z ČR. Pro ilustraci uvedeme data z USA. Incidence této nemoci upravená na věk je 0,34/100 000 mužů a 0,17/100 000 žen. Incidence se zvyšuje s věkem. U mužů do 45 let je jen 0,01/100 000, zatímco u mužů nad 75 let již dosahuje 3,63/100 000. U žen ve věku do 45 let je incidence jen 0,01/100 000 a ve věku nad 75 let je již 1,64/100 000 [2]. V Anglii je uváděna vyšší incidence 1,03/100 000 [3,4]. Zvyšující se incidenci Waldenströmovy makroglobulinemie ve stáří lze interpretovat jako výsledek alterace imunity u starších osob [5].
Existuje zřejmě určitá familiární predispozice ke vzniku maligních lymfoproliferací. Při analýze 257 rodin, v nichž jeden příslušník měl Waldenströmovu makroglobulinemii, bylo v 18,7 % rodin nalezen alespoň jeden příbuzný prvního řádu s některou B-lymfoproliferací (Waldenströmova makroglobulinemie u 5,1 %, maligní nehodgkinský lymfom u 3,5 %, mnohočetný myelom u 3,1 %, chronická lymfatické leukemie u 2,7 % a monoklonální gamapatie nejistého významu u 1,9 %, akutní lymfatická leukemie u 1,2 % a Hodgkinova choroba u 1,2 %), což je více než činí výskyt v průměrné populaci [6].
Příznaky nemoci
Příznaky nemoci lze obecně dělit na:
- projevy cytopenie způsobené infiltrací kostní dřeně,
- patologické projevy způsobené monoklonálním imunoglobulinem,
- projevy extramedulární proliferace lymfoplazmocytárního lymfomu (lymfadenopatie, splenomegalie).
K nespecifickým příznakům patří únavový syndrom (patologická únava - fatigue, která nemizí ani po dostatečném odpočinku), pocit slabosti a nevýkonnosti, úbytek hmotnosti, subfebrilie či febrilie. Příznaky shrnuje tab. 1.

Projevy způsobené monoklonálním imunoglobulinem IgM
Projevy hyperviskozity - spontánní krvácení, neurologické symptomy a retinopatie
Patofyziologie hyperviskózního syndromu
Molekuly imunoglobulinů se liší svojí distribucí v organismu. Molekuly typu IgG či IgA jsou přítomny jak v intravaskulárním, tak v extravaskulárním prostoru, zatímco molekuly typu IgM se nacházejí z 80 % intravaskulárně. Molekuly imunoglobulinu IgM díky své velikosti a asymetrii zvyšují viskozitu krve podstatně více než stejná koncentrace molekul ostatních imunoglobulinů. Závislost koncentrace imunoglobulinu typu IgM a viskozity plazmy i krve není lineární, ale spíše exponenciální. Strmější vzestup křivky závislosti viskozity na koncentraci imunoglobulinu IgM souvisí mimo jiné s intenzivnější agregací IgM molekul při vyšší koncentraci.
Vzestup koncentrace monoklonálního IgM z 20 na 30 g/l způsobuje nárůst viskozity o méně než 2 centipoise (cP), zatímco vzestup koncentrace z 40 na 50 g/l zvýší viskozitu asi o 5 cP. Tento poznatek je důležitý pro léčebnou plazmaferézu, neboť výměna jednoho objemu plazmy sníží koncentraci monoklonálního IgM asi o 35 - 40 %, ale viskozitu sníží až o 60 % [3,4].
Udává se, že klinické projevy zvýšené viskozity (hyperviskózní syndrom) začínají při viskozitě plazmy dosahující zhruba 4násobku viskozity vody, což obvykle bývá při koncentracích monoklonálního IgM nad 30 g/l, dle jiných údajů až nad 50 g/l [7-9].
Klinické příznaky hyperviskózního syndromu
Zvýšená viskozita plazmy a dále snížená deformovatelnost stěn erytrocytů vlivem vyšší koncentrace monoklonálního imunoglobulinu IgM zhoršují dominantně mikrocirkulaci. Výsledkem je spontánní krvácení ze sliznic nosu a dásní.
Oční pozadí je jediné místo v organizmu, kde lze pozorovat cévní změny, vyvolané hyperviskozitou (přeplněné kapiláry a mikrokrvácení), případně i trombózu vena centralis retinea. To vše je důvodem, proč nemocní udávají zhoršování zraku [10]. Hyperviskozita ale poškozuje nejen zrak, ale i sluchové ústrojí, ba dokonce byl popsán případ náhlé hluchoty.
Prvním příznakem hyperviskózního syndromu může být ale také chronická bolest hlavy, a to ještě v době, kdy na očním pozadí není patologický nález. Tato bolest se vysvětluje zvýšeným nitrolebním tlakem. Ten může být důsledkem zvětšení objemu plazmy vlivem vyšší koncentrace monoklonálního imunoglobulinu typu IgM. Nitrolební změny mohou vést i ke změně povahy. Zhoršuje se výbavnost, paměť. Psychika se může v důsledku hyperviskozity postupně změnit až do obrazu demence. Starší literatura zde popisuje coma paraproteinemicum.
Hyperviskozita a zvýšený plazmatický objem může vest také k srdečnímu selhání, dušnosti a k městnavé srdeční slabosti [11].
Z vlastních zkušeností musíme říci, že hodnota viskozity, od níž nastupují příznaky, je velmi individuální a vždy je nutno řídit se subjektivními údaji pacienta a klinickým nálezem a nejen laboratorními údaji. Neexistuje fixní závislost mezi hodnotou viskozity a klinickým obrazem. Dopady hyperviskozity na organizmus výrazně ovlivňuje stav cév a zřejmě i další vlivy.
Kryoglobulinemie
Kryoglobulinemie je termín pro přítomnost bílkovin, které v cévním řečišti při poklesu teploty pod fyziologické rozmezí kryoprecipitují.
Kryoglobulinemie se klasifikuje dle komponenty, která precipituje. Klinické příznaky závisí na teplotě, při níž kryoprotein gelifikuje.
Klasifikace a patofyziologie kryoglobulinemie
Kryoglobulin I. typu je tvořen monoklonálním imunoglobulinem IgM, nebo případně jiným typem monoklonálního imunoglobulinu, který samostatně precipituje při ochlazení, aniž by se specificky vázal na jiné bílkoviny. Patofyziologickým podkladem klinických příznaků je intravaskulární precipitace kryoglobulinu a narušení cirkulace vlivem gelifikace. Kryoglobulinemie I. typu může mít těžké projevy, ale může být také pouhým laboratorním fenoménem bez klinických projevů. Závisí to na teplotě, při níž nastává kryoprecipitace. V případě makroglobulinemie se vyskytuje nejčastěji právě tento typ.
Kryoglobulin II. typu je definován jako monoklonální imunoglobulin vázající se v chladu na Fc fragmenty polyklonálních imunoglobulinů jiných tříd. Typicky je to monoklonální imunoglobulin typu IgM, vázající se na Fc fragmenty polyklonálních imunoglobulinů typu IgG. Monoklonální IgM má v tomto případě charakter revmatoidního faktoru. Tato nemoc je nazývána smíšená kryoglobulinemie (mixed cryoglobulinaemia).
Kryoglobulin III. typu (synonymem polyklonální kryoglobulin) je tvořen pouze polyklonálními imunoglobuliny s tepelnou charakteristikou kryoglobulinů. Je pozorována nejčastěji v souvislosti s hepatitidou C [12].
Klinické příznaky
Typickými příznaky kryoglobulinemie I. typu je zbělení akrálních částí končetin nebo lividní zbarvení těchto okrsků pro prochlazení. V některých případech způsobuje chladovou urtiku, a také purpuru. Porucha prokrvení tkání na pokladě kryoglobulinu může vést i k trvalému poškození, nejtěžšími projevy jsou kožní ulcerace a nekrózy a případně poškození ledvin [13,14].
Kryoglobulinemie II. typu je imunokomplexová choroba. Způsobuje vaskulitidy dominantně malých cév kůže, ledvin, jater a periferních nervů. Typickými klinickými projevy je purpura [15,16]. Frekvenci jednotlivých příznaků kryoglobulinemie II. typu uvádí tab. 2 [12,17,18].
![Příznaky kryoglobulinemie II. typu [17].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/88f7b20cf6545246ec5de56657f3f062.jpg)
Kryoglobulinémie III. typu je způsobena polyklonálními imunoglobuliny s charakterem kryoglobulinu. Často bývá asociovaná s chronickou hepatitidou C, která vyvolává lymfoproliferativní odpověď. Ta se může transformovat do maligní lymfoproliferace, frekvence přechodu dosahuje až 10 % [19]. Typické projevy této formy jsou artralgie a kožní purpura, podobně jako u II. typu kryoglobulinemie.
U Waldenströmovy makroglobulinemie se frekvence výskytu kryoglobulinemie pohybuje kolem 10 % [11]. Dimopoulos [16] popisuje nález monoklonálního kryoglobulinu u 20 %, ale klinicky významné příznaky kryoglobulinemie pouze u 5 %.
Koagulopatie s krvácivými projevy
Monoklonální imunoglobulin může interferovat s jednotlivými fázemi hemostázy. Makroglobulin může také vazbou na trombocyty narušit jejich adhezi a agregaci.
Stalnikiewicz popisuje 3% výskyt závažnějších koagulačních poruch. V hodnoceném souboru diagnostikovali 2 případy získané von Willebrandovy nemoci a 1 případ Glanzmannovy trombastenie, 1 případ trombocytopenie autoimunitního původu a 1 případ koagulopatie s cirkulujícím antikoagulans [11].
Projevy způsobené depozity monoklonálního IgM
Také monoklonální imunoglobulin typu IgM se může ukládat v různých orgánech a poškozovat je.
Nejen lehké řetězce, ale také kompletní molekula monoklonálního IgM může poškozovat ledviny. V případě s klinicky zjevnou nefropatií se vychytává a precipituje monoklonální imunoglobulin typu IgM v glomerulárních kličkách, kde tvoří subendoteliální depozita, poškozující glomeruly. Tento typ poškození glomerulů může zapříčinit neselektivní proteinurii, dehydrataci a selhání ledvin. K těmto pochodům přispívá hyperviskozita a naopak plazmaferéza vede k rychlému zlepšení renálních funkcí.
U některých osob s Waldenströmovou makroglobulinemií se v průběhu nemoci objevují uzly (noduly) a papuly na kůži. Histologické a histochemické vyšetření těchto morf prokázalo depozita monoklonálního imunoglobulinu typu IgM ve formě amorfní hmoty bez současné maligní infiltrace okolí [15,20].
Monoklonálního imunoglobulin IgM se ukládá a tvoří depozita výjimečně i v jiných orgánech. V ojedinělých případech se v průběhu nemoci objevily průjmy, malabsorpce a případně gastrointestinální krvácení. Biopsie střeva v těchto případech prokázala depozita imunoglobulinu IgM v lamina propria či submukóze [21].
AL-amyloidóza může být primární, tedy bez průkazu maligního klonu, tak také sekundární, provázející mnohočetný myelom anebo Waldenströmovu makroglobulinemii. Amyloid byl detekován u 2 % osob s monoklonálním imunoglobulinem typu IgM. Analýza případů AL-amyloidózy provázející Waldenströmovu makroglobulinemii uvádí následující frekvenci tkáňového poškození: srdce 44 %, periferní nervy 38 %, ledviny 32 %, měkké tkáně 18 %, játra 14 %, plíce 10 %. Incidence kardiopulmonální amyloidózy byla vyšší než u AL-amyloidózy, provázející jiné choroby. Přítomnost amyloidových depozit výrazně zkracuje přežití pacientů s Waldenströmovou makroglobulinemií, medián jejich přežití byl jen 25 měsíců [22-28].
Bostonský registr amyloidóz obsahoval 812 nově diagnostikovaných nemocných s AL-amyloidózou. Z nich jen 16 osob mělo jiný podklad nemoci než MGUS či mnohočetný myelom, u 12 osob to byla Waldenströmova makroglobulinemie [29]. Těmito čísly jen ilustrujeme skutečnosti, že AL-amyloidóza jako komplikace Waldenströmovy makrolgobulinemie je extrémně vzácná.
Neuropatie způsobená monoklonálním imunoglobulinem IgM s protilátkovou aktivitou namířenou proti antigenům nervových vláken
Neuropatie je prokazatelná asi u 20 % nemocných s Waldenströmovou makroglobulinemií.
Neuropatie má nejčastěji charakter distální senzorické demyelinizační neuropatie. Častá je ataxie, případně třes. Slabost jako dominující projev neuropatie u IgM gamapatie je spíše výjimečná, provází spíše neuropatie při gamapatii typu IgA nebo IgG. Motorické nervy bývají u IgM gamapatie postiženy jen zcela výjimečně [30].
Patofyziologickým podkladem je monoklonální protilátka typu IgM, zaměřená proti některému antigenu nervových vláken. Nejčastěji (v 50 %) je prokazována anti-MAG protilátka (anti-myelin associated glykoprotein). Je však známo nejméně 5 různých antigenů nervových struktur, na něž se váže monoklonální imunoglobulin typu IgM [19,31-33].
Neuropatie, provázející gamapatii typu IgM, má elektrofyziologický (EMG) korelát, odpovídající chronické zánětlivé demyelinizační polyneuropatii. Závažná neuropatie sama o sobě může být invalidizující a tedy i indikací k léčbě.
Nemoc chladových aglutininů (cold aglutinin disease)
Chladové aglutininy jsou lidské autoprotilátky, které se váží na antigeny erytrocytů. Monoklonální chladová protilátka typu IgM se naváže na erytrocytární antigeny při teplotě pod 37 °C. Erytrocyty s navázanými protilátkami jsou pak hemolyzovány dominantně v buňkách monocyto-makrofágového systému, takže hemolýza je v těchto případech extravaskulární a zhoršuje se po prochlazení.
Dočasné polyklonální chladové aglutininy způsobující mírnou hemolytickou anémii mohou vzniknout po mykoplazmové infekci. Pacienti s nemocí chladových aglutininů mají většinou MGUS, ale někteří mohou mít i Waldenströmovu makroglobulinemii [19].
Poškození jiných orgánů vazbou monoklonálního imunoglobulinu na jejich antigeny
Ve výjimečných případech měl monoklonální imunoglobulin IgM charakter protilátky proti bazální membráně glomerulů, proti strukturám kůže či retiny. Důsledkem byla glomerulonefritida, paraneoplastický pemphigus či retinitida [34]. Výjimečný je popis Fanconiho syndromu (porucha proximálního tubulu s výraznými ztrátami fosfátů, kyseliny močové, glukózy, aminokyselin, nízkomolekulárních bílkovin typu β-2-mikroglobulinu a prealbuminu. Fanconiho syndrom byl v těchto případech často spojený s proximálním typem renální acidózy a osteomalacií [35].
Příznaky způsobené maligní infiltrací
Lymfadenopatie, případně extralymfatické infiltráty
Lymfadenopatii popisuje Stalnikiewitz [11] jenom u 7 % nemocných. Dimopoulos [16] uvádí, že pouze 1/3 nemocných má zřetelnou lymfadenopatii, splenomegalii či hepatomegalii.
Příznaky z extralymfatické infiltrace jsou vzácné, jsou však možné a mohou postihnout kterýkoliv orgán či kůži [36].
Mezi vzácné komplikace patří kožní projevy, makuly, papuly, v nichž lze histologicky prokázat lymfoplazmocytární infiltráty [37].
U 3 - 5 % nemocných jsou detekovány infiltrace v plicích (noduly či difuzní infiltráty) nebo plicní výpotky [38,39].
Ojediněle se vyskytuje difuzní infiltrace CNS. Předpokládá se, že dlouhotrvající hyperviskozita poškodí vaskulární permeabilitu v mozku a umožní perivaskulární infiltraci lymfoplazmocytoidními buňkami, což může vést ke zmatenosti, ztrátě paměti, dezorientaci, poruchám motoriky a rozvoji komatózního stavu [40-43].
Anémie, trombocytopenie a vícečetná cytopenie (cytopenie postihující 2 a více linií)
Stalnikiewicz [11] definuje ve své studii multilineární cytopenii poklesem koncentrace hemoglobinu pod 120 g/l, poklesem počtu trombocytů pod 150 × 109/l a poklesem počtu bílých krvinek pod normu. Celkem 16 % pacientů splnilo tato kritéria. Výjimečně může být vstupním příznakem vysoce závažná multilineární cytopenie, kdy pacient přichází s hodnotami odpovídajícími závažnému útlumu krvetvorby.
Cytopenie, postihující více linií, může být primárním projevem nemoci v době stanovení diagnózy, ale většinou se vyvíjí později, v průběhu nemoci.
Osteolýza
Osteolytické defekty, které jsou typické pro mnohočetný myelom, výjimečně provázejí i jiné lymfoproliferativní onemocnění a tedy i Waldenströmovu makroglobulinemii [44-46].
Komplikace v průběhu nemoci
V průběhu této nemoci je popisován zvýšený výskyt sekundárních maligních chorob, Stalnikiewicz udává 11 % [11]. Garcia-Sanz [56] zaznamenal v průběhu choroby další maligní onemocnění u 7,8 % léčených a 5,3 % zatím neléčených osob.
Nemoc samotná způsobuje defekt imunity a ten je dále prohlubován léčbou, takže jsou možné nejen běžné, ale i oportunní infekce, a dokonce byla popsána progresivní multifokální leukoencefalopatie, způsobené JC polyomavirem [47].
Definice Waldenströmovy makroglobulinemie a stanovení diagnózy
Definice nemoci
Waldenströmova makroglobulinemie je dnes přesně pojmenovanou histopatologickou jednotkou, definovanou přítomností lymfoplazmocytárního lymfomu a monoklonální IgM gamapatie. Na rozdíl od mnohočetného myelomu není v definici Waldenströmovy makroglobulinemie uváděna koncentrace monoklonálního imunoglobulinu typu IgM (tab. 3). Ta totiž neodráží masu maligních buněk, neboť ty jej zřejmě někdy produkují více a jindy méně. Proto i závěry několika větších klinických studií ukázaly, že koncentrace monoklonálního imunoglobulinu má malý nebo žádný prognostický význam [48-56]. Dříve tradovaná diskriminační hranice 30 g/l monoklonálního IgM ztratila s přibývajícím poznáním svůj význam. Je sice zřejmé, že u pacientů s MGUS nebo jinými lymfoproliferativními chorobami nepřekračuje koncentrace monoklonálního IgM obvykle 30 g/l, nicméně mnozí pacienti s koncentrací monoklonálního IgM pod 30 g/l mohou mít symptomatickou formu Waldenströmovy nemoci [4].
![Diagnostická kritéria Waldenströmovy makroglobulinemie [54].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/4083137d7267bc1de89f16bf358c1079.jpg)
Histologický průkaz infiltrace kostní dřeně je zásadní pro stanovení této diagnózy, neboť je přítomna ve všech případech. Je tvořena malými lymfocyty se zřetelnou diferenciací směrem k plazmacytoidním buňkám a plazmocytům. Ve většině případů jde o intertrabekulární typ infiltrace. Pokud je infiltrace pouze paratrabekulární, je to neobvyklé a v těchto případech je nutno diferenciálně diagnosticky odlišit folikulární lymfom, zvláště pokud je přítomna lymfadenopatie. Kombinace cytomorfologie, typu infiltrace a imunofenotypu umožňuje dobře definovat tuto jednotku.
Biopsie kostní dřeně je proto obligátním vyšetřením pro stanovení této diagnózy, zatímco exstirpace lymfatické uzliny je doporučena pouze v těch případech, kdy je lymfadenopatie patrná a dobře dostupná. Lymfadenopatie bez infiltrace kostní dřeně činí diagnózu této nemoci málo pravděpodobnou.
Biopsie kostní dřeně je proto doporučována u všech nemocných s monoklonálním imunoglobulinem typu IgM, u nichž klinické nebo laboratorní parametry odpovídají přítomnosti lymfoproliferativního onemocnění, nebo u těch pacientů, kteří mají přítomno poškození organizmu monoklonálním imunoglobulinem, zvláště je-li zvažována cytoredukční léčba. Přínos případného provedení trepanobiopsie u zcela asymptomatického člověka je méně jasný [57].
Morfologický nález v kostní dřeni a imunofenotyp patologických buněk
Infiltrace dřeně lymfoplazmocytárním lymfomem může nabývat difuzní nebo intersticiální formu. Pouze paratrabekulární infiltrace je podezřelá z folikulárního lymfomu.
Infiltráty se skládají ze směsi malých lymfocytů, lymfoplazmocytoidních buněk a zralých plazmatických buněk. Autoři histopatologických studií uvádějí, že ve 39 % vzorků bylo rovnoměrně zastoupeno celé spektrum, od malých lymfocytů přes plazmocytoidní buňky po plazmatické buňky, v dalších 39 % byla predominance malých lymfocytů s malým počtem plazmocytů a plazmocytoidních buněk a ve 22 % případech se jednalo o směs malých lymfocytů a plazmocytů s malým počtem plazmocytoidních buněk. Počet mastocytů byl zvýšen u 26 % vyšetřených vzorků [58]. Dle současných poznatků mastocyty podporují lymfoplazmocytární proliferaci. Aktivita angiogeneze v kostní dřeni byla u nemocných s aktivní nemocí zvýšená [59].
Imunofenotyp
Ve většině případů lymfocyty vykazují silnou expresi povrchového imunoglobulinu IgM, dále exprimují pan-B znaky: CD19, CD20, CD22 a CD79. Plazmocytární komponenta exprimuje znak CD38. Patologické lymfocyty obvykle neexprimují CD5, CD10, CD23 nebo IgD [54], nicméně v literatuře lze nalézt popisy odchylek s přítomností CD10, CD23, CD79b, CD11c, CD25. Přítomnost znaku CD5, případně CD10 a CD23 je velmi neobvyklá, nevylučuje však tuto jednotku, nicméně v těchto případech je nutno velmi pečlivě odlišit chronickou lymfatickou leukemii a lymfom plášťové zóny [60,61]. Dalšími často se vyskytujícími znaky je CD25+, CD27+, FMC7+, BCL-2+ a CD52, zatímco znaky CD103 a CD138 jsou velmi, velmi vzácné [62]. Britské guidelines uvádí typický imunofenotyp následovně: CD5-, CD10-, CD19+, CD20+, CD22+, CD23-, CD25+, CD27+, CD75-, CD79+, CD103-, CD138-, FMC7+, BCL-1+, BCL-6-, PAX-5+ [3,4].
Míra plazmocelulární diferenciace může velmi kolísat, v některých případech může být počet plazmocytů velmi vysoký. V těchto případech je důležité prokázat povrchový imunoglobulin a/nebo B-buněčné antigeny. Pokud je infiltrát tvořen pouze plazmatických buňkami (cytoplazmatický IgM+, CD20-, CD138+), tak se nemůže jednat o lymfoplazmocytární lymfom a zřejmě se jedná o plazmocytom či mnohočetný myelom.
K diferenciální diagnostice myelomu a lymfomu pomůže cytogenetické vyšetření.
Vzhledem k tomu, že je sice definován typický imunofenotyp lymfoplazmocytárního lymfomu, ale zároveň byly v literatuře publikovány četné odchylky od tohoto typického genotypu, je morfologie základem pro stanovení diagnózy.
Imunofenotyp buněk odpovídá nejvíce postgerminálním paměťovým B-buňkám. Tato hypotéza je podporována analýzou genu pro těžký řetězec imunoglobulinu, který sice vykazuje somatickou hypermutaci, ale nevykazuje u většiny vyšetřených případů intraklonální diverzitu [3,4,63-66].
Cytogenetika
Při cytogenetickém vyšetření byly popsány poměrně četné numerické a strukturální abnormality, ale zatím nebyla popsána změna, která by jednoznačně definovala tuto chorobu.
Klasické cytogenetické studie jsou málo výtěžné pro nízký mitotický index nádorových buněk. Takže do roku 2006 bylo publikováno jen 5 studií analyzujících karyotyp, v rámci nichž bylo vyšetřeno 165 osob [67-73]. Frekvence cytogenetických abnormalit v těchto studiích byla malá, v průměru 35 % (17 - 37 %) a nejčastější byla delece 6q s výskytem v 6 - 16 %. Další nalezené abnormality byly delece 13q, častěji detekované u osob s pokročilou chorobou, výskyt trizomie chromozomu 5 a 8 byl < 10 % [71].
Konvenční cytogenetické vyšetření má u pacientů s IgM gamapatií malý či žádný význam, neboť většina nemá touto metodou detekován patologický karyotyp pro nízký proliferativní index klonálních B-buněk.
Delece 6q
Pro nízký mitotický index byla použita v dalších studiích fluorescenční in situ hybridizace (FISH) [71-77]. Delece dlouhého raménka chromozomu 6 byla pozorována až u 63 % jako nejčastější strukturální abnormalita [71]. I v dalších studiích byla prokázána nejméně u poloviny nemocných.
Prognostický význam delece 6q je uváděn některými autory jako nejasný [71], zatímco jiní je hodnotí jako nepříznivý prognostický faktor. Ocio et al [78] tuto abnormalitu detekoval v 7 % s pomocí klasické cytogenetiky, v 34 % při použití FISH a v 54 % při použití FISH s barvením cytoplazmatického imunoglobulinu. V jeho souboru 102 nemocných měla delece 6q nepříznivý prognostický význam. Pacienti s bezpříznakovou formou makroglobulinemie, mající deleci 6q, měli rychlejší přechod do symptomatické formy nemoci [78]. V další práci bylo prokázáno, že delece 6q nebyla přítomna u MGUS typu IgM, zatímco byla přítomna u 55 % pacientů s Waldenströmovou gamapatií. Přítomnost delece 6q tedy může tak odlišit Waldenstromövou makroglobulinemii od IgM MGUS. U nodální formy lymfoplazmocytárního lymfomu však tato delece nebyla nalezena.
Další typická aberace pro B-buněčné neoplazie, jako je ztráta RB1 nebo přestavba genu pro těžký řetězec (IgH přestavba), byly nalézány jen vzácně.
Nepřítomnost translokace zasahující IgH lokus
Pomocí fluorescenční interfázové in situ hybridizace (FISH) bylo zjištěno, že pro Waldenströmovu makroglobulinemii je charakterická nepřítomnost translokací, zasahujících lokus 14q32, obsahující genetickou informaci pro těžký řetězec imunoglobulinu.
Shop [70] vyšetřoval u 48 osob s Waldenströmovou makroglobulinemií přítomnost translokace t(9;14), tedy translokaci v oblasti genu pro těžký řetězec imunoglobulinu (14q32), která je přítomna u většiny B-lymfoproliferací. Žádná z těchto vyšetřovaným osob neměla translokaci t(9;14), v žádném vzorku nebyl přítomen další signál z oblasti lokusu 14q32 [72).
Pomocí fluorescenční in situ hybridizace (FISH) bylo prokázáno, že translokace postihující IgH lokus 14q32, kde se nachází gen pro těžký imunoglobulinový řetězec IgH, nebývají u Waldenströmovy makroglobulinemie přítomné.
Proto je možné použít vyšetření prokazující t(14;18) a t(11;14) v diagnosticky obtížných případech k odlišení vzácného případu IgM myelomu od Waldenströmovy makroglobulinemie. U IgM myelomu jsou naopak translokace postihující lokus 14q32 časté, konkrétně t(11;14)(q13;q32) byly prokázány u 7 z 8 případů a stejné zjištění je uvedeno v dalších pracích [79-87].
Analýzy genové exprese
První zprávy o analýzách genové exprese (gene-expression profilig) prokázaly poměrně homogenní genovou expresi nezávislou na přítomnosti či nepřítomnosti delece 6q a blíží se spíše genové expresi B-CLL anebo normálním buňkám. Pouze expresí několika genů se liší. Zvýšená exprese byla nalezena u genu pro IL6, který je důležitou součástí MAPK signální dráhy [75,76].
Základní vyšetření vhodná při podezření na Waldenströmovu makroglobulinemii
Vstupním základním vyšetřením je průkaz monoklonálního imunoglobulinu typu IgM v séru a případných volných lehkých řetězců v moči a jeho kvantitativního stanovení a sledování jeho kvantitativního vývoje. Je nutné, aby sledování kvantitativních změn probíhalo v jedné laboratoři, neboť na rozdíl od většiny biochemických hodnot, které jsou dnes standardizované, kvantitativní stanovení monoklonálního imunoglobulinu v sobě zahrnuje i subjektivní prvek, a proto se kvantity monoklonálního imunoglobulinu, stanovené v různých laboratořích, obvykle neshodují. Obvykle se stanovuje nejen monoklonální imunoglobulin, ale i polyklonální imunoglobuliny typu IgM IgG a IgA a CRP [88-95].
Další doporučená vyšetření:
- trepanobiopsie lopaty kosti kyčelní,
- jaterní enzymy, bilirubin, urea, kreatinin,
- vyšetření protilátek proti erytrocytům a při jejich pozitivitě doplnit vyšetření chladových aglutininů,
- vyšetření přítomnosti kryoglobulinu,
- β-2-mikroglobulin,
- CT vyšetření je vhodné u symptomatických nemocných před léčbou, ale není zcela nezbytné u asymptomatických nemocných,
- u pacientů s neuropatií EMG vyšetření. Pouze v případě dostupnosti se vyšetřují protilátky proti glykoproteinu asociovanému s myelinem, anti-MAG protilátky (anti - myelin asssociated glykoprotein antibodies), případně proti dalším antigenům. Tato vyšetření získávají na důležitosti, pokud jsou prováděna před případnou cytoreduktivní léčbou indikovanou pro neuropatii.
Diferenciální diagnóza monoklonální IgM gamapatie
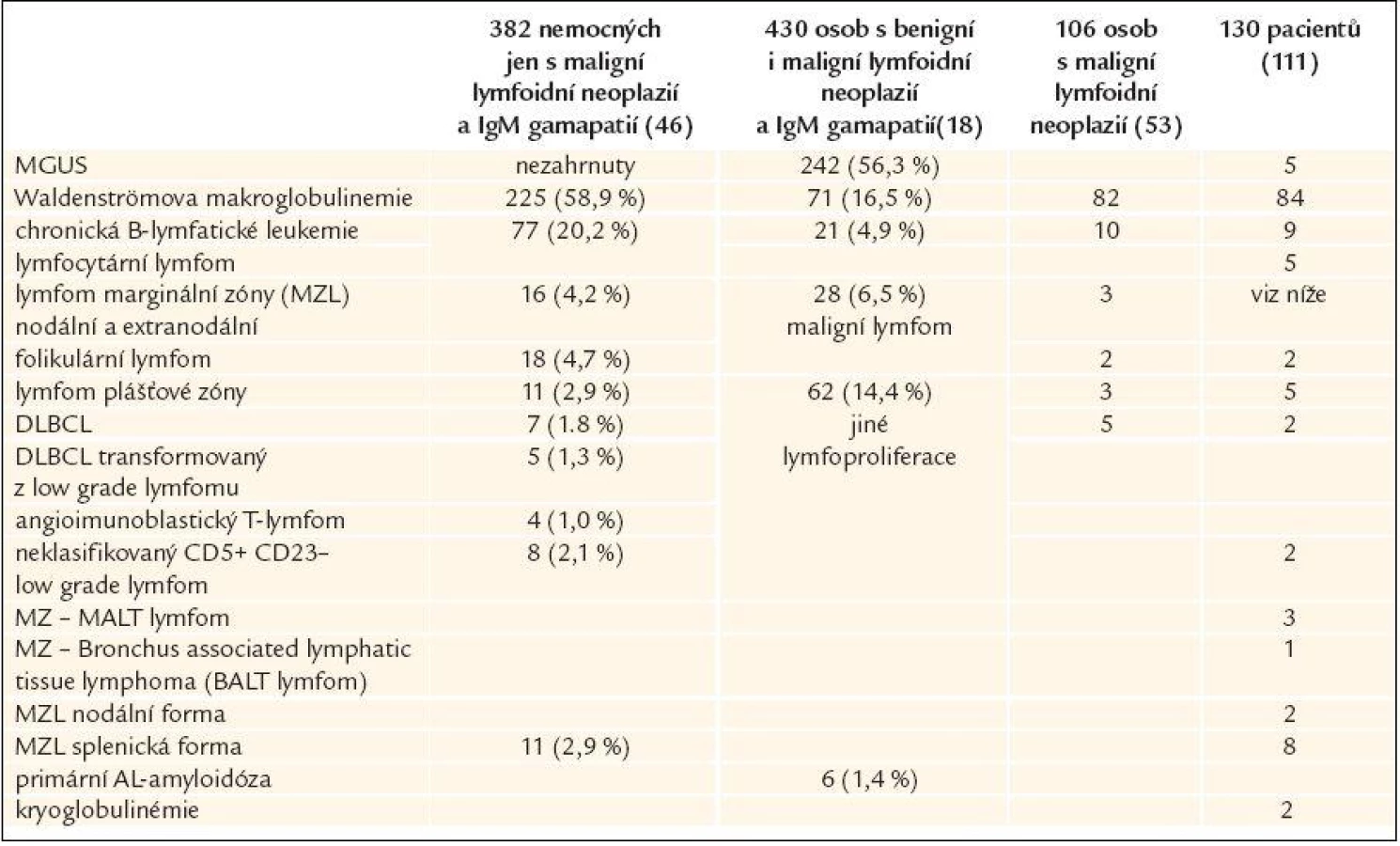
Monoklonální imunoglobulin typu IgM může provázet většinu lymfoproliferací, obvykle však ze skupiny nízce agresivních chorob (chronická B-lymfatická leukemie, vlasatobuněčná leukemie, splenický lymfom z vlasatých lymfocytů, mnohočetný myelom). Ve všech těchto případech však obvykle nepřesahuje koncentrace monoklonálního IgM 30 g/l. IgM koncentrace nad 30 g/l byla výlučně u pacientů s Waldenströmovou makroglobulinemií, ale většina nemocných s Waldenströmovou makroglobulinemií měla IgM pod 30 g/l. Přehled zastoupení jednotlivých stavů s průkazem monoklonálního IgM uvádí tab. 4 [96].
Monoklonální gamapatie nejistého významu typu IgM
Pokud je přítomen monoklonální imunoglobulin typu IgM a není prokázána žádná maligní choroba (negativní nález v biopsii kostní dřeně a nepřítomna lymfadenopatie) a nejsou příznaky poškození organizmu monoklonálním imunoglobulinem, tak v těchto případech mluvíme o monoklonální gamapatii nejistého významu typu IgM. Tyto odchylky od normy jsou náhodným nálezem při vyšetření prováděném z jiných příčin [88-92,95].
Nemoci způsobené monoklonálním imunoglobulinem
Pokud není prokazatelná maligní lymfoproliferace histologickým vyšetřením kostní dřeně (případně lymfadenopatie či jiné infiltrace), je přítomen monoklonální imunoglobulin a je diagnostikována kryoglobulinemie, nemoc chladových aglutininů, neuropatie nebo amyloidóza, řadíme tyto případy do skupiny nazvané nemoci způsobené monoklonálním imunoglobulinem (Monoclonal IgM related disorders). Monoklonální imunoglobulin je v těchto případech produkován malým množstvím klonálních buněk, které nejsou ještě detekovatelné morfologicky.
Asymptomatická forma Waldenströmovy makroglobulinemie
Pokud je histologicky potvrzena infiltrace kostní dřeně lymfoplazmocytárním lymfomem, jde o Waldenströmovu makroglobulinemii, nehledě na výši koncentrace monoklonálního imunoglobulinu. Pokud nejsou přítomny žádné známky nemoci vyjma infiltrace kostní dřeně a přítomnosti monoklonálního IgM, hovoříme o asymptomatické formě Waldenströmovy makroglobulinemie. Rozlišení od MGUS je možné pouze pomocí trepanobiopsie a histologického hodnocení kostní dřeně.
Symptomatická forma Waldenströmovy makroglobulinemie
Je přítomen typický nález v kostní dřeni a jsou projevy, způsobené infiltrací kostní dřeně (cytopenie a organomegalie) a/nebo poškozováním organismu monoklonálním imunoglobulinem (hyperviskozita, kryoglobulinemie, amyloidóza) a/nebo autoimunitní fenomény (periferní neuropatie, nemoc chladových gamaglobulinů). Vše shrnuje tab. 5.

Diferenciální diagnostika Waldenströmovy makroglobulinemie a mnohočetného myelomu
Klasická morfologická diferenciální diagnostika se nemusí vždy zdařit. Pacienti s mnohočetným myelomem typu IgM mívají méně intenzivní osteolýzu, takže ani absence osteolytických změn nelze brát jako jednoznačný argument pro Waldenströmovu makroglobulinemii. Průkaznější jsou zřejmě některé genetické znaky, jako je translokace zahrnující gen pro těžký řetězec imunoglobulinu v oblasti 14q32, jak je výše zmíněno v části o cytogenetice. V případech IgM myelomu je nejčastěji gen IgH translokován do oblasti 11q13 a vzniká tak translokace t(11;14)(q13;q32), která je jinak častá u lymfomů plášťové zóny (mantle cell lymphoma).
Prognóza symptomatické formy Waldenströmovy makroglobulinemie
Průměrné přežití pacientů s touto nemocí je 5 let, ale alespoň 20 % nemocných žije déle než 20 let a 10 - 20 % nemocných s touto nemocí umírá z jiné příčiny. Ve vzácných případech, kdy monoklonální IgM gamapatie je komplikována současně probíhající AL-amyloidózou, je průměrné přežití jen 11,1 měsíce. V průběhu nemoci může dojít ke zvratu v lymfoproliferativní onemocnění vyššího stupně malignity [48,56,97-100].
Poslední publikovanou analýzou prognostických faktorů je studie Mayo clinic. V letech 1960 a 2001 registrovali 337 pacientů. Medián přežití od stanovení diagnózy byl 6,4 roku. Medián „disease specific surfoval“ byl ale 11,2 roky. Univariantní analýza identifikovala následující nepříznivé faktory:
- věk nad 65 let,
- organomegalie (hepatomegalie, splenomegalie, ale ne lymfadenopatie),
- zvýšený β-2-mikroglobulin,
- anémie pod 100 g/l, leukopenie pod 4,0 × 109/1, trombocytopenie pod 150 × 109/1, snížený albumin pod 40 g/1‚
- koncentrace monoklonálního M-IgM vyšší než 40 g/l byla v této studii prognostickým faktorem, i když v jiných koncentrace monoklonálního Ig neměla prognostický význam.
Multivariantní analýza prokázala dva signifikantní negativní prognostické znaky: věk nad 65 let a organomegalie. Pacienti, kteří neměli ani jeden z nich, měli medián přežití 10,6 roku, pacienti s jedním 4,6 a pacienti se dvěma 3,1 let. Pokud byl β-2-mikroglobulin nad 4,0 mg/l, zvýšila se pravděpodobnost smrti 3krát [100].
Velmi zajímavým zjištěním z Mayo clinic je, že ač medián celkového přežití byl 6,4 roku, tak medián disease specific survival byl 11,2 roky. To znamená, že přežití těchto nemocných je podstatně delší než popisovaly starší studie. Úmrtí na Waldenströmovu makroglobulinemii pozorovali u 125 ze 237 pacientů (53 %). Myelodysplazie a leukemie postihly 7 % z 237 nemocných, tyto nemoci mohou být důsledkem léčby alkylačními cytostatiky.
Prognostické faktory byly analyzovány také v mnoha předchozích studií, neboť analýza prognostických faktorů je poměrně vděčné téma vědeckých prací. Také další studie potvrdily, že mezi nepříznivé prognostické faktory patří vyšší věk, anémie a cytopenie, snížená koncentrace albuminu, zvýšená koncentrace β-2-mikroglobulinu. Koncentrace monoklonálního imunoglobulinu má malý či žádný prognostický význam [48,49,52,98]. Prognostické indexy vytvořené z těchto ukazatelů uvádí tab. 6.

Bezpříznakové a celkové přežití excelentně koreluje s výší β-2-mikroglobulinu v době stanovení diagnózy, zatímco samotné dosažení léčebné odpovědi (alespoň PR) nebo časový interval do dosažení léčebné odpovědi nekoreloval s dobou bezpříznakového a celkového přežití [6]. Podobně v britské analýze nekorelovalo dosažení léčebné odpovědi na chlorambucil s délkou přežití [13], zatímco jiní autoři tuto souvislost popisují.
Internacionální prognostický index platný pro mnohočetný myelom má dle Dimopouluse [108] také platnost pro pacienty s myelomem. Medián přežití v době publikace nebyl dosažen pro I. stadium, u nemocných II. stadia byl 116 měsíců, u III. klinického stadia jen 54 měsíců.
Prognóza asymptomatické formy Waldenströmovy makroglobulinemie a IgM monoklonální gamapatie nejistého významu
Doba, po kterou může pacient zůstat bez symptomů nemoci, je delší než např. u asymptomatického myelomu [101] a udává medián do progrese 6,9 roku a popisuje 3 nepříznivé faktory související s rychlostí přechodu asymptomatické do symptomatické formy Waldenströmovy makroglobulinemie (koncentrace hemoglobinu pod 115 g/l, β-2-mikroglobulin 3 mg/l a více, koncentraci IgM nad 30,0 g/l). Pokud nebyl přítomen žádný z nepříznivých faktorů, byl interval do progrese 10 let, přítomnost jednoho rizikového faktoru zkrátila medián do progrese na 2 roky, přítomnost 2 již na půl roku [101]. V další analýze medián intervalu od stanovení asymptomatické formy Waldenströmovy makroglobulinemie do vzniku symptomů byl 7,8 roku [102].
Mora popsal následující nepříznivé faktory v univariantní analýze pro transformaci asymptomatické formy monoklonální IgM gamapatie, ale i IgM MGUS:
- zvyšující se míra infiltrace kostní dřeně lymfoplazmocytárními buňkami (nad 10 %),
- vysoká sedimentace (nad 40/hod),
- snižující se koncentrace hemoglobinu (pod 120 g/l),
- zvyšují se koncentrace monoklonálního IgM,
- lymfocytóza periferní krve nad 4 × 104/l,
- detekovatelná Benceova-Jonesova bílkovina v moči.
V multivariantní analýze pak výši koncentrace IgM a lymfocytóza v periferní krvi [103,104].
Společný prognostický index pro pacienty s MGUS a symptomatickou formou Waldenströmovy makroglobulinemie popsal Baldini [106] (tab. 7).

Závěr
Waldenströmova makroglobulinemie se vyznačuje velmi pestrým spektrem projevů a variabilitou průběhu. Považovali jsme za vhodné přinést přehled těchto klinických projevů a dále probrat otázky diferenciálně diagnostického odlišení této nozologické jednotky od dalších nemocí ze skupiny nízce maligních B-lymfoproliferací a mnohočetného myelomu typu IgM. Popsané imunohistochemické znaky této nemoci a výsledky cytogenetického případně molekulárně biologického vyšetření této nemoci umožní odlišit tuto nemoc od podobných jednotek s monoklonálním IgM imunoglobulinem.
Podporováno grantem LC06027 Masarykovy univerzity, Česká republika.
Prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 18. 7. 2007
Přijato po recenzi: 26. 9. 2007
Sources
1. Waldenström J. Incipient myelomatosis or essential hyperglobulinemia with fibrinogenopenia. A new syndrome? Acta Med Scand 1944; 117 : 217-247.
2. Desikan KR, Li Z, Jaganath S. Waldenströms makroglobulinemia: current therapy and future approaches. Bio Drugs 2002; 16 : 201-207.
3. Johnson SA, Birchall J, Luckie C et al. Guidelines on management of Waldenström’s macroglobulinemia. Brit J Haematol 2006; 132 : 687-697.
4. Johnson SA. Advances in the treatment of Waldenström’smacroglobulinemia. Exp Rev Anticancer Treatment 2006; 6: 329-334.
5. Klán J, Doležalová I, Pelíšková D et al. Waldenströmova makroglobulinémie jako model imunopatologie ve stáří. Geriatria 2004; 10 : 18-27.
6. Treon SP, Hunter ZR, Aggarwal A et al. Characterization of familiar Waldenström’smacroglobulinemia. Ann Oncol 2006; 17 : 488-494.
7. Tichý M, Urban P, Matěja P et al. Laboratorní analýza souboru 3 049 monoklonálních imunoglobulinů. Klin Biochem Metabol 2002; 10 : 257-261.
8. Tichý M. Viscosity of paraproteinemic séra. Acta Med 1996; 39 : 41-43.
9. Tichý M. Laboratorní analýza monoklonálních imunoglobulinů (paraproteinů). Český Těšín: Finidr 1997.
10. Rencová E, Malý J, Bláha M et al. Dynamika sítnicových změn v závislosti na léčbě Waldenströmovy choroby. Čs Oftal 1993; 49 : 3-7.
11. Stalnikiewicz L, Carrote-Lefebvre I, Detourmignies L et al. Prognostic factors in Waldenström’smacroglobulinemia. Description of the complication during the evolution - preliminary results on 101 patients. Semin Oncol 2003; 30 : 216-219.
12. Shihabi ZK Cryoglobulins: An important but neclected clinical test. Ann Clin Lab Science 2006; 36: 395-408.
13. Tichý M, Hrnčíř Z, Urban P et al. Monoklonální imunoglobuliny. Klin Biochem Metabol 2004; 12 : 84-87.
14. Tichý M. Monoklonální gamapatie. Labor Aktuel CS 2000; 7-10.
15. Daoud MS, Lust JA, Kyle RA. Monoclonal gammapaties and associated skin disorders. J Amer Acad Dermatol 1999; 40 : 507-535.
16. Dimopoulos MA, Kyle RA, Anagnastopoulos A et al. Diagnosis and management of Wadenström’s macroglobulinemia. J Clin Oncol 2005; 23 : 1564-1577.
17. Bryce AH, Kyle RA, Dispenzieri A et al. Natural history of therapy of 66 patients with mixed cryoglobulinemia. Amer J Hematol 2006; 81: 511-518.
18. Kyle RA, Gahrton JP. The spektrum of IgM monoclonal gammopathy in 430 cases. Mayo Clin Proc 1987; 62 : 719-731.
19. Stone MJ, Merlini G, Pascal V. Autoantibody activity in Waldenströms macroglobulinaemia. Clin Lymphoma 2005; 5 : 225-229.
20. Bonnetblanc JM, Bedane CH, Fazol J et al. Tumoral macroglobulinosis of the skin in situ release of IgM. Acta Dermatovenereol 2006; 86 : 63-64.
21. Dimopoulos MA, Panayiotis P, Moulapoulos LA et al. Waldenström’s makroglobulinemia: Clinical features, complication and management. J Clin Oncol 2000; 18 : 214-226.
22. Gertz MA, Kyle RA. Amyloidosis with IgM gammapathies. Semin Oncol 2003; 30: 325-328.
23. Gertz MA. Natural history and therapy of 66 patients with mixed cryoglobulinemia. Amer J Hematol 2006; 81 : 511-518.
24. Tichý M. Primární amyloidóza. Lék zpr Lék Fak Univ Karlovy Hr Králové 1999; 44 : 99-107.
25. Schimonová M, Zatloukal P, Havlíček F et al. Amyloidóza plic při Waldenstromövě chorobě. Stud Pneumol Phtiseol 1998; 58 : 72-74.
26. Okuda M, Okuda Y, Obuta T et al. Primary lung involvement with amyloid deposition in Waldenström macroglobulinaemia. Respirology 2004; 9: 414-418.
27. Gardyn J, Schwartz A, Gal R et al. Waldenström’s macroglobulinaemia associated with AA amyloidosis. Int J Hematol 2001; 74 : 76-78.
28. Gisseroth O, Landais C, Cremades S et al. Amyloid arthropathy and Waldenström’s macroglobulinemia. Joint Bone Spine 2006; 73 : 456-458.
29. Sanchorawala V, Blanchard E, Seldin DC et al. AL-amyloidosis associated with B cell lymphoproliferative disorders: frequency and treatment outcomes Amer J Hematol 2006; 81: 692-695.
30. Cesana C, Barbarami L, Miqueleiz S et al. Clinical characteristics and outcome of imunoglobulin-M related disorders. Clin Lymphoma 2005; 5: 261-264.
31. Levine T, Pestron A, Florence J et al. Peripheral neuropaties in Waldenström’s macroglobulinemia. J Neurol Neurosurg Psychiatry 2006; 77 : 224-228.
32. Pestronk A. Peripheral neuropathy in Waldenström’s macroglobulinemia. J Neurol Neurosurg Psychiatr 2006; 77 : 224-228.
33. Simovic D, Gorson, KC, Ropper AH. Comparison of IgM-MGUS and IgG MGUS polyneuropathy. Acta Neurol Scand 1998; 97 : 194-200.
34. Garcia-Pacheco H, Khan A, Venka KK Rapidly progressive glomerulonefritis in a patients with Waldenström’smacroglobulinemia. Clin Nephrol 2005; 64 : 396-399.
35. Bridoux F, Sirac Ch, Hugue V et al. Fanconi syndrome induced by monoclonal Vkappa3 light chain in Waldenström’s macroglobulinemia. Amer J Kidny disease 2005; 45: 749-757.
36. Yokote T, Akioka T, Oka S et al. Cutaneous infiltration with Waldenström macroglobulinemia. Leukemia Res 2006; 30 : 1207-1210.
37. Autier J, Buffet M, Pinquier L et al. Cutaneeous Waldenströms macroglobulinaemia with deck chair sign treated with cyclophosphamide. J Am Acad Dermatol 2005; 52 : 45-47.
38. Cerna M, Risik K, Jakubikova N. Lymfolazmocytoidní imunocytom - Waldenströmova choroba s postihnutím pluc. Stud Pneumol Phtiseol CS 1989; 49 : 198-202.
39. Pozdechova A, Virsik K, Tichá M et al. Intrathorakalne prejavy Waldenströmovej makroglobulinémie. Stud Pneumol Phtiseol CS 1980; 40: 599-604.
40. Kyrtsonis MC, Angelopoulou MK, Kontopidou FN et al. Primary lung involvement in Waldenström’s macroglobulinemia. Acta Hametol 2001; 105 : 92-96.
41. Kyrtsonis MC, Vassilakopoulos TP, Angelopoulou MK et al. Waldenstöm’s macroglobulinemia: clinical course and prognostic factors in 60 patients. Ann Hematol 2001; 80: 722-727.
42. Malý J, Tichý M, Blaha M et al. A case of acute Waldenström macroglobulinemia. Sbor Ved Praci Lék Fak Univ Karlovy Hrad Kr 1983; 26 : 165-173.
43. Civit T, Coulbouis S, Baylac F et al. Waldenström macroglobulinaemia and cerebral lymphoplamocytic proliferation. Neurochirurgie 1997; 43: 245-249.
44. Jondeau K, Alterescu R, Franc B et al. Unusual evolution of Waldenström’s macroglobulinemia into ostolytic myeloma. Eur J. Haematol 2006; 77 : 74-79.
45. Lin P, Buesco R, Wilson C et al. Wandenström macroglobulinaemia involving extramedulary sites. Amer J Surg Patol 2003; 27 : 1104-1113.
46. Lin P, Hao S, Handy BC et al. Lymphoid neoplasms associated with IgM paraprotein. Am J Clin Pathol 2005; 123 : 200-205.
47. Slavin ChM, Seymour J Progressive multifocal leucoencefalopahty complicating Waldenström’s macroglobulinemia. Leukemia Lymphoma 2003; 44 : 1819-1821.
48. Facon T, Brouillard M, Duhamel A et al. Prognostic factors in Waldenström’s macroglobulinaemia: a report of 167 cases. J Clin Oncol 1993; 11 : 1553-1558.
49. Gobi PG, Bettini R, Montecucco C et al. Study of prognosis in Waldenström’s macroglobulinaemia. Blood 1994; 83 : 1452-1459.
50. Morel P, Monconduit M, Jacomy D et al. Prognostic factors in Waldenström’s macroglobulinaemia. A report on 232 patients with the description of a new scoring system and its validation on 253 other patients. Blood 2000; 96 : 852-858.
51. Morel, P, Duhamel A, Gobbi P et al. International Prognostic Scoring system for Waldenström’s Macroglobulinemia. Blood 2006; 108 : 42a.
52. Owen RG, Barrans SL, Richards SJ et al. Waldenström’s macroglobulinemia: Developement of diagnostic criteria and identification of prognostic factors. Am J Clin Pathol 2001; 116 : 420-428.
53. Owen RG, Parapia LA, Higginson J et al: Clinicopathological correlates of IgM paraproteinemias. Clin Lymphoma 2000; 1 : 39-43.
54. Owen RG, Treon SP, Al-Katib A et al. Clinicopathological definition of Waldenström’s macroglobulinaemia. Consensus panel recommendation from the Second International Workshop on Waldenström’smacroglobulinemia. Semin Oncol 2003; 30 : 567-584.
55. Owen RG, Treon SP, AL-Katib A et al. Clinicopathological definition of Waldenström’s macroglobulinemia. Semin Oncol 2003; 30 : 110-115.
56. Garcia-Sanz R, Montoso S, Torrequebrada A et al. Waldenström’s macroglobulinemia. Presenting features and outcome in a series of 217 patients. Br J Haematol 2001; 115 : 575-582.
57. Rajkumar SV Monoclonal gammopathy of unknown significance, Waldenström’s macroglobulinaemia, AL-amyloidosis and related plasma cell disorders. Diagnosis and treatment. Mayo Clin Proc 2006; 81 : 893-703.
58. Remstein ED, Hanson CA, Kyle RA et al. Despite apparent morphologic and immunophenotypic heterogenity Waldenoström´s macroglobuliniemia is consistently composed of cell showing morpholic continuum of small lymphocytes, plasmocytoid lymphocytes and plasma cells. Semin Oncol 2003; 30: 182-186.
59. Rajkumar V, Haman S, Greip FR. Angiogenesis in Waldenström’s macroglobulinaemia. Semin Oncol 2003; 30 : 262-264.
60. Konoplev S, Madeiro J, Bueso-Ramos CE et al. Immunophenotypic profile of lymfoplasmocytic lymphoma/Waldenström’s macroglobulinaemia. Am J Clin Pathol 2005; 124: 414-420.
61. Hunter ZR, Branagan AR, Mannin R et al. CD5, CD10 and CD23 expression in Waldenström’smacroglobulinaemia. Clin Lymphoma 2004; 5: 246-249.
62. San Miguel JF, Vidriales MB, Ocio E et al. Immunopfenotypic analysis of Waldenström’s macroglobulinaemia. Semin Oncol 2003; 30: 187-195.
63. Sahota SS, Forconi F, Ottensmeier CH et al. Origins of the malignant clone in typical Wandenström´s macroglobulinaemia. Semin Oncol 2003; 30 : 136-141.
64. Kriangkum J, Taylor BJ, Strachan E et al. Impaired class switch recombination in Waldenström’smacroglobulinemia despite apparently norme CSR machinery. Blood 2006; 107 : 2920-2927.
65. Kriangkum J, Tailor BJ, Reimann T et al. Origins of Waldenström’s macroglobulinemia: does it arise from a unusual B cell precursor? Clinical Lymphoma 2005; 5 : 217-219.
66. Rollett RA, Wilkinson EJ, Gonzalez D et al. Immunoglobulin Haevy Chain sequence analysis in Waldenström’s macroglobulinemia and immunoglobulin M monoclonal gammapathy of undetermined significance. Clin Lymphoma Myeloma 2006; 7 : 70-72.
67. Calasanz MJ, Cigudosa JC, Odero MD et al. Cytogenetic analysis of 280 patients with multiple myeloma and releated disordes. Genes chromosomes Cancer 1997; 18 : 84-93.
68. Louviaux I, Michaux L, Hagemeier A et al. Cytotenetic abnormalities in Waldenström’sisease. Blood 1998; 92 (Suppl 1): Abstr. 3776.
69. Mansoor A, Medeiros LHJ, Weber DM et al. Cytogenetic findings in lymfoplasmocytic lymphoma. Chromosomal abnormalities are associated with polymorphous subtype and an aggressive clinical course. Amer J Clinic Pathol 2001; 116 : 543-549.
70. Schop RFJ, Jalal SM, Van Wier SA et al. Deletions of 17p13.1 and 13q14 are uncomon in Waldenström macroglobulinemia clonal cells and mostly seen at the time of disease progression. Cancer Genetics Cytogenetics 2002; 132 : 55-60.
71. Schop RFJ, Krehl WM, Van Wier SA et al. Waldenström’s macroglobulinemia: neoplastic cells lack IgH translocation but have frequent 6q deletions. Blood 2002; 100 : 2996-3001.
72. Schop RFJ, Van Wier SA, Xu R et al. 6q deletion discriminates Waldenström’s macroglobulinemia from IgM monoclonal gammapathy of undetermined significance. Cancer Genetics Cytogenetics 2006; 169: 150-153.
73. Treon SR, Morton C, Braverman E et al. Deletion in 6q21-22 are commonly present in patients with familiar and non-familiar Waldenström’s macroglobulinemia. And IgM monoclonal gammapathy of unknown significance. Blood 2003; 102: Abstr. 2526.
74. Shaw GR, Kronberg DL. P53 deletion but not trisomy 12 are adverse in B cell lymphoproliferative disorders. Cancer Genetics Cytogenetics 2000; 119 : 146-154.
75. Chang H, Saymour S, Li D et al. Analysis of IgH translocation chromosome 13q14 and 17p13.1 (pp3) deletions by fluorescent in situ hybridization in Waldenström’s makroglobulinemia. Leukemie 2004; 18 : 1160-1162.
76. Chang WJ, Schop RF, Price-Troska T et al. Gene expression profiling of Waldenström’smacrogobulinemia reveals phenotype more similar to chronic lymphocytic leukemia then multiple myeloma. Blood 2006; 108 : 2755-2763
77. Wong KF. Waldenström’s macroglobulinaemia with karyotypic aberrations involving both homologous 6q. Cancer Genet Cytogenet 2001; 124: 137-139
78. Ocio, EM, Hernánez JH, Mateo G et al. Immunophenotypic and cytogenetic comparison of Waldenström’s macroglobulinemia and splenic marginal zone lymphoma. Clin Lymphoma 2005; 5 : 241-245.
79. Avet-Loiseau H, Garand R, Lodé L et al. 14g32 translocations discriminate IgM multiple myelom from Waldenström’s macroglobulinemia. Semin Hematom 2003; 30 : 153-155.
80. Johnson SA. Waldenström macroglobulinaemia. Rev Clin Exp Hematol 2002; 6 : 421-334.
81. Avet-Loiseau H, Facon T, Grosbois B et al. Oncogenesis in multiple myeloma: 14q32 and 13q chromosom abnormalities are non randomly distributed but correlate with natural history immunological features and clinical presentation. Blood 2002; 99 : 2185-2191.
82. Konduri K, Sahota SS, Babbage G et al. Imunoglobulin M myeloma Evaluation of molecular features and cytokine expression. Clin Lymphoma 2005; 5: 285-289.
83. Pilarski L. Impaired recombination in Waldenström’s macroglobulinemia. Blood 2006; 107 : 2920-2927
84. Ocio EM, Schop RFJ, Gonzales B et al. 6q deletion in Waldenström macroglobulinaemia is associated with features of adverse prognosis. Brit J Haematol 2006; 136 : 80-86.
85. Owen RG. Immunoglobulin heavy chain sequences analysis in Waldenström’smacroglobulinemia and imunoglobulin M monoclonal gammopathy of undetermined significance. Clin Lymphoma Myeloma 2006; 7 : 70-72.
86. Ackroyd S, O’Connor SJM, Owen RG Rarity of IgH translocations in Waldenström’smacroglobulinaemia. Cancer Genet Cytogenet 2005; 163 : 77-80.
87. Cook JR, Aguilera NI, Reshmi S et al. Deletion of 6q is not a characteristic marker of nodal lymphoplasmocytic lymphoma. Cancer Genet Cytogenet 2005; 162 : 85-88.
88. Ščudla V. Význam sérových hladin volných lehkých řetězců imunoglobulinů v diagnostice a hodnocení aktivity mnohočetného myelomu. Vnitř Lék 2005; 51 : 1249-1259.
89. Tichý M, Friedecký B, Vávrová J et al. Standardizace biochemických laboratorních vyšetření u mnohočetného myelomu. Klin Biochem Metabol 2006; 14 : 8-13.
90. Tichý M, Gregor J, Holečková J et al. C-reaktivní protein. Labor Aktuell CS 2002; 8-12.
91. Tichý M, Hrnčír M, Mráček J. Simultaneous occurrence of IgM lambda wit pyroprecipitation properties and cryoglobulin in the serum of patient with Waldenström’smacroglobulinemia. Neoplasma 1978; 25 : 741-744.
92. Tichý M, Stulík M, Kovářová H et al. Použití dvojrozměrné chromatografie k analýzy monoklonálních lehkých řetězců imunoglobulinů v moči. Voj Zdrav Listy 1994; 63 : 79-83.
93. Tichý M. Paraproteinémie nejasného původu. Klin Biochem Metabol 1998; 6 : 172-175.
94. Tichý M. Stanovení monoklonálních imunoglobulinů a jejich fragmentů u monoklonálních gamapatií. Vnitř Lék 2005; 51 : 1225-1227.
95. Sakalová A, Mistrík M, Škultétyová D et al. Waldenströmova makroglobulinémie, aký je súčasný stav? Hemat Transfuz 1999; 9 : 56-60.
96. Kyle RA, Rajkumar S, Therneau TM et al. Prognostic factors and prediction of outcome of immunoglobulin M monoclonal gammopathy of undetermined significance. Clin Lymphoma 2005; 5 : 257-260.
97. Kyle RA, Rajkumar SV. Monoclonal Gammapaties of undertermined significance. Hematol Oncol Clin North Amer 1999; 13 : 1181-1201.
98. Kyle RA, Treon SP, Alexanian R et al. Prognostic markers and criteria to initiate therapy in Waldenström’smacroglobulinemia. Consensus panel recommendation from the second international Workshop on Waldenström’smacroglobulinemia. Semin Oncol 2003; 30: 116-120.
99. Klán J, Topinková E, Pelíšková D et al. Waldenströmova makroglobulinémie v geriatrické praxi. Čes Geriat Rev 2004; 2 : 40-44.
100. Ghobrial IR, Fonseca R, Gertz MA et al. Prognostic model for disease specific and overall mortality newly diagnosed symptomatic patients with Waldenström’smacroglobulinaemia. Brit J Haematol 2006; 133 : 158-1654.
101. Alexanian R, Weber D, Delasalle F et al. Asymptomatic Waldenström’s macroglobulinemia. Semin Oncol 2003; 30 : 206-210.
102. Cesana C, Miquelei S, Bernuzzi P et al. Smouldering Waldenstrom macroglobulinaemia: Factors predicting evolution to symptomatic disease. Semin Oncol 2003; 30: 206-210.
103. Morra E, Cesana C, Klersy C et al. Predictive variables for malignant transformation on 452 patients with asymptomatic IgM monoclonal gammopathy. Semin Oncol 2003; 30 : 172-177.
104. Morra E, Cesana C, Klersy C et al. Prognostic factors for transformation in asyptomatic imunoglobulin IgM monoclonal gammapathies Clinical Lymphoma 2005; 5: 256-269.
105. Baldini L, Goldaniga M, Guffanti A et al. Imunoglobulin M monoclonal gammapathy of undetermined significance and indolent Waldenström’s macroglobulinaemia recognize the same determinants of evolution into symptomatic lymphoid disorders: A proposal for a commmon prognostic scoring system. J Clin Oncol 2005; 23 : 4662-4668.
106. Baldini L, Goldaniga M, Guffanti A et al. Imunoglobulin M monoclonal gammapathy of undetermined significance and indolent Waldenström’s macroglobulinaemia recognize the same determinants of evolution into symptomatic lymphoid disorders: A proposal for a commmon prognostic scoring system. J Clin Oncol 2005; 23 : 4662-4668.
107. Dhodapkar MV, Jacobson JL, Gertz MA et al. Prognostic factors and response to fludarabine therapy in Waldenström’s macroglobulinemia: an update of a US Intergroup trial. Semin Oncol 2003; 30 : 220-225.
108. Dimopoulos MA, Gika D, Zervas K et al. The international staging system for multiple myeloma is applicable in symptomatic Waldenström’s macroglobulinaemia. Leukemia Lymphoma 2004; 45 : 1809-1813.
109. Merlini G, Baldini L, Broglia C et al. Prognostic factors in symptomatic Waldenström’s macroglobulinaemia. Semin Oncol 2003; 30 : 211-215.
110. Nagan S, Rohatiner AZS, Matthews J et al. Waldenström’s macoglobulinaemia: A restrospective analysis of 40 patients from 1972-2001. Semin Oncol 2003; 30 : 236-238.
111. Pangalis GA, Kyrtsonis MCh, Kontopidu FN et al. Differential diagnosis of Waldenström’s macroglobulinemia and other B cell disorders. Clin Lymphoma 2005; 5: 235-240.
112. Schnitzler L, Hurez D, Verret JL et al. Chronic urtica and hyperostosis in macroglobulinemia. Ann Dermatol Venereol 1989; 116: 547-550.
113. Tichý M. Turbidimetrické stanovení lehkých řetězců imunoglobulinů. Labor Aktuel CS 1998; 6-10.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2007 Issue 12
Most read in this issue
- Current view on the risks of artificial pulmonary ventilation
- Recurrent arrhythmias after catheter ablation of originally paroxysmal atrial fibrillation and results of repeat ablation
- Waldenström macroglobulinemia – clinical manifestations and differential diagnosis and prognosis of the disease
- Diabetes mellitus and microalbuminuria