
Glukokortikoidy a osteoporóza
Glucocorticoids and osteoporosis
Treatment with glucocorticoids (GC) has no alternative in many medical disciplines for their anti-inflammatory and immunosuppressive effect. However, osteoporosis and the related fractures are a serious complication brought about by longterm GC therapy. The risk of fractures, especially of the vertebras and the ribs, becomes higher as early as in the first months of oral GC therapy. It grows in proportion to the daily dose of GC, and is present even if low doses are administered (2.5–7.5 mg of prednisone per day). Decreasing bone density (BMD) is not accountable for the higher risk of fractures in GC therapy and fractures occur with higher values of BMD than in primary osteoporosis. There is still no tool that we could use to quantify the changes in the bone quality and the increased risk of fracture in clinical practice. The principal mechanism by which GC induces osteoporosis is inhibition of bone formation caused by the suppression of osteoblastogenesis as well as the activity of functional osteoblasts, with accelerated osteocyte and osteoblast apoptosis. There are significant differences between individuals in terms of GC sensitivity, the reasons of which have not yet been explained. Prior to planned long-term GC therapy (> 3 months) with daily doses higher than 2.5 mg of prednisone p.o. (or higher doses of inhaled GC), it is recommended to perform a densitometry exam using dual-energy X-ray absorptiometry (DXA) in the lumbar region of the spine and femoral collum to evaluate additional risk factors of osteoporosis and fractures for a more precise estimate of the risk of fracture in the specific patient. Sufficient intake of calcium (1,000–1,500 mg of elementary calcium per day) and of the vitamin D (800 IU per day) should be assured in all patients treated by GC. Endogenous production of sexagens should be evaluated and possible substitution therapy should be considered in premenopausal women and younger men. Today, bisphosphonates can be given to patients with a high risk of fracture, the effects of which in preventing the decrease of BMD and vertebral fractures have been documented in randomised clinical studies, even though the evaluation of the risk of fractures was not the primary endpoint of those studies. However, in view of the antiremodelling effect of bisphosphonates, it is clear that this therapy does not eliminate the cause of GC induced osteoporosis and drugs with stimulating effect on osteoblasts will certainly be preferred in the future. Very promising are the first clinical studies of injection parathormone (PTH 1-34) which stimulated bone formation in a continuing GC treatment.
Key words:
glucocorticoids – osteoporosis – bone metabolism – bone formation – bone resorption – individual sensitivity to glucocorticoids – calcium – D vitamin – estrogens – testosterone – bisphosphonates – osteoanabolic treatment
Authors:
doc. MUDr. Vít Zikán, Ph.D.
Authors‘ workplace:
III. interní klinika 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Štěpán Svačina, DrSc., MBA
Published in:
Vnitř Lék 2007; 53(7-8): 831-840
Category:
Overview
Léčba glukokortikoidy (GK) je v mnoha medicínských oborech nezastupitelná vzhledem k jejímu protizánětlivému a imunosupresivnímu účinku. Vážnou komplikací dlouhodobé léčby GK je však osteoporóza a s ní spojené zlomeniny. Riziko zlomenin, zejména obratlů a žeber se zvyšuje již v prvních měsících po zahájení perorální léčby GK. Riziko zlomenin se zvyšuje v závislosti na denní dávce GK, ale rizikové je užití i nízkých dávek (2,5-7,5 mg prednisonu/den). Zvýšení rizika zlomenin není vysvětlitelné poklesem kostní denzity (BMD) při léčbě GK a ke zlomeninám dochází při vyšších hodnotách BMD než u primární osteoporózy. Zatím není k dispozici nástroj, který by dokázal tyto změny kvality kosti a zvýšené riziko zlomenin v klinické praxi kvantifikovat. Hlavní mechanizmus, kterým GK navozují osteoporózu, je útlum kostní novotvorby způsobený potlačením osteoblastogeneze i aktivity funkčních osteoblastů s urychlením apoptózy osteocytů a osteoblastů. Mezi jednotlivci existují významné rozdíly v citlivosti na GK, jejichž příčiny se zatím nepodařilo objasnit. Před plánovanou dlouhodobou léčbu GK (> 3 měsíce) v denních dávkách vyšších něž 2,5 mg prednisonu p.o. (nebo u vyšších dávek GK v inhalační formě) je doporučeno provést denzitometrické vyšetření pomocí dvouenergiové RTG absorpciometrie (DXA) v oblasti bederní páteře a krčku stehenní kosti a zhodnotit další rizikové faktory osteoporózy a zlomenin, které upřesní odhad rizika zlomeniny u jednotlivého pacienta. U všech pacientů léčených GK je vhodné zajistit dostatečný příjem vápníku (1 000 - 1 500 mg elementárního vápníku/den) a vitamin D (800 IU/den). U premenopauzálních žen a mladších mužů je vhodné zhodnotit endogenní produkci sexaenů a případně zvážit substituční léčbu. U pacientů s vysoký rizikem zlomeniny můžeme v současné době užít bisfosfonáty, jejichž účinky v prevenci úbytku BMD a snížení rizika zlomenin obratlů byly dokumentovány v randomizovaných klinických studiích, i když hodnocení rizika zlomenin nebylo primárním cílem provedených studií. Vzhledem k antiremodelačnímu účinku bisfosfonátů je ale zřejmé, že tato léčba neodstraňuje příčinu GK indukované osteoporózy a v budoucnosti budou jistě upřednostňovány léky se stimulujícím účinkem na osteoblasty. Velmi slibné jsou první klinické studie s injekčním parathormonem (PTH 1-34), který i při pokračující léčbě GK stimuluje kostní novotvorbu.
Klíčová slova:
glukokortikoidy - osteoporóza - kostní metabolizmus - kostní novotvorba - kostní resorpce - individuální citlivost na glukokortikoidy - vápník - vitamin D - estrogeny - testosteron - bisfosfonáty - osteoanabolická léčba
Úvod
Léčba glukokortikoidy (GK) je v mnoha medicínských oborech nezastupitelná vzhledem k jejímu protizánětlivému a imunosupresivnímu účinku. Dlouhodobější nadbytek GK ať již endogenní (Cushingův syndrom) nebo exogenní (dlouhodobá léčba GK) má ale pro organizmus řadu nepříznivých následků. Zvláště závažnou komplikací dlouhodobé expozice nadbytku GK je osteoporóza a následné zlomeniny, zejména v oblastech s vyšším zastoupením trámčité kosti (obratlová těla, žebra a krček stehenní kosti). Zlomeniny snižují kvalitu života, invalidizují a ve svých důsledcích zkracují život postižených osob. Porozumění komplexní patofyziologii GK indukované osteoporózy je východiskem pro nalezení vhodné prevence a léčby této závažné komplikace dlouhodobé léčby GK. Cílem tohoto článku je stručné shrnutí současných znalostí z patofyziologie glukokortikoidy indukované osteoporózy a jejich využití pro preventivní a léčebná opatření.
Epidemiologie
Odhaduje se, že GK užívá 0,5-0,9 % dospělé populace, avšak prevalence se zvyšuje s věkem a ve skupině osob nad 70 let věku dosahuje podle různých analýz 2,5-5,2 % [1,2]. GK indukovaná osteoporóza se vyvine přibližně u 30-50 % pacientů, kteří jsou léčeni orálními GK alespoň 6 měsíců [3,4]. Inhalační kortikoidy také nejsou zcela bez negativního vlivu na skelet, jak prokazují některé studie hodnotící změny kostní denzity [5].
Epidemiologické studie svědčí pro nejméně dvojnásobné zvýšení rizika zlomeniny obratle a proximálního femoru u pacientů léčených orálními GK [1,4,6,7]. Největší epidemiologická studie, která zpracovala data z rozsáhlé databáze lékařů ve Velké Británii (General Practice Research Database), zahrnula více než 240 000 pacientů léčených orálními GK a kontrolní skupinu neléčených pacientů odpovídajícího věku a pohlaví [4]. Bylo zjištěno, že riziko zlomeniny závisí na užité denní dávce GK. Riziko klinických zlomenin obratlů u pacientů s denní dávkou 2,5-7,5 mg prednisonu je zvýšeno 2,6krát, při denní dávce prednisonu nad 7,5 mg je riziko zlomeniny obratle zvýšeno 5,2krát a zlomeniny krčku femoru 2,3krát [4]. Klinicky důležité je zjištění, že riziko zlomenin se zvyšuje poměrně rychle během 3-6 měsíců po zahájení léčby GK a k nárůstu rizika zlomenin dochází již při relativně nízkých dávkách prednisonu. Po přerušení léčby GK se riziko zlomenin snižuje [4,7]. Prevalence zlomenin obratlů u pacientů dlouhodobě léčených glukokortikoidy je však v epidemiologických studiích částečně podhodnocena, jelikož byly hodnoceny pouze klinické (symptomatické) zlomeniny obratlů. Je přitom dokumentováno, že pouze 30 % zlomenin obratlů je symptomatických. Angeli et al [8] zjistili na souboru 551 pacientů léčených GK, že prevalence asymptomatických (radiologických) zlomenin obratlů je vyšší než 37 %, z toho > 14 % pacientů mělo 2 a více asymptomatických zlomenin obratlů.
Mechanizmy působení glukokortikoidu na kostní metabolizmus
Působení GK na trámčitou kost je hlavním predisponujícím faktorem zlomenin obratlových těl, kde tvoří trámčitá kost přibližně 66 %, zlomenin žeber a zlomenin krčku stehenní kosti (podíl trámčité kosti v krčku femoru je přibližně 25 %). Histomorfometrické studie kosti dokumentují snížení šířky osteoidních lemů, nízkou rychlost apozice minerálu a ztenčení kostních trámců a prokazují tak, že útlum kostní novotvorby je hlavním mechanizmem, kterým dlouhodobé podávání GK způsobuje úbytek kostní hmoty [9]. Účinky GK na kostní metabolizmus jsou měřitelné velmi brzy po zahájení léčby. Charakteristickým rysem nadbytku GK je potlačení markerů kostní novotvorby, zejména osteokalcinu, jak je dobře dokumentováno u pacientů s Cushingovým syndromem [10,11]. Ton et al také prokázali, že u zdravých postmenopauzálních žen i nízké dávky prednisonu 5 mg/den vedou během 4 týdnů k významnému snížení markerů kostní formace osteokalcinu, C - a N-terminální propeptidu prokolagenu I v séru [12].
Kostní denzita (BMD) klesá u zdravých mužů o 2-4,5 % během 6 měsíců léčby GK [13], později se úbytek BMD zpomaluje [14]. Předpokládá se, že počáteční rychlý pokles BMD je důsledkem rychle navozené negativní rovnováhy kostní remodelace s převahou kostní resorpce nad novotvorbou kosti a pozdější pomalejší fáze úbytku BMD je způsobená celkovým útlumem kostní remodelace [14,15]. Po odstranění zdroje autonomní nadprodukce kortizolu nebo po přerušení léčby GK se upravuje porušená rovnováha kostní remodelace, BMD se vrací v některých případech až k normálním hodnotám [16]. Léčba GK ale zvyšuje riziko zlomenin nezávisle na změnách kostní denzity (BMD). Změny kostní denzity prokazovaly závislost na kumulativní dávce GK na rozdíl od rizika zlomeniny, které záviselo spíše na denní než na kumulativní dávce GK [17,18].
Rozlišujeme přímé účinky GK na kostní buňky (osteoblasty, osteocyty a osteoklasty) a nepřímé účinky GK na kost, které vyplývají z jejich zásahu do fosfokalciového metabolizmu, inhibici hypotalamo-hypofýzo-gonadální osy a somatotropní osy. Nepříznivý účinek na kostní novotvorbu a riziko zlomenin a pádů má také GK navozená myopatie (steroidní myopatie). Nezanedbatelný je rovněž vliv vlastního chronického onemocnění, pro které je léčba GK indikována, a odlišení účinků samotných GK od účinků vlastního onemocnění na skelet může být obtížné. Riziko zlomeniny závisí také na přítomnosti dalších rizikových faktorů primární osteoporózy [19]. Komplexní patofyziologie GK indukované osteoporózy a zlomenin je znázorněna na schématu 1.

Přímé účinky glukokortikoidů na kostní buňky
Kostní novotvorba
Histomorfometrické studie kosti prokazují, že útlum kostní novotvorby je hlavním mechanizmem, kterým GK ovlivňují kostní metabolizmus [9]. GK tlumí osteoblastogenezi, aktivitu již diferencovaných osteoblastů a rovněž urychlují programovanou buněčnou smrt (apoptózu) osteoblastů a osteocytů [20]. Snížený počet osteoblastů je tedy výsledkem jak jejich zvýšeného zániku apoptózou, tak snížené osteoblastogeneze. Prokázalo se, že GK jsou schopny ovlivňovat diferenciaci mezenchymálních buněk stromatu kostní dřeně směrem adipocytové linie, a tento proces může probíhat v neprospěch osteoblastogeneze [21,22]. GK snižují expresi některých klíčových transkripčních faktorů nutných pro diferenciaci mezenchymálních buněk kostní dřeně směrem k osteoblastům, jako je transkripční faktor Runx2/Cbfa1 (core binding factor a1/Runt related transcription factor-2) [23], a naopak zvyšují expresi transkripčních faktorů nutných pro diferenciaci směrem adipocytové linie, jako jsou PPARγ2 (Peroxisome Proliferator Activated Receptor-γ2) a CCAAT/enhancer binding proteins [24]. V procesu diferenciace směrem adipocytové linie působí GK také na další signální cesty. Zjistilo se, že kortizol např. indukuje expresi mRNA transmembránového proteinu Notch1 v osteoblastech [25]. Notch proteiny jsou transmembránové receptory, které kontrolují procesy diferenciace buňky [26] a zvýšená exprese proteinu Notch 1 v osteoblastech a buňkách stromatu kostní dřeně napodobuje některé účinky GK, poškozuje zrání osteoblastů a podporuje adipocytogenezi [25]. Zvýšena exprese Notch1 také tlumí Wnt/β-catenin signální cestu, která jinak aktivuje transkripční faktory Tcf (T cell factor) z Lef rodiny (Lymphoid enhancer-binding factor) pro geny s osteoanabolickou aktivitou [27]. Na útlumu Wnt/β-katenin signální cesty se podílejí také další mechanizmy, jako je GK navozená zvýšená expresí Dkk-1, extracelulárního antagonisty Wnt signální cesty z Dickkopf rodiny [28].
GK významně tlumí metabolickou aktivitu zralých osteoblastů a potlačují syntézu řady růstových faktorů, jako např. klíčového růstového faktoru kosti IGF 1 [29] a jeho vazebných proteinů (IGFBP 3,4,5) a také syntézu kolagenu I a dalších proteinů kostní matrix, jako je osteokalcin [30].
Přímé působení GK na osteoblasty bylo experimentálně potvrzeno in vivo na modelu transgenní myši se zvýšenou expresí 11-β-hydroxysteroid dehydrogenázy (11-β-HSD2) v diferencovaných osteoblastech a osteocytech [31]. Specifická exprese 11-β-HSD2 v osteoblastech a osteocytech byla dosažena vložením cDNA 11-β-HSD2 do promotorové oblasti genu pro osteokalcin, který se exprimuje pouze ve zralých osteoblastech a osteocytech. 11-β-HSD2 zajišťuje na prereceptorové úrovni konverzi biologicky aktivních GK na jejich neaktivní 11-ketometabolity, tedy zvýšená exprese tohoto enzymu v osteoblastech je činí prakticky necitlivé na působení GK. O´Brien et al zjistili, že u těchto transgenních myší nedochází po zátěži GK k urychlení apoptózy osteoblastů a osteocytů a dále prokázali, že mechanická odolnost kosti hodnocená v oblasti obratlového těla byla u těchto transgenních myší zachována, a to i přes obdobný pokles kostní denzity (BMD) jako u kontrolní skupiny zdravých myší. Tyto experimentální výsledky tedy potvrzují hypotézu, že viabilita osteocytů a osteoblastů významně ovlivňuje pevnost kosti, a to nezávisle na kostní denzitě. Zvýšený počet apoptotických osteocytů je nalézán u pacientů s GK indukovanou osteoporózou [32] a také pro klinicky závažnou komplikaci léčby GK „osteonekrózu“ hlavice kosti stehenní je typický nález hojné akumulace apoptotických osteocytů [33]. Osteocyty jsou nejhojnější kostní buňky (je jich přibližně 10krát více než osteoblastů), které mezi sebou vzájemně komunikují hustou sítí výběžků. Předpokládá se, že osteocyty přenášejí informace o nutnosti reparace kosti z místa poškození kosti dalším osteocytům, osteoblastům a kostním povrchovým (hraničním) buňkám (lining cells). Zvýšený zánik osteocytů apoptózou při léčbě GK tedy znamená ztrátu této důležité signální cesty a nemožnost cílené reparace kosti. Zvýšený zánik osteocytů může být vysvětlením pro epidemiologické zjištění nárůstu rizika zlomeniny již v prvních měsících léčby GK.
Kostní resorpce
Význam osteoklastů a kostní resorpce pro rozvoj GK indukované osteoporózy a zlomenin zatím nebyl zcela objasněn. Bylo zjištěno, že GK potlačují osteoblastogenezi i osteoklastogenezi, ale nesnižují počet zralých osteoklastů (jejich počet se nemění nebo se zvyšuje), zatímco počet osteoblastů se rychle snižuje [20]. Na počátku léčby GK tak vzniká nerovnováha mezi počtem osteoblastů a osteoklastů [15]. Osteoresorpce je tak pravděpodobně zodpovědná za počáteční rychlejší pokles BMD pozorovaný v prvních měsících po zahájení léčby GK. Odlišení přímých účinku GK na osteoklasty od jejich nepřímých účinků in vivo umožnil model transgenní myši se zvýšenou expresí genu pro 11-β-HSD2 v osteoklastech [34]. Specifické exprese tohoto enzymu v osteoklastech bylo dosaženo vložením cDNA 11-β-HSD2 do promotorové oblasti genu pro tartarát rezistentní kyselou fosfatázu, enzymu, který se exprimuje pouze v osteoklastech. Zvýšená exprese 11-β-HSD2 v osteoklastech zajistila necitlivost těchto buněk na působení GK. Podávání GK mělo srovnatelné účinky na histomorfometrické parametry funkce osteoblastů u transgenních i kontrolních myší, avšak u transgenních myší došlo ve srovnání s kontrolní skupinou k signifikantnímu poklesu počtu osteoklastů. BMD páteře transgenních myší neklesala na rozdíl od kontrolní skupiny myší, kde podávání GK navodilo očekávané snížení BMD. Tyto výsledky prokazují, že GK působí přímo na osteoklasty, a že tento účinek GK je alespoň částečně zodpovědný za počáteční úbytek kostního minerálu.
Ke zvýšení kostní resorpce přispívá nepřímo také snížená produkce estrogenů a androgenů navozená léčbou GK a účinky GK na fosfokalciový metabolizmus (snížení střevní absorpce vápníku a zvýšené vylučování vápníku močí). Sekundární hyperparatyreóza se v patogenezi zvýšené osteoresorpce ale pravděpodobně neuplatňuje [35]. Ke zvýšené osteoresorpci ale může nepřímo přispívat zvýšená produkce RANKL (ligand receptoru aktivující nukleární faktor χB) a snížené produkce sekrečního glykoproteinu osteoprotegerinu buňkami stromatu kostní dřeně a osteoblasty [36]. GK také zvyšují expresi kolonie stimulujícího faktoru 1 (CSF-1), který společně s RANKL indukuje osteoklastogenezi [37]. Účinek GK na kostní resorpci závisí pravděpodobně také na užité dávce. Podávání GK v nízké dávce (5 mg prednisonu denně) nenavodilo změnu koncentrací markerů osteoresorpce [12]. V jiné studii u mladých dospělých osob s roztroušenou sklerózou navodila pulzní léčba vysokou dávkou metylprednisonu zvýšení markeru kostní resorpce C-terminálního telopeptidu kolagenu I (βCTX) 7.-9. den léčby (zvýšení o 149 ± 63 %) s tendencí k poklesu markeru 10. den léčby [38]. Je ale dobře dokumentováno, že při dlouhodobější léčbě GK dochází k celkovému útlumu kostní remodelace, primárně kostní novotvorby a vzhledem k přerušení signalizace osteoblastů směrem k osteoklastům, dochází též k útlumu kostní resorpce [39].
Nepřímé účinky glukokortikoidů na kostní metabolizmus
GK nepřímo ovlivňují kostní metabolizmus působením na extracelulární metabolizmus minerálů. GK ovlivňují absorpci vápníku ve střevě a v ledvinách. V duodenu inhibují aktivní transcelulární transport vápníku, snižují syntézu Ca vázajícího proteinu (calbindin) a zvyšují rychlost degradace 1,25-(OH)2-vitaminu D a jeho vazebných míst [40]. Nadbytek GK vede k hyperkalciurii, kdy se pravděpodobně uplatňuje přímý inhibiční účinek GK na zpětnou tubulární absorpci vápníku [40]. Novější práce nesvědčí pro rozvoj sekundární hyperparatyreózy při akutní nebo dlouhodobé léčbě GK, jelikož koncentrace PTH u pacientů léčených GK jsou v normálním rozmezí a pro dlouhodobou léčbu GK je také charakteristický útlum kostní remodelace, na rozdíl od hyperparatyreózy, kde je kostní remodelace zvýšená [35]. Bylo však zjištěno, že GK významně ovlivňují sekreční dynamiku parathormonu (PTH) [41]. U zdravých mladých osob se rozlišují dvě hlavní sekreční komponenty PTH:
- a) stálá tonická sekrece PTH a
- b) pulzatilní složka sekrece (vyšší frekvence pulsů PTH, přibližně po 15-20 minutách, o nízké amplitudě).
Tato pulzatilní komponenta tvoří přibližně 25 % celkové denní sekrece PTH. Bylo zjištěno, že GK potlačují tonickou komponentu sekrece a zvýrazňují pulzatilní složku sekrece PTH. Mechanizmus, jakým GK mění sekreční dynamiku PTH, zatím není přesně znám.
GK tlumí hypotalamo-hypofýzo-gonadální osu [42]: snižují sekreci gonadoliberinu (GnRH), snižují odpověď LH na LHRH, snižují vazebná místa pro gonadotropiny v gonádách, a také přímo potlačují periferní produkci estrogenů a testosteronu [43].
GK inhibují dále somatotropní osu a sekreci růstového hormonu (GH), pravděpodobně stimulací syntézy a sekrece somatostatinu [44]. Podávání GH u pacientů léčených GK vede k významnému vzestupu koncentrací markerů kostní novotvorby, osteokalcinu a C-terminálního propeptidu prokolagenu I [45]. GK mohou však také přímo působit na IGF-1 a jeho vazebné proteiny na úrovni kosti. Kortizol snižoval transkripci a koncentrace IGF-1 a IGF BP5 v kosti [46,47].
Rozdíly v individuální citlivosti na glukokortikoidy
Účinky GK na organizmus však nezávisí pouze na denní dávce a délce léčby, ale současně i na individuální citlivosti, resp. rezistenci buněk na GK. I když jsou rozdíly v individuální odpovědi na GK z klinické praxe dobře známy, příčiny této rozdílné senzitivity buněk na GK se zatím nepodařilo zcela objasnit. Uvažuje se, že tato rozdílná senzitivita je alespoň částečně způsobena odlišnou citlivostí glukokortikoidních receptorů. Bylo nalezeno několik polymorfizmů genu pro glukokortikoidní receptor a zjištěny asociace mezi těmito polymorfizmy a tělesným složení a některými metabolickými parametry [48] Větší význam by mohl mít rozdílný lokální metabolizmus GK v kosti, zejména rozdíly v expresi a/nebo aktivitě enzymu 11-β-hydroxysteroid dehydrogenázy (izoenzym 11-β-HSD1 a 11-β-HSD2). V kosti převažuje exprese izoenzymu 11-β-HSD1, která byla nalezena v osteoblastech i v osteoklastech [49]. 11-β-HSD1 katalyzuje aktivaci neaktivních 11-ketoforem GK (kortizon) na jejich aktivní 11-hydroxylované metabolity (kortizol) a na tkáňové úrovni tak zesiluje působení GK [50] (obr. 2). Prereceptorová regulace se uplatňuje nejen u GK, ale také u dalších steroidních hormonů androgenů a estrogenů, kde je jejich účinek významně ovlivněn působením lokálních enzymů 5-α-reduktázy, hydroxysteroiddehydrogenázy a aromatázy. Dědičné nebo získané změny v aktivitě 11-β-HSD1 by mohly vysvětlit odlišnou senzitivitu různých tkání na endogenní nebo exogenní GK. Vrozený defekt tohoto enzymu byl již popsán u několika pacientů s Cushigovou nemocí, u kterých chyběli charakteristické klinické příznaky hyperkortizolizmu [51]. Cooper et al dále dokumentovali, že aktivita 11-β-HSD1 v kultuře lidských osteoblastů se lineárně zvyšuje s věkem dárce a také při expozici GK [52]. Zvýšení aktivity 11-β-HSD1 s věkem by tak mohlo přispívat k rozvoji involuční osteoporózy.
Prevence a léčba GK indukované osteoporózy
Preventivní nebo léčebná opatření proti úbytku kostní hmoty by měla být zvážena u každého pacienta, u kterého se plánuje dlouhodobá léčba glukokortikoidy (déle než 3 měsíce) v denních dávkách vyšších než 2,5 mg prednisonu per os (nebo vyšších dávek GK v inhalační formě). Před rozhodnutím o vhodném preventivním nebo léčebném postupu by měly být zhodnoceny rizikové faktory zlomenin (tab. 2) a indikována kostní denzitometrie bederní páteře a proximálního konce stehenní kosti. Hodnotu kostní denzity (bone mineral density - BMD) je doporučeno stanovit dvouenergiovou RTG absorpciometrií (dual energy X-ray absorpciometry - DXA). Tato metoda hodnotí množství kostního minerálu na jednotku plochy (g/cm2) a v současné době je považována za standard v diagnostice osteoporózy. Je ale nutné zdůraznit, že současné diagnostické guidelines WHO na základě hodnocení BMD (T-skóre -2,5 SD) byly vytvořeny pro diagnostiku postmenopauzální osteoporózy a nikoli pro GK indukovanou osteoporózu. U pacientů léčených GK dochází k netraumatickým zlomeninám při vyšších hodnotách BMD než u pacientů s postmenopauzální osteoporózou. Doporučení odborných společností proto stanovují práh BMD pro vznik zlomenin výše, již na úroveň T-skóre -1 SD [53] nebo T-skóre -1,5 SD [54,55]. Je však zřejmé, že při léčbě GK dochází k výrazným změnám kvality kosti, kterou metoda DXA nemůže rozpoznat a ke zlomeninám dochází u pacientů léčených GK nezávisle na BMD. Rozhodnutí o léčbě by proto nemělo záviset pouze na hodnotě BMD (T-skóre), ale měly by být zohledněny také další rizikové faktory osteoporotických zlomenin (tab. 1) [19,56].
![Rizikové faktory osteoporotických zlomenin [56].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/4afe01668a1494ac518e8b611dfd2efd.jpg)

Vápník a vitamin D
Jelikož GK snižují střevní absorpci vápníku a zvyšují vylučování vápníku ledvinami, je dostatečný příjem vápníku (1 500 mg elementárního vápníku/den) společně s vitaminem D (800 IU/den) nezbytnou prevencí sekundární hyperparatyreózy. Vitamin D3 (cholekalciferol) i aktivní analoga vitaminu D3 působí přes společný biologicky aktivní metabolit D-hormon kalcitriol [1α-25(OH)2D3 ]. Suplementace samotným cholekalciferolem je užitečná zejména u pacientů s nedostatkem vitaminu D a s normální aktivitou 1α-hydroxylázy (CYP27B1) v ledvinách. Aktivní syntetická analoga vitaminu D působí i při dostatečné hladině 25OHD3 a zvláště užitečné budou při snížené aktivitě 1α-hydroxylázy např. při zánětlivých chorobách. Alfakalcidiol (1α-hydroxyvitamin D3) je hydroxylován na aktivní kalcitriol 25-hydroxylázou nejen v játrech, ale také přímo v osteoblastech [57].
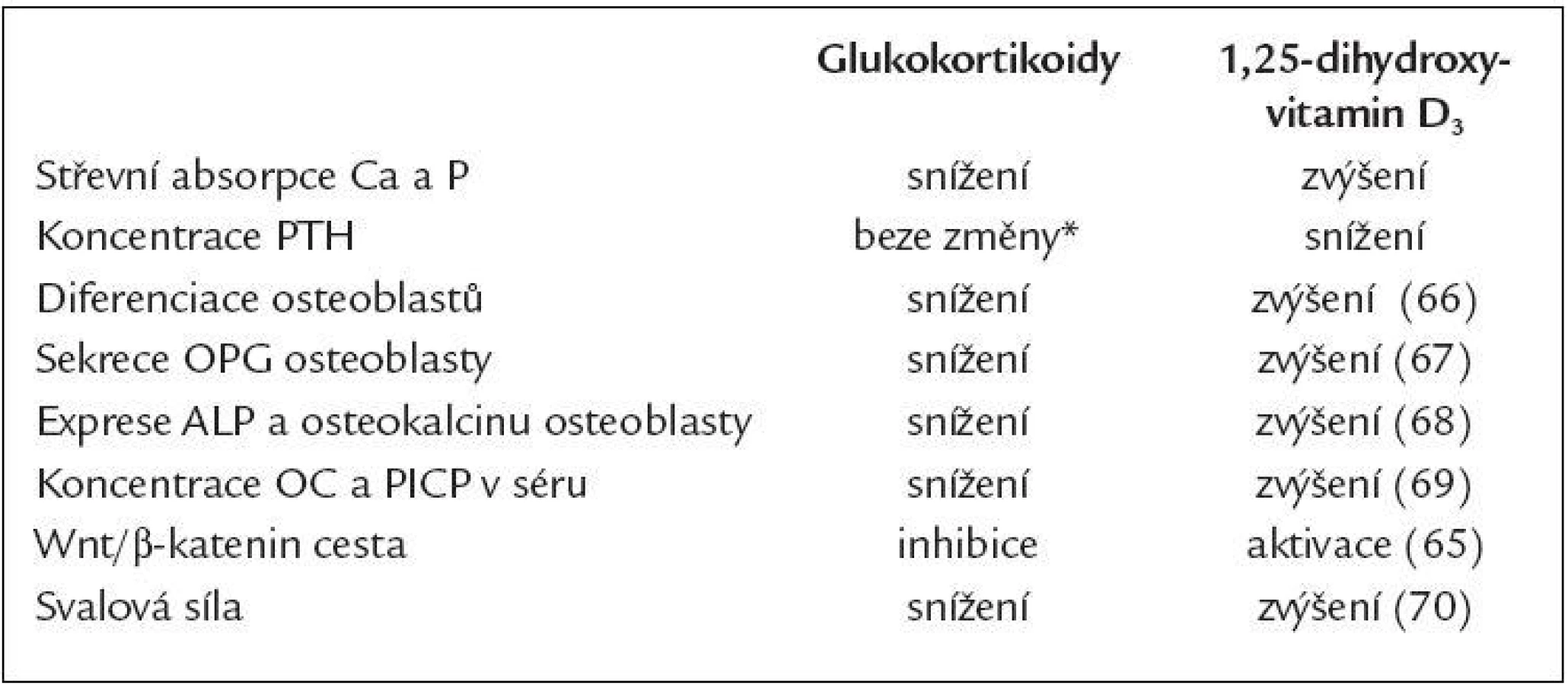
Aktivní metabolity vitaminu D zvyšují střevní absorpci vápníku [58], snižují vylučování vápníku ledvinami [59], inhibují syntézu PTH [60], zlepšují svalovou funkci [61] a pozitivním způsobem také modulují imunitní systém při zánětlivých chorobách, kdy snižují tvorbu zánětlivých cytokinů TNFα a IL-6 [62]. Význam má rovněž přímé působení aktivních metabolitů na kostní buňky. Alfakalcidiol stimululuje kostní novotvorbu a inhibuje kostní resorpci [63,64]. Zdůvodnění pro použití aktivních forem vitaminu D u pacientů léčených GK je uvedeno také v tab. 2 [65-70].
Estrogeny a testostosteron
Léčba GK tlumí hypotalamo-hypofýzo-gonadální osu na více úrovních a může tak vést k rozvoji hypogonadizmu u žen i u mužů. Deficit sexagenů stimuluje kostní resorpci a může tak přispívat alespoň zpočátku k úbytku BMD, při dlouhodobé léčbě GK však převažují antiremodelační účinky GK [71,72]. Deficit estrogenů i androgenů, ale podobně jako léčba GK, urychluje apoptózu osteoblastů a osteocytů [73, 15]. Substituční léčba estrogeny a androgeny tak může u pacientů léčených glukokortikoidy bránit alespoň na počátku léčby GK úbytku kostní hmoty a zlepšit mechanické vlastnosti kosti vzhledem k antiapoptotickém působení estrogenů a androgenů na osteoblasty a osteocyty [73-75].
Podobně by mohly účinkovat i selektivní modulátory estrogenních receptorů, syntetické nehormonální sloučeniny, které působí rovněž na estrogenní receptory na osteoblastech a osteocytech jako agonisté estradiolu a brání tak rovněž jako estradiol nadměrné osteoresorpci a mohou rovněž bránit apoptóze osteocytů [76].
U premenopauzálních žen je při léčbě glukokortikoidy vhodné vždy kontrolovat endogenní produkci estrogenů a při jejich nedostatku zajistit vhodnou substituční léčbu. Podobně u mužů mladších 50 let je vhodné kontrolovat endogenní produkci testosteronu a při jeho nedostatku zvážit substituční léčbu. Hormonální substituční léčba je vhodná u žen po předčasné menopauze a také u žen v prvních letech po menopauze, kdy hormonální substituce brání úbytku kostní hmoty a zvyšuje kostní denzitu i u pacientů léčených GK [77]. U mužů léčených GK roční substituční léčba testosteronem rovněž vedla ke zvýšení BMD v páteři a ke zlepšení svalové síly [78]. Studie hodnotící účinky selektivních modulátorů estrogenních receptorů (raloxifenu) u pacientů léčených GK zatím nejsou k dispozici.
Bisfosfonáty
Bisfosfonáty jsou stabilní analoga pyrofosfátu s vysokou afinitou ke kostnímu minerálu, jejichž hlavním mechanizmem účinku je urychlení apoptózy zralých osteoklastů s následným potlačením kostní resorpce a útlumem kostní remodelace. Klinické studie potvrzují, že u pacientů léčených GK aminobisfosfonáty potlačují kostní remodelaci a zvyšují kostní denzitu (BMD) během prvních 2 let léčby. Zatím chybějí prospektivní studie, u nichž by primárním cílem bylo hodnocení rizika zlomeniny. Snížení rizika vertebrálních zlomenin ale bylo dokumentováno v klinických studiích s aminobisfosfonáty (alendronát a risedronát) jako sekundární cíl a v rámci post hoc analýz. Jelikož dosavadní klinické studie zahrnuly nízký počet premenopauzálních žen a mladších mužů léčených GK, je účinek a efektivita aminobisfosfonátů u těchto skupin pacientů zatím není jistá. Z hlediska mechanizmu působení GK na kost vyplývá, že užití antiremodelační (antiresorpční) léčby např. bisfosfonátů bude nejúčinnější v prvních měsících po zahájení léčby GK, kdy se v úbytku kostní hmoty uplatňuje osteoresorpce [79]. Pacienti, kteří jsou již delší dobu léčeni GK, mohou být méně citliví na antiremodelační léčbu vzhledem k utlumené kostní remodelaci (útlum kostní novotvorby i kostní resorpce). V poslední době jsou diskutovány také jiné mechanizmy působení bisfosfonátů na kost. Ochranný účinek aminobisfosfonátů na kost nelze vysvětlit pouze účinkem na kostní resorpci, o čemž svědčí např. disproporční účinek aminobisfosfonátů na BMD a riziko zlomeniny u pacientek s postmenopauzální osteoporózou [80]. Jedním s diskutovaných mechanizmů působení aminobisfosfonátů je inhibice apoptózy osteocytů a osteoblastů [81]. Při léčbě aminobisfosfonáty je nutné respektovat jejich známé kontraindikace. Zvláště opatrní musíme být také při užití aminobisfofonátů u premenopauzálních žen, u kterých není vyloučeno otěhotnění. Aminobisfosfonáty pronikají placentou a mohou se tak akumulovat ve skeletu plodu [82]. Vzhledem k dlouhému poločasu aminobisfosfonátu, který je způsoben jeho silnou vazbou na kostní minerál, zatím není známá bezpečná doba po vysazení léku, kdy by bylo riziko expozice plodu bisfosfonátem eliminováno. Mezi jednotlivými bisfosfonáty jsou významné rozdíly v jejich poločasu, zejména vzhledem k rozdílům v afinitě ke kostnímu minerálu, kdy nejvyšší afinitu má zoledronát a nejnižší klodronát (zoledronát > alendronát > ibandronát > risedronát > etidronát > klodronát). U premenopauzálních žen je proto doporučováno užití bisfosfonátu s kratším poločasem v kosti [83].
Osteoanabolická léčba
Vzhledem k mechanizmu účinku glukokortikoidů na kostní metabolizmus s výrazným potlačením kostní novotvorby je jistě výhodné léčebné užití přípravků, které mají potenciál stimulovat osteoblasty a kostní novotvorbu. Mezi tyto osteoanabolické přípravky patří parathormon (PTH). Léčba PTH stimuluje proliferaci a diferenciaci buněk osteoblastické řady [84] a brání apoptóze osteoblastů [85]. PTH stimuluje syntézu klíčových růstových faktorů kosti IGF-I a TGFβ-I [86] a také aktivuje Wnt/β-katenin signální cestu [87]. Srovnání účinků PTH a GK na kost je uvedeno v tab. 3. Prvním registrovaným osteoanabolickým přípravkem, který prokazatelně snižuje riziko zlomenin u žen s postmenopauzální osteoporózou, je rekombinantní aminoterminální fragment lidského parathormonu hPTH [1-34], genericky teriparatid. U pacientů s glukokortikoidy indukovanou osteoporózou bylo zatím prokázáno, že PTH může skutečně překonat útlum kostní novotvorby navozený glukokortikoidní léčbou. Roční léčba PTH [1-34] u postmenopauzalních žen s osteoporózou substituovaných estrogeny a léčených glukokortikoidy (v dávce 5-20 mg prednisonu denně) navodila významné zvýšení markerů kostní novotvorby (osteokalcinu a propeptidu prokolagenu I) a nárůst kostní denzity v bederní páteři (zvýšení o 11 %) [88]. Zatím chybějí studie, které by potvrdily, že toto zvýšení BMD u pacientů léčených glukokortikoidy je asociováno se snížením rizika zlomeniny.

Závěr
Glukokortikoidy významně zasahují do kostního metabolizmu a způsobují hluboký útlum kostní remodelace, zejména kostní novotvorby. Léčba GK i v relativně nízkých dávkách zvyšuje riziko zlomenin, zejména v místech s vyšším zastoupením trámčité kosti (obratlová těla). Ke zvýšení rizika zlomenin dochází již v prvních měsících po zahájení léčby GK. Zvýšení rizika zlomenin není vysvětlitelné poklesem BMD a zlomeniny vznikají při vyšších hodnotách BMD než u primární osteoporózy. Zatím není k dispozici nástroj, který by dokázal tyto změny kvality kosti a zvýšené riziko zlomenin při léčbě GK v klinické praxi spolehlivě kvantifikovat. Mezi jednotlivci také existují významné rozdíly v citlivosti na GK, jejichž příčiny se dosud nepodařilo objasnit. Porozumění patofyziologii glukokortikoidy indukované osteoporózy přináší nové možnosti prevence a léčby GK indukované osteoporózy. Z hlediska mechanizmu účinku GK na kost je výhodné užití osteoanabolických léků, jako je PTH [1-34], který stimuluje kostní novotvorbu a působí tak přímo proti katabolickým účinkům GK. Osteoporóza a s ní spojené zlomeniny je velmi závažnou komplikací dlouhodobé léčby GK, proto snaha o její prevenci a léčbu v každodenní klinické praxi je velmi důležitá. I přes řadu nevyřešených otázek a nejistot můžeme u pacienta včasným preventivním nebo léčebným opatřením omezit devastující účinek GK na kostní metabolizmus. Nové poznatky z patofyziologie GK indukované osteoporózy jistě v budoucnosti přinesou i nové možnosti prevence a léčby.
MUDr. Vít Zikán, Ph.D.
www.vfn.cz
e-mail: vitzikan@hotmail.com
Doručeno do redakce: 23. 5. 2007
Sources
1. Kanis JA, Johansson H, Oden A et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res 2004; 19 : 893-899.
2. van Staa TP, Leufkens HG, Abenhaim L et al. Use of oral corticosteroids in the United Kingdom. QJM 2000; 93 : 105-111.
3. Reid IR Glucocorticoid-induced osteoporosis: assessment and treatment. J Clin Densitom 1998; 1 : 65-73.
4. van Staa TP, Leufkens HG, Abenhaim L et al. Use of oral corticosteroids and risk of fractures. J Bone Miner Res 2000; 15 : 993-1000.
5. Israel E, Banerjee TR, Fitzmaurice GM et al. Effects of inhaled glucocorticoids on bone density in premenopausal women. N Engl J Med 2001; 345 : 941-947.
6. Steinbuch M, Youket TE, Cohen S. Oral glucocorticoid use is associated with an increased risk of fracture. Osteoporos Int 2004; 15 : 323-328.
7. Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with systemic and topical corticosteroids. J Intern Med 2005; 257 : 374-384.
8. Angeli A, Guglielmi G, Dovio A et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone 2006; 39 : 253-259.
9. Dalle Carbonare L, Arlot ME, Chavassieux PM et al. Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 2001; 16 : 97-103.
10. Francucci CM, Pantanetti P, Garrapa GG et al. Bone metabolism and mass in women with Cushing's syndrome and adrenal incidentaloma. Clin Endocrinol (Oxf) 2002; 57 : 587-593.
11. Cortet B, Cortet C, Blanckaert F et al. Quantitative ultrasound of bone and markers of bone turnover in Cushing's syndrome. Osteoporos Int 2001; 12 : 117-123.
12. Ton FN, Gunawardene SC, Lee H et al. Effects of low-dose prednisone on bone metabolism. J Bone Miner Res 2005; 20 : 464-470.
13. Pearce G, Ryan PF, Delmas PD et al. The deleterious effects of low-dose corticosteroids on bone density in patients with polymyalgia rheumatica. Br J Rheumatol 1998; 37 : 292-299.
14. LoCascio V, Bonucci E, Imbimbo B et al. Bone loss in response to long-term glucocorticoid therapy. Bone Miner 1990; 8 : 39-51.
15. Weinstein RS, Jilka RL, Parfitt AM et al. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of the deleterious effects on bone. J Clin Invest 1998; 102 : 274-282.
16. Manning PJ, Evans MC, Reid IR Normal bone mineral density following cure of Cushing's syndrome. Clin Endocrinol (Oxf) 1992; 36 : 229-234.
17. van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13 : 777-787.
18. van Staa TP, Leufkens HG, Abenhaim L et al. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxf) 2000; 39 : 1383-1389.
19. van Staa TP, Geusens P, Pols HA et al. A simple score for estimating the long-term risk of fracture in patients using oral glucocorticoids. QJM 2005; 98 : 191-198.
20. Weinstein RS, Chen JR, Powers CC et al. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest 2002; 109 : 1041-1048.
21. Nuttall ME, Gimble JM Is there a therapeutic opportunity to either prevent or treat osteopenic disorders by inhibiting marrow adipogenesis? Bone 2000; 27 : 177-184.
22. Nuttall ME, Patton AJ, Olivera DL et al. Human trabecular bone cells are able to express both osteoblastic and adipocytic phenotype: implications for osteopenic disorders. J Bone Miner Res 1998; 13 : 371-382.
23. Chang DJ, Ji C, Kim KK et al. Reduction in transforming growth factor beta receptor I expression and transcription factor CBFa1 on bone cells by glucocorticoid. J Biol Chem 1998; 273 : 4892-4896.
24. Li X, Jin L, Cui Q et al. Steroid effects on osteogenesis through mesenchymal cell gene expression. Osteoporos Int 2005; 16 : 101-108.
25. Sciaudone M, Gazzerro E, Priest L et al. Notch 1 impairs osteoblastic cell differentiation. Endocrinology 2003; 144 : 5631-5639.
26. Deregowski V, Gazzerro E, Priest L et al. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/β-catenin but not bone morphogenetic protein signaling. J Biol Chem 2006; 281 : 6203-6210.
27. Smith E, Frenkel B. Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3β-dependent and -independent manner. J Biol Chem 2005; 280 : 2388-2394.
28. Ohnaka K, Taniguchi H, Kawate H et al. Glucocorticoid enhances the expression of dickkopf-1 in human osteoblasts: novel mechanism of glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun 2004; 318 : 259-264.
29. Delany AM, Canalis E. Transcriptional repression of insulin-like growth factor I by glucocorticoids in rat bone cells. Endocrinology 1995; 136 : 4776-4781.
30. Cui Q, Wang GJ, Balian G. Steroid-induced adipogenesis in a pluripotential cell line from bone marrow. J Bone Joint Surg Am 1997; 79 : 1054-1063.
31. O’Brien CA, Jia D, Plotkin LI et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 2004; 145 : 1835-1841.
32. Weinstein RS Manolagas SC. Apoptosis and osteoporosis. Am J Med 2000; 108 : 153-164.
33. Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab 2000; 85 : 2907-2912.
34. Jia D, O’Brien CA, Stewart SA et al. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology 2006; 147 : 5592-5599.
35. Rubin MR, Bilezikian JP. Clinical review 151: The role of parathyroid hormone in the pathogenesis of glucocorticoid-induced osteoporosis: a re-examination of the evidence. J Clin Endocrinol Metab 2002; 87 : 4033-4041.
36. Hofbauer LC, Gori F, Riggs BL et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999; 140 : 4382-4389.
37. Rubin J, Biskobing DM, Jadhav L et al. Dexamethasone promotes expression of membrane-bound macrophage colony-stimulating factor in murine osteoblast-like cells. Endocrinology 1998; 139 : 1006-1012.
38. Dovio A, Perazzolo L, Osella G et al. Immediate fall of bone formation and transient increase of bone resorption in the course of high-dose, short-term glucocorticoid therapy in young patients with multiple sclerosis. J Clin Endocrinol Metab 2004; 89 : 4923-4928.
39. Dempster DW, Moonga BS, Stein LS et al. Glucocorticoids inhibit bone resorption by isolated rat osteoclasts by enhancing apoptosis. J Endocrinol 1997; 154 : 397-406.
40. Gennari C. Differential effect of glucocorticoids on calcium absorption and bone mass.Br J Rheumatol 1993; 32(Suppl 2): 11-14.
41. Bonadonna S, Burattin A, Nuzzo M et al.Chronic glucocorticoid treatment alters spontaneous pulsatile parathyroid hormone secretory dynamics in human subjects. Eur J Endocrinol 2005;152 : 199-205
42. Doga M, Bonadonna S, Giustina A. Glucocorticoids and bone: cellular, metabolic and endocrine effects. Hormones (Athens) 2004; 3 : 184-190.
43. Manelli F, Giustina A. Glucocorticoid-induced osteoporosis. Trends Endocrinol Metab 2000; 11 : 79-85.
44. Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 1998; 19 : 717-797.
45. Giustina A, Bussi AR, Jacobello C et al. Effects of recombinant human growth hormone (GH) on bone and intermediary metabolism in patients receiving chronic glucocorticoid treatment with suppressed endogenous GH response to GH-releasing hormone. J Clin Endocrinol Metab 1995; 80 : 122-129.
46. Delany AM, Durant D, Canalis E. Glucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol 2001; 15 : 1781-1789.
47. Gabbitas B, Pash JM, Delany AM et al. Cortisol inhibits the synthesis of insulin-like growth factor-binding protein-5 in bone cell cultures by transcriptional mechanisms. J Biol Chem 1996; 271 : 9033-9038.
48. van Rossum EF, Lamberts SW Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res 2004; 59 : 333-357.
49. Cooper MS, Walker EA, Bland R et al. Expression and functional consequences of 11β-hydroxysteroid dehydrogenase activity in human bone. Bone 2000; 27 : 375-381.
50. Draper N, Stewart PM 11β-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol 2005; 186 : 251-271.
51. Tomlinson JW, Draper N, Mackie J et al. Absence of Cushingoid phenotype in a patient with Cushing's disease due to defective cortisone to cortisol conversion. J Clin Endocrinol Metab 2002; 87 : 57-62.
52. Cooper MS, Rabbitt EH, Goddard PE et al. Osteoblastic 11β-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res 2002; 17 : 979-986.
53. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44 : 1496-1503.
54. Royal College of Physicians. Glucocorticoid-Induced Osteoporosis: guidelines for prevention and treatment. London: Royal College of Physicians 2002.
55. Růžičková O, Bayer M, Pavelka K et al. Doporučení pro prevenci a léčbu glukokortikoidy indukované osteoporózy u pacientů s revmatickým onemocněním (Společné stanovisko České revmatologické společnosti a Společnosti pro metabolická onemocnění skeletu). Čes Revmatol 2004; 12 : 163-174.
56. Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet 2002; 359 : 1929-1936.
57. Ichikawa F, Sato K, Nanjo M et al. Mouse primary osteoblasts express vitamin D3 25-hydroxylase mRNA and convert 1-α-hydroxyvitamin D3 into 1-α, 25 dihydroxyvitamin D3. Bone 1995; 16 : 129-135.
58. Bouillon R, Van Cromphaut S, Carmeliet G. Intestinal calcium absorption: Molecular vitamin D mediated mechanisms. J Cell Biochem 2003; 88 : 332-339.
59. Hoenderop JG, Muller D, Van Der Kemp AW et al. Calcitriol controls the epithelial calcium channel in kidney. J Am Soc Nephrol 2001; 12 : 1342-1349.
60. Russell JD, Lettieri D, Sherwood LM Suppression by 1,25-(OH)2D of transcription of the parathyroid hormone gene. Endocrinology 1985; 119 : 2864-2866.
61. Schacht E, Richy F, Reginster JY. The therapeutic effects of alfacalcidol on bone strength, muscle metabolism and prevention of falls and fractures. J Musculoskelet Neuronal Interact 2005; 5 : 273-284.
62. Mathieu C, Adorini L. The coming of age of 1,25-dihydroxyvitamin D(3) analogs as immunomodulatory agents. Trends Mol Med 2002; 8 : 174-179.
63. Li M, Healy DR, Simmons HA et al. Alfacalcidol restores cancellous bone in ovariectomized rats. J Musculoskelet Neuronal Interact 2003; 3 : 39-46.
64. Shiraishi A, Takeda S, Masaki T et al. Alfacalcidol inhibits bone resorption and stimulates formation in an ovariectomized rat model of osteoporosis: distinct actions from estrogen. J Bone Miner Res 2000; 15 : 770-779.
65. Ohnaka K, Tanabe M, Kawate H et al. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 2005; 329 : 177-181.
66. Scharla SH, Strong DD, Mohan S et al. 1,25-Dihydroxyvitamin D3 differentially regulates the production of insulin-like growth factor I (IGF-I) and IGF-binding protein-4 in mouse osteoblasts. Endocrinology 1991; 129 : 3139-3146.
67. Hofbauer LC, Dunstan CR, Spelsberg TC et al. Osteoprotegerin production by human osteoblast lineage cells is stimulated by vitamin D, bone morphogenetic protein-2, and cytokines. Biochem Biophys Res Commun 1998; 250 : 776-781.
68. van Driel M, Koedam M, Buurman CJ et al. Evidence for auto/paracrine actions of vitamin D in bone: 1α-hydroxylase expression and activity in human bone cells. FASEB J 2006; 20 : 2417-2419.
69. de Nijs RN, Jacobs JW, Lems WF et al. Alendronate or alfacalcidol in glucocorticoid-induced osteoporosis. N Engl J Med 2006; 355 : 675-684.
70. Scharla SH, Schacht E, Lempert UG. Alfacalcidol versus plain vitamin D in inflammation induced bone loss. J Rheumatol Suppl 2005; 76 : 26-32.
71. Weinstein RS, Jia D, Powers CC et al. The skeletal effects of glucocorticoid excess override those of orchidectomy in mice. Endocrinology 2004; 145 : 1980-1987.
72. Pearce G, Tabensky DA, Delmas PD et al. Corticosteroid-induced bone loss in men. J Clin Endocrinol Metab 2005; 83 : 801-806.
73. Kousteni S, Chen JR, Bellido TM et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 2002; 298 : 843-846.
74. Gu G, Hentunen TA, Nars M et al. Estrogen protects primary osteocytes against glucocorticoid-induced apoptosis. Apoptosis 2005; 10 : 583-595.
75. Plotkin LI, Aguirre JI, Kousteni S et al. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal-regulated kinase activation. J Biol Chem 2005; 280 : 7317-7325.
76. Mann V, Huber C, Kogianni G et al. The antioxidant effect of estrogen and Selective Estrogen Receptor Modulators in the inhibition of osteocyte apoptosis in vitro. Bone 2007; 40 : 674-684.
77. Lukert BP, Johnson BE, Robinson RG. Estrogen and progesterone replacement therapy reduces glucocorticoid-induced bone loss. J Bone Miner Res 1992; 7 : 1063-1069.
78. Crawford BA, Liu PY, Kean MT et al. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J Clin Endocrinol Metab 2003; 88 : 3167-3176.
79. Saag KG, Emkey R, Schnitzer TJ et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339 : 292-299.
80. Cummings SR. How drugs decrease fracture risk: lessons from trials. J Musculoskelet Neuronal Interact 2002; 2 : 198-200.
81. Plotkin LI, Manolagas SC, Bellido T. Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone 2006; 39 : 443-452.
82. Patlas N, Golomb G, Yaffe P et al. Transplacental effects of bisphosphonates on fetal skeletal ossification and mineralization in rats. Teratology 1999; 60 : 68-73.
83. Franchimont N, Canalis E. Management of glucocorticoid induced osteoporosis in premenopausal women with autoimmune disease. Autoimmun Rev 2003; 2 : 224-228.
84. Nishida S, Yamaguchi A, Tanizawa T et al. Increased bone formation by intermittent parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone 1994; 15 : 717-723.
85. Jilka RL, Weinstein RS, Bellido T et al. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest 1999; 104 : 439-446.
86. Pfeilschifter J, Laukhuf F, Muller-Beckmann B et al. Parathyroid hormone increases the concentration of insulin-like growth factor-I and transforming growth factor β 1 in rat bone. J Clin Invest 1995; 96 : 767-774.
87. Tobimatsu T, Kaji H, Sowa H et al. Parathyroid hormone increases β-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology 2006; 147 : 2583-2590.
88. Lane NE, Sanchez S, Modin GW et al. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. J Clin Invest 1998; 102 : 1627-1633.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2007 Issue 7-8
Most read in this issue
- Hypopituitarizmus: substituční terapie
- Autoimunitní polyglandulární syndromy: klinické aspekty
- Aspirační cytologie štítné žlázy
- Náhodně zjištěné expanze v selární oblasti