
Izolovaná forma srdeční amyloidózy v podobě počínající infiltrativní kardiomyopatie bez restriktivní fyziologie
The isolated form of cardiac amyloidosis in the form of beginning infiltrative cardiomyopathy without restrictive physiology
The authors describe an interesting case of isolated cardiac manifestation of AL-amyloidosis manifesting as an incipient infiltrative cardiomyopathy with heart failure symptoms due to moderate left ventricular diastolic dysfunction. Restrictive cardiomyopathy with severe diastolic dysfunction is considered as the characteristic manifestation of fully developed cardiac amyloidosis. However, the organ deposition of amyloid is progressive and left ventricular filling worsens continuously, starting with less advanced forms of diastolic dysfunction; the restrictive physiology is characteristic only for advanced phases of the disease. Therefore, the possibility of the incipient infiltrative cardiomyopathy due to the amyloidosis should be considered in patients with heart failure symptoms and echocardiographic findings of unexplained left ventricular hypertrophy with only mild or moderate diastolic dysfunction.
Key words:
heart failure – echocardiography – cardiomyopathy – amyloidosis
Authors:
P. Kuchynka 1; T. Paleček 1; S. Šimek 1; J. C. Lubanda 1; M. Elleder 2; I. Špička 3; P. Jansa 1; A. Linhart 1
Authors‘ workplace:
II. interní klinika – klinika kardiologie a angiologie 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Aleš Linhart, DrSc.
1; Ústav dědičných a metabolických poruch 1. lékařské fakulty UK a VFN Praha, přednosta prof. MUDr. Milan Elleder, DrSc.
2; I. interní klinika – klinika hematologie 1. lékařské fakulty UK a VFN Praha, přednosta doc. MUDr. Marek Trněný, DrSc.
3
Published in:
Vnitř Lék 2008; 54(10): 1010-1013
Category:
Case Report
Overview
Autoři popisují zajímavý případ izolované kardiální manifestace AL-amyloidózy v podobě počínající infiltrativní kardiomyopatie, vedoucí k symptomům srdečního selhávání v důsledku středně těžké diastolické dysfunkce levé komory. Charakteristickým obrazem rozvinutého srdečního postižení na podkladě amyloidózy je restriktivní kardiomyopatie vyznačující se těžkou poruchou diastolické funkce. Orgánová infiltrace amyloidem je však postupným procesem, a ke zhoršování plnění levé komory tak dochází kontinuálně, počínaje méně výraznými formami diastolické dysfunkce; restriktivní fyziologie je typická až pro pokročilá stadia onemocnění. U nemocných se symptomy srdečního selhávání, a s echokardiografickým obrazem nevysvětlitelné hypertrofie levé komory doprovázené jen mírnou či středně těžkou diastolickou dysfunkcí je proto v našich geografických podmínkách v diferenciální diagnostice nutno vždy zvažovat také možnost počínající kardiomyopatie v důsledku amyloidózy.
Klíčová slova:
srdeční selhání – echokardiografie – kardiomyopatie – amyloidóza
Popis případu
Dosud zdravá, 62letá žena, s anamnézou přibližně rok trvající námahové dušnosti, bez další symptomatiky ve smyslu stenokardií, palpitací, synkopálních stavů nebo otoků dolních končetin, byla na naše pracoviště odeslána spádovým kardiologem k vyšetření etiologie plicní hypertenze, zjištěné echokardiograficky v místě bydliště.
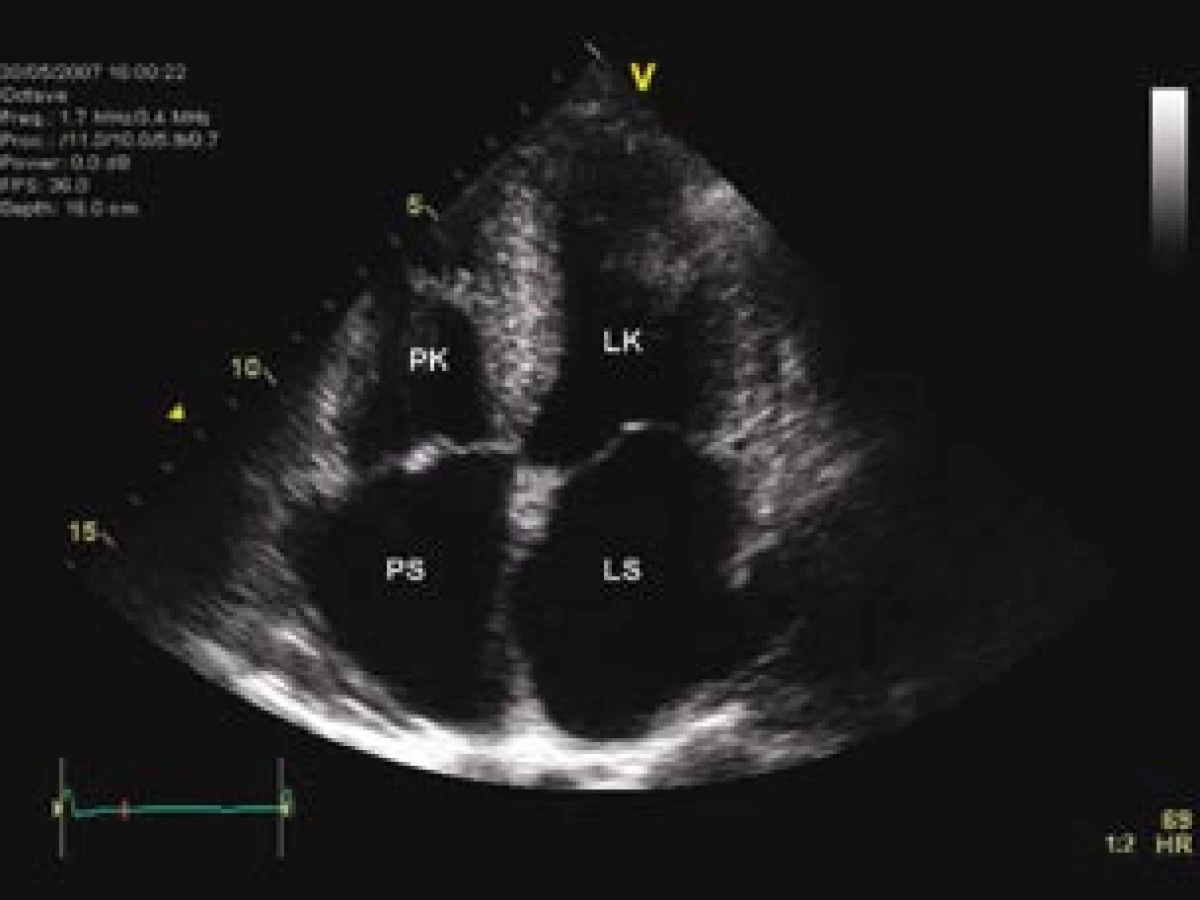
Při fyzikálním vyšetření byla pacientka eupnoická, normotenzní (TK 120/80 mm Hg), tepová frekvence byla přiměřená (66/min). Patrna byla zvýšená náplň krčních žil. V prekordiu byl slyšitelný systolický šelest s akcentací 2. ozvy nad plícnicí. Dýchání nad oběma plícemi bylo čisté, sklípkové. V oblasti břicha nebyla játra poklepově ani pohmatem zvětšena. Dolní končetiny byly bez otoků. Na EKG byl zaznamenán sinusový rytmus s normálním atrioventrikulárním převodem a obrazem kombinace levé přední hemiblokády s inkompletní blokádou pravého raménka Tawarova. Echokardiograficky byla zjištěna nezvětšená, mírně koncentricky hypertrofická levá komora (LK) (tloušťka mezikomorového septa 13 mm) s normální regionální i celkovou systolickou funkcí (ejekční frakce 67 %) – obr. 1. Na základě hodnocení dopplerovských parametrů transmitrálního toku, toku v plicních žilách a tkáňové dopplerovské echokardiografie byla popsána tzv. pseudonormalizace plnění LK, charakteristická pro její středně těžkou dia-stolickou dysfunkci, spojenou s mírným navýšením plnicích tlaků LK (obr. 2, 3). Obě síně byly mírně dilatovány, pravá komora zvětšena nebyla a měla normální systolickou funkci hodnocenou podle amplitudy a rychlosti pohybu trikuspidálního anulu. Tenze v plícnici byla lehce zvýšená (odhadovaný systolický tlak v plícnici činil 43–48 mm Hg). Cípy mitrální a trikuspidální chlopně byly lehce zesíleny, významná chlopenní vada však přítomna nebyla. Na základě echokardiografického nálezu bylo vysloveno podezření na postkapilární etiologii plicní hypertenze v důsledku diastolické dysfunkce LK. V diferenciální diagnostice středně těžké diastolické dysfunkce mírně koncentricky hypertrofické LK byla na prvém místě, při absenci arteriální hypertenze a aortální stenózy, zvažována možnost počínající restriktivní, nejspíše infiltrativní kardiomyopatie se zatím nekompletně vyjádřeným obrazem restriktivního charakteru plnění. Jelikož nejčastější příčinou restriktivní kardiomyopatie je v naší geografické oblasti amyloidóza, byl následný diagnostický proces směřován ke stanovení či vyloučení její přítomnosti. V laboratorních vyšetřeních byla přítomna hraničně vysoká hodnota sedimentace (20 mm za 1. hod), lehce zvýšená hodnota C‑reaktivního proteinu (9 mg/l), nebyly patrny odchylky v krevním obrazu či v diferenciálním rozpočtu leukocytů. Hladiny celkové bílkoviny, kalcia či kyseliny močové nebyly zvýšeny. Také hodnoty renálních funkcí a jaterního enzymatického souboru byly bez odchylek. Vyšetření moči neprokázalo patologické nálezy. Ultrasonograficky byla zjištěna normální velikost obou ledvin, bez patologických změn jejich morfologie. RTG vyšetření skeletu neprokázalo ložiska osteolýzy.


Imunoelektroforetickým vyšetřením byla jak v séru, tak v moči zjištěna přítomnost Bence-Jonesovy bíl-koviny (BJB) typu λ. Byla proto indikována biopsie rektální sliznice, která však depozita amyloidu neprokázala. Vzhledem k velmi suspektnímu echokardio-grafickému nálezu incipientní kar-diomyopatie bylo proto následně provedeno invazivní kardiologické vyšetření zahrnující kromě komplexního zhodnocení hemodynamických poměrů i endomyokardiální biopsii. Koronarograficky byl přítomen normální nález na epikardiálních věnčitých tepnách, při pravostranné katetrizaci byla potvrzena lehká postkapilární plicní hypertenze, nebyl přítomen restriktivní charakter tlakových křivek ani vyrovnání diastolických tlaků v srdečních oddílech (střední tlak v plícnici 30 mm Hg, tlak v zaklínění 20 mm Hg, střední tlak v pravé síni 11 mm Hg). Byly odebrány 4 vzorky endomyokardiální biopsie (EMB). Histologické vyšetření, omezené rozsahem vzorku, prokázalo amyloidní depozita s typickým dichroizmem po barvení kongo-červení. Maximum amyloidu mělo vazbu na cévy. Elektronová mikroskopie potvrdila přítomnost amyloidních fibril. Ve spolupráci s hematology byla následně provedena trepanobiopsie kosti kyčelní. Pomocí imunofenotypizace bylo detekováno 1 % plazmatických buněk a při histologickém vyšetření bylo zjištěno nahrazení 10–20 % krvetvorby λ pozitivním plazmocelulárním infiltrátem. Nález byl patologem popsán jako tzv. doutnající myelom, amyloid nebyl v kostní dřeni prokázán. Na základě všech výše uvedených vyšetření celý případ uzavíráme jako počínající infiltrativní kardiomyopatii na podkladě izolované srdeční AL-amyloidózy při tzv. doutnajícím myelomu, s dosud nevyjádřeným restriktivním charakterem poruchy diastolického plnění. Nemocná zůstává ve sledování kardiologů i hematologů, kterými je nyní léčena vysokodávkovanou kortikoterapií. V plánu je dle odpovědi onemocnění na léčbu zvážení autologní transplantace kmenových buněk (autologous stem cell transplantation – ASCT).
Diskuze
Amyloidóza je poměrně vzácné onemocnění způsobené extracelulárním ukládáním amyloidu v různých orgánech či tkáních lidského těla [1]. Depozice může být lokalizovaná pouze na jeden orgán, většinou však bývá postiženo více orgánů současně. Vzácně se jedná o hereditární formu onemocnění, která se přenáší autozomálně dominantně a je podmíněna např. mutacemi genu pro transthyretin či ně-kte-ré ze skupiny apolipoproteinů. Častěji se setkáváme se získanou formou, u níž odlišujeme primární AL-amyloidózu, sekundární AA-amyloidózu a amyloidózu na podkladě depozice divokého transthyretinu (prealbuminu), ke které dochází velmi často v pokročilém věku („senilní amyloidóza“), na rozdíl od mutantního transthyretinu, který vede k závažnější amyloidóze v nižších věkových kategoriích [2,3]. Srdeční postižení je úzce spojeno zejména s AL-amyloidózou. Tento typ amyloidózy je podmíněn klonální proliferací plazmocytů s produkcí monoklonálního proteinu či jeho částí (AL-amyloidóza z lehkých řetězců imunoglobulinů). Vyskytuje se častěji u mužů a nejčastěji bývá pozorována v 6. dekádě života [4]. Kromě srdce se onemocnění manifestuje i postižením ledvin, jater, vzácně makroglosií a purpurou [5]. Infiltrace srdeční tkáně amyloidem vede k postupné deterioraci diastolické funkce LK [6], kdy při plně vyjádřeném srdečním postižení je patrný obraz restriktivní kardiomyopatie [7]. Ve více než 50 % případů bývá na EKG patrna snížená voltáž komplexů QRS, v řadě případů bývají přítomny i kmity QS, které mohou napodobovat EKG obraz jizvy po infarktu myokardu [8]. Echokardiografie, která je základním diagnostickým vyšetřením, prokazuje u typicky vyvinutých případů srdeční amyloidózy difuzní zesílení stěn LK s těžkou alterací její diastolické funkce do podoby restriktivního plnění [9], normální či jen lehce sníženou celkovou systolickou funkci, dilataci obou síní, zesílení chlopenních cípů, velmi častá je přítomnost malého množství perikardiálního výpotku [10]. Pravostranná srdeční katetrizace potvrdí restriktivní typ plnění LK s typickým vyrovnáním diastolických tlaků ve všech srdečních oddílech a tzv. obrazem dip-plateau na tlakové křivce LK. Klíčovým vyšetřením v průkazu srdeční amyloidózy však nadále zůstává přímý průkaz amyloidu v tkáňové biopsii. Vzhledem k častému multiorgánovému postižení je zpravidla prováděno bioptické vyšetření nejprve submukózy rekta, bukání sliznice či kostní dřeně a teprve v případech, kdy jsou tato vyšetření negativní a přetrvává podezření na srdeční amyloidózu, doplňujeme ještě EMB [11].
Námi referovaná kazuistika poukazuje na význam správné interpretace echokardiografického nálezu u nemocného se symptomatologií srdečního selhání, především parametrů dia-stolické funkce LK a nutnost komplexní diagnostiky nejasného postižení srdečního svalu. Echokardiografie dokáže s vysokou přesností zhodnotit nejen morfologii a systolickou funkci, ale také diastolickou funkci LK a tlakové poměry v malém oběhu [12]. U naší nemocné byla přítomna středně těžká diastolická dysfunkce (tzv. pseudonormalizovaný charakter plnění) v terénu lehce koncentricky hypertrofické, nezvětšené LK, která měla normální systolickou funkci. Dále byla prokazována dilatace obou síní a lehká, nejspíše postkapilární plicní hypertenze. Tento nález ve spojení s námahovou dušností ukazoval na přítomnost diastolického srdečního selhávání. Vzhledem k absenci arteriální hypertenze v anamnéze a aortální stenózy nebylo možné vysvětlit koncentrickou hypertrofii levé komory jejím chronickým tlakovým přetížením, a proto byla v diferenciální diagnóze zvažována na prvém místě možnost počínající infiltrativní kardiomyopatie, s ohledem na zbytnění stěn LK především amyloidóza. Dále bylo možné spekulovat o hypertrofické kardiomyopatii, tato se však jevila málo pravděpodobnou vzhledem ke kombinaci jen mírné hypertrofie stěn LK s již středně těžkou poruchou dia-stolické dysfunkce, která není pro hypertrofickou kardiomyopatii typickou, a dále pro absenci EKG známek hypertrofie LK, jež je charakteristicky nacházena právě u srdeční amyloidózy.
Klinická manifestace srdeční amyloidózy je v literatuře klasicky uváděna jako restriktivní kardiomyopatie charakterizovaná těžkou diastolickou dysfunkcí LK. Restriktivní postižení myokardu je však až konečným stadiem srdeční amyloidózy a k jeho vývoji dochází postupně spolu s progresí infiltrace myokardu substrátem. Případ naší nemocné názorně dokumentuje, že v počínajících fázích postižení srdečního svalu vede depozice amyloidu nejprve k mírnějším až středním formám diastolické dysfunkce LK. V diferenciální diagnostice mírné koncentrické hypertrofie LK s vyjádřenou poruchou jejího plnění, pro níž není podklad v dlouhodobém tlakovém přetížení, je proto nutné na možnost incipientní srdeční amyloidózy myslet. Skutečnost, že pokročilá diastolická dysfunkce LK nemusí být zpočátku plně vyjádřena a echokardiografickému obrazu dominuje zesílení stěn, reflektuje i nově navržená klasifikace kardiomyopatií Evropské kardiologické společnosti, kde je srdeční amyloidóza uváděna jak ve skupině nemocných s fenotypem hypertrofické, tak i restriktivní kardiomyopatie [13].
Nejčastější prvotní orgánovou manifestací infiltrace amyloidem bývá postižení ledvin [2]. Na příkladu naší nemocné je však patrné, že zejména v počínajících fázích onemocnění může být izolované postižení myokardu jedinou známkou choroby. Bioptické vyšetření jiných orgánů, které bývá při diagnostice amyloidózy běžně prováděno, proto nemusí patologická depozita amyloidu odhalit. Proto je nutné při silném podezření na možnost infiltrativního postižení srdce (v našem případě echokardiografický obraz a přítomnost Bence-Jonesovy bílkoviny v séru i moči) přistoupit k provedení EMB. Jelikož EMB je výkonem, který je zatížen určitým rizikem komplikací, a diagnostika kardiomyopatií je procesem komplexním, je vhodné nemocné s nejasným nálezem srdečního postižení vyšetřovat v centrech, která se problematikou kardiomyopatií zabývají a jsou schopna ve spolupráci s dalšími lékařskými specializacemi, v tomto případě s hematology, postižení myokardu amyloidem nejen diagnostikovat, ale také adekvátně léčit.
Také z hlediska léčby základního onemocnění by mělo platit pravidlo o centralizaci pacientů na specializovaných pracovištích. Platí to zejména pro ty nemocné, u kterých připadá v úvahu indikace zatím, podle většiny autorů, nejúčinnějšího terapeutického postupu u AL-amyloidózy – vysokodávkované chemoterapie s podporou ASCT. Rozhodnutí o indikaci této léčby je nelehké, neboť peritransplantační mortalita je 4–8krát vyšší než např. u příbuzného onemocnění – mnohočetného myelomu. Navíc hlavním faktorem, který (kromě rozsahu onemocnění, resp. počtu postižených orgánů) riziko mortality dále zvyšuje, je právě symptomatická amyloidóza srdce [14]. V případě námi referované nemocné sice chybí ně-kte-ré negativní prognostické faktory pro indikaci ASCT (známky závažnější kardiální nedostatečnosti, ejekční frakce levé komory < 40 %), uspokojivý klinický efekt úvodní kortikoterapie a postoj pacientky nás zatím k tomuto postupu nevedl. Pro konzervativnější indikaci vysokodávkované terapie svědčí i výsledky nedávné randomizované studie, podle kterých ASCT nemusí být jednoznačně efektivnějším postupem proti konvenční léčbě [15].
Tato práce byla podpořena Výzkumným záměrem Univerzity Karlovy v Praze č. MSM 00 21620817, uděleným Ministerstvem školství, mládeže a tělovýchovy České republiky.
MUDr. Tomáš Paleček
www.lf1.cuni.cz
e‑mail: kardiomyopatie@seznam.cz
Doručeno do redakce: 27. 4. 2008
Přijato po recenzi: 3. 6. 2008
Sources
1. Pepys MB. Amyloidosis. Annu Rev Med 2006; 57 : 223–241.
2. Adam Z, Sčudla V. Clinical maniofestation of AL-amyloidosis and some other types of amyloidosis. Vnitř Lék 2001; 47 : 36–45.
3. Shah BK, Inoue Y, Mehra MR. Amyloidosis and the heart. Arch Intern Med 2006; 166 : 1805–1813.
4. Dubrey SW, Cha K, Anderson J et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. Q J Med 1998; 91 : 141–157.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 patients. Semin Hematol 1995; 32 : 45–49.
6. Palka P, Lange A, Donnelly E et al. Dop-pler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr 2002; 15 : 1353–1359.
7. Brychta T, Pařenica J, Zatočil T et al. Restrictive cardiomyopathy as a manifestation of primary amyloidosis. Vnitř Lék 2004; 50 : 66–71.
8. Murtagh B, Hammill SC, Gertz MA et al. Electrocardiographic findings in primary systemic amyloidosis and biopsy proven cardiac involvement. Am J Cardiol 2005; 95 : 535–537.
9. Klein AL, Hatle LK, Burstow DJ et al. Doppler characterization of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 1989; 13 : 1017–1026.
10. Linhart A, Paleček T, Aschermann M. Echokardiografie pro praxi. Praha: Audioscan 2002 : 117–118.
11. Pellikka PA, Holmes DR, Edwards WD et al. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med 1988; 148 : 662–666.
12. Oh JK, Hatle LK, Tajik AJ et al. Diastolic heart failure can be diagnose by comprehensive two-dimensional and Doppler echocardiography. J Am Coll Cardiol 2006; 47 : 500–506.
13. Elliott P, Anderson B, Arbustini E et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29 : 270–276.
14. Comenzo RL, Gertz MA. Autologous stem cell transplantation for primary systemic amyloidosis. Blood 2002; 99 : 4276–4282.
15. Jaccard A, Moreau P, Leblond V et al. High‑dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007; 357 : 1083–1093.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2008 Issue 10
Most read in this issue
- Duální protidestičková léčba
- Srdeční arytmie při obstruktivní spánkové apnoe
- Hemoeliminační metody v léčbě sepse: současný stav
- Hemofilie