
Histiocytóza z Langerhansových buněk u osob dospělého věku – nemoc s mnoha tvářemi.
Zkušenosti jednoho pracoviště a přehled projevů nemoci
Langerhans cell histiocytosis in adult patients – a disease with many faces
Experience of a centre and an overview of the disease symptoms
Over a period of 18 years, 17 patients with proven Langerhans cell histiocytosis (LCH) were treated at the Haematological Clinic in Brno. In 13 of them, the disease was diagnosed at adult age, and 4 patients were referred to the centre with LCH diagnosed at early child age. One of these 4 patients suffered from repeated recurrences of the disease at adult age and was diagnosed with progressive neurodegenerative damage of the CNS at the age of 25 which in its terminal phase resulted in the patient’s immobility, loss of sphincter control, incapacity to communicate and death at the age of 32. LCH was diagnosed at adult age in 13 patients. The form with primary bone involvement was detected in 8 out of 13 patients (62%). Only 2 of 13 patients (15%) had multiple bone lesions upon diagnosis, the remaining 6 patients (46%) had only one lesion at the time of diagnosis. Repeated recurrence of bone involvement was only recorded in 3 out of 13 patients (23%). The combination of recurrent bone involvement and the development of lung affection (dyspnoea, irritating cough, nodularities and cysts in HRCT images) were documented in 2 out of 13 patients (15%). One of the patients diagnosed with LCH at the age of 37 had repeated recurrence of bone involvement, which was also treated by 2 cycles of high-dose chemotherapy and autologous transplantation. He died of bronchopneumonia due to the affection of the lungs by LCH at 48 years of age. Primary extraoseal (extamedular) involvement was diagnosed in 5 out of 13 patients (38%) (mandibular gum infiltration, single cervical node infiltration, hand skin infiltration, infiltration of the perineal region and infiltration of the hypophysial infundibular and primary lung form of LCH). In the 1st case, excision was the solution applied to the infiltration of the lingual side of the gums, without further recurrence. In the 2nd case, the infiltrated region of skin over the metacarpophalangeal joint was irradiated and the infiltration disappeared. In the 3rd case, the first sign of the disease was diabetes insipidus in a 34-year old man, and an infiltrate in the anal region similar to condylomata acuminata. The diagnosis was confirmed 2 years after the development of diabetes insipidus from perianal infiltrates. After treatment with leustatin in 4 cycles (10 mg a day for 5 consecutive days), control MR showed that the infiltration in the hypophysial infundibular had disappeared, while the finding in the perianal region only regressed by 50% after therapy with leustatin, the reason for subsequent application of radiotherapy (20 Gy). The finding in the perianal region is normal one year after therapy, but substitution therapy with adiuretin is still necessary. The 4th patient was a case of LCH with primary pulmonary involvement diagnosed on the basis of HRCT and lavage with an immunohistochemical proof (expression of CD1 and of protein S-100) of a high number of Langerhans cells. The occurrence of LCH at adult age is rare and the disease may affect the skeleton as well as other organs. Therefore each new osteolytic lesion should be submitted for histological exam, as well as each pathologic formation, because diagnosing the disease without a microscopic and immunohistochemical exams is not possible. In the case of occurrence of diabetes insipidus at adult age, LCH should be considered as one of the possible underlying diseases. LCH pulmonary involvement should be considered in patients with an interstitial pulmonary process and the examinations should be focused accordingly (thoracoscopy with sampling for histological exams or bronchoalveolar lavage) plus the indispensable immunohistochemical examination.
Key words:
Langerhans cell histiocytosis – eosinophillic bone granuloma – osteolysis – diabetes insipidus – multiple myeloma – 2-chlordeoxyadenosin – high-dose chemotherapy with autologous transplantation
Authors:
Z. Adam 1; L. Pour 1; M. Krejčí 1; J. Neubauer 2; J. Vaníček 3; V. Vašků 4; R. Hájek 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, zastupující přednosta prof. MUDr. Jiří Mayer, CSc.
1; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil A. Válek, CSc.
2; Klinika zobrazovacích metod Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Petr Krupa, CSc.
3; I. dermatovenerologická klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MUDr. Vladimír Vašků, CSc.
4
Published in:
Vnitř Lék 2008; 54(11): 1063-1080
Category:
Review
Overview
V průběhu 18 let bylo na Interní hematoonkologické klinice v Brně léčeno a sledováno celkem 17 pacientů s prokázanou histiocytózou z Langerhansových buněk (LCH). U 13 z nich byla nemoc diagnostikována v dospělém věku, 4 pacienti k nám byli předáni s LCH diagnostikovanou již v raném dětském věku. U jednoho z těchto 4 pacientů nemoc v dospělosti opakovaně recidivovala a ve věku 25 let bylo diagnostikováno progredující neurodegenerativní poškození CNS, které v terminální fázi vedlo k nehybnosti nemocného, neovládání svěračů, neschopnosti komunikace a ke smrti v 32 letech. V dospělosti byla LCH diagnostikována u 13 nemocných. Primárně kostní formu mělo 8 ze 13 (62 %). Mnohočetná kostní ložiska při stanovení diagnózy měli pouze 2 ze 13 (15 %) nemocných, ostatních 6 (46 %) nemocných mělo pouze 1 ložisko v době stanovení diagnózy. Opakované kostní recidivy nemoci byly zaznamenány u 3 ze 13 (23 %). Kombinace recidivující kostní formy s rozvojem plicního postižení LCH (dušnost, dráždivý kašel, nodularity a cysty při HRCT zobrazení) bylo dokumentováno u 2 z 13 (15 %) osob. Jeden z těchto nemocných s LCH diagnostikovanou ve 37 letech měl opakované kostní recidivy, léčené mimo jiné 2 cykly vysokodávkované chemoterapie s autologní transplantací, a nakonec ve 48 letech života zemřel na broncho-pneumonii při postižení plic LCH. U 5 ze 13 (38 %) byla diagnostikována primárně extraoseální forma (infiltrát dásní, infiltrace jedné krční uzliny, infiltrace kůže ruky, infiltrace perianální krajiny a stopky hypofýzy a primárně plicní forma LCH). V 1. případně infiltrace linguální strany dásní byla řešena excizí bez další recidivy. V 2. případě infiltrovaná oblast kůže nad metakarpofalangeálním skloubením byla ozářena a morfa vymizela. Ve 3. případě byl prvním projevem nemoci diabetes insipidus, vniklý u 34letého muže, a infiltrát v anální krajině podobný akuminátním kondylomatům. Diagnóza byla ověřena po 2 letech od vzniku diabetes insipidus z perianálních infiltrátů. Po léčbě leustatinem 4 cykly (10 mg denně 5 dní po sobě) vymizela na kontrolním MR infiltrace v oblasti stopky hypofýzy, morfy v perianální krajině však ustoupily po ukončení léčby leustatinem jen z 50 %, a proto následovala radioterapie (20 Gy). Rok po ukončení léčby je normální nález v perianální krajině, ale trvá nutnost substituce adiuretinu. Ve 4. případě se jednalo o primární plicní LCH, diagnostikovanou na základně HRCT a laváže s imunohistochemickým průkazem (exprese CD1 a proteinu S-100) vysokého počtu Langerhansových buněk. LCH v dospělosti je výjimečnou nemocí, která může poškodit nejen skelet, ale i jiné orgány. Proto by každé nové osteolytické ložisko mělo být podrobeno histologickému vyšetření, stejně tak jako každé nejasné patologické morfy, protože bez mikroskopického a imunohistochemického vyšetření nelze tuto nemoc rozpoznat. Pokud v dospělosti vznikne diabetes insipidus, je nutno mimo jiné hledat etiologii v LCH. U nemocných s intersticiálními plicními procesy je nutno myslet na plicní formu LCH a provádět odpovídající vyšetření (torakoskopie s odběrem materiálu na histologii nebo bronchoalveolární laváž) s neopomenutelným imunohistochemickým vyšetřením.
Klíčová slova:
histiocytóza z Langerhansových buněk – eozinofilní kostní granulom – osteolýza – diabetes insipidus – mnohočetný myelom – 2-chlordeoxyadenosin – vysokodávkovaná chemoterapie s autologní transplantací
Úvod
Histiocytóza z Langerhansových buněk (LCH) je nozologická jednotka o velmi širokém spektru forem a závažnosti klinických projevů, lze říci, že tato nemoc představuje chameleona mezi krevními nemocemi. Projevy LCH u dospělých počínají náhodným RTG nálezem jednoho osteolytického ložiska, ale mohou mít podobu i generalizované systémové choroby. Dříve, vzhledem ke své tajuplnosti, nejasné etiologii a rozpakům, zdali toto onemocnění řadit k nádorovým nebo infekčním onemocněním či lipidovým tezaurismózám, bylo nazýváno histiocytosis X.
Incidence LCH v ČR zcela přesně neznáme. Nicholson uvádí incidenci 5/1 milion všech osob s tím, že u dětí je tato nemoc podstatně častější [1]. V České republice je dětskými hematology a onkology ročně registrováno asi 15 dětí s nově diagnostikovanou LCH, počet dětí s izolovanou kostní formou LCH, která nevyžaduje jinou než chirurgickou léčbu, není znám. Němečtí autoři uvádějí incidenci LCH u dospělých 1–2 případy/1 milion obyvatel [2]. Manifestaci u dětí shrnuje tab. 1. Díky nízké incidenci je nutné získávat informace o této nemoci z popisů jednoho či více případů, klinické studie se u takto vzácné nemoci organizují velmi obtížně [3–11].

Pokud se nemoc projeví až v dospělosti, dominuje kostní manifestace. Méně častý je souběh kostního a plicního postižení. Podle analýzy provedené Baumgartnerem [2] je postižení hypofýzy, lymfatických uzlin a jater v dospělém věku vzácné, ale vyskytuje se. Při analýze projevů této nemoci u dospělých byla získána následující čísla: v 80 % bývají postiženy kosti, v 60 % kůže, ve 33 % játra, slezina a uzliny, ve 30 % kostní dřeň, ve 25 % plíce, ve 25 % orbita, ve 20 % ucho a orodentální oblast, kde nemoc může zapříčinit uvolňování zubů. U dospělých pacientů má onemocnění velmi různorodý průběh. U některých postihne jenom jedno ložisko a po léčbě se již neobjeví, u jiných má recidivující charakter, u pacientů jsou objevována stále nová a nová ložiska a choroba může být příčinou omezené hybnosti či může dokonce přivodit smrt [9,12]. Formy LCH u dospělých shrnuje tab. 2.
![Klasifikace Langerhansovy histiocytózy u dospělých [13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/36d69ee86b3395e5b66d4347288e834b.png)
Zásadním vyšetřením, nutným pro sta-novení diagnózy, je samozřejmě bioptická excize. Pro nepatologa je vhodné jen vědět, že v době akutní choroby jsou v ložisku přítomny četné Langerhansovy buňky, exprimující proliferační markery a eozinofily. Později v ložisku ubývá Langerhansových buněk, přibývá makrofágů a fibrocytů a nakonec obraz odpovídá postnekrotické fibróze. Někdy může být obtížné rozlišení osteomyelitidy od kostních ložisek histiocytózy, stejně jako odlišení plicní formy histiocytózy od jiných granulomatózních plicních procesů. Histologický vývoj ložiska Langerhansovy histiocytózy lze tedy rozdělit do následujících fází:
- proliferativní stadium (převažují Lan-gerhansovy buňky)
- granulomatózní stadium (pestrá cytologie)
- xantomatózní stadium a tvorba jizev
K potvrzení diagnózy Langerhansovy histiocytózy se proto používají další znaky, jejichž přítomnost je typická:
- protein S-100
- cytoplazmatická ATP-áza a D-manosidáza
- průkaz Birbeckových granulí v cytoplazmě elektronovou mikroskopií
- přítomnost antigenu CD1a
- přítomnost antigenu CD45
- antigen CD68 se vyskytuje nepravidelně
Pro Langerhansovu histiocytózu je patognomický nález antigenů II. HLA třídy, přítomnost CD1a antigenu a často i přítomnost antigenů fyziologicky se nacházejících na fagocytujících histiocytech: CD11c a CD14. Dále jsou uváděny následující znaky CD1a++, CD 11c++, CD44++ ,CD54++, CD58++. Znaky CD2, CD11a, CD11b jsou na těchto patologických buňkách výjimečně a ve slabé intenzitě.
WHO klasifikace uvádí dále termín Langerhans cell sarkoma, který je vyhrazen pro maligní proliferaci Langerhansových buněk, které dle svojí morfologie mají známky dediferenciace a obvykle také agresivnější sarkomatózní růst a přitom imunofenotyp Langerhansových buněk.
Morfologické změny v ložisku v průběhu času, k nimž nutno přihlížet při interpretaci histologického nálezu, a morfologické znaky LCH shrnuje tab. 3.

Popisy případů
Dospělé osoby s LCH diagnostikovanou v dětském věku
Pacientka nar. 1978. Diagnóza LCH byla stanovena v 10 měsících (nemoc Lettererova a Siweho) s dominující kožní formou. Pacientka byla léčena v letech 1979–1981 prednisonem a vinkristinem. Poslední kontrola u nás byla v roce 2004 – bez recidivy. Je to jediná pacientka předaná k nám z pediatrického pracoviště, která zůstává stále bez recidivy nemoci.
U druhého pacienta, muže, nar. 1975, se nemoc projevila také kožní formou v 1. roce života a od počátku nemoci byl zjištěn diabetes insipidus. K 2. atace nemoci zřejmě došlo v roce 1993, kdy náhle vznikly neurologické kmenové příznaky, centrální vestibulární syndrom, neúplný neocerebelární syndrom vpravo a naznačená pravostranná pyramidová symptomatologie. Tyto problémy spontánně téměř ustoupily a mladý muž je dále u nás sledován bez recidivy nemoci, od dětství je však nutná trvalá substituce adiuretinu. V době příznaků bylo sice provedeno zobrazení CNS metodou CT, ale žádná patologie není v popisu CT uvedena. V literatuře jsou popisovány podobné ataky LCH s uvedenými neurologickými příznaky [13].
Také třetí pacientka, nar. 1974, měla diagnózu LCH stanovenu ve 2 letech (1976), kdy se nemoc manifestovala kožními příznaky a diabetes insipidus. Po léčbě vinkristinem a prednisonem byla dlouhodobě bez známek recidivy, ale s nutností trvalé substituce adiuretinu. Na rozdíl od předchozích však v roce 1996 (ve 20 letech) byla zjištěna indurace v okcipitální oblasti s defektem kosti, zřetelným na RTG snímku. Pacientka byla ozářena ložiskovou dávkou 20 Gy. K další recidivě do roku 2008 nedošlo. Pacientka se po provdání pokoušela otěhotnět pomocí in vitro fertilizace, která byla opakovaně neúspěšná. Endokrinologové vysvětlili neúspěchy in vitro fertilizace deficitem somatotropinu, bez jehož dostačující substituce jsou pokusy o in vitro fertilizaci zbytečnou investicí ze strany nemocné. Pacientka má panhypopituarizmus s deficitem nejen adiuretinu, ale také TSH a STH a gonadotropinů.
Čtvrtý pacient, nar. 1976, s LCH dia-gnostikovanou v dětském věku, měl téměř chronický průběh této nemoci. Prvním příznakem nemoci byl diabetes insipidus (1981) zjištěný v 5. roce života. V roce 1985 bylo zjištěno zpomalení růstu. Diagnóza LCH byla stanovena až v roce 1988. V té době měl exoftalmus, diabetes insipidus, kostní defekt v oblasti levé orbity, čili klasický obraz Handovy-Schillerovy-Christianovy nemoci.
V roce 1993 byla zjištěna infiltrace kůže perianální krajiny – později histologicky ověřena LCH této kožní oblasti. Při předání z pediatrického pracoviště v roce 1996 došlo k recidivě v mandibule, což se projevilo bolestivým zduřením. Histologicky byla potvrzena další ataka LCH. Lokální recidiva v mandibule byla řešena radioterapií a na ni navázala léčba vinblastinem 10 mg i.v. v týdenních intervalech, celkem 8krát, ale od 4. aplikace byla redukována dávka na 5 mg pro neutropenii. Další progrese byla zjištěna v roce 1998, na scintigrafii skeletu i RTG snímku bylo nové ložisko v os parietalis a pouze na scintigrafii ložisko v pravém akromiu. Následovala chemoterapie etoposid + dexametazon a lokální ozáření kalvy cíleně na kostní ložiska.
V září roku 2001 došlo k výraznému zhoršení, které se ohlásilo zduřením nad levým okem. Zduření a infiltrace v této oblasti někdy ohlašuje současné postižení CNS. Zduření nad levým okem se zřetelným ložiskem na kalvě bylo léčeno lokální radioterapií a pulsy dexametazonu.
Po měsíci se objevila diplopie, ataxie a další neurologické problémy. O pár týdnů později prudce vzestoupily veškeré standardně vyšetřované jaterní enzymy, aniž by byla prokázána infekční hepatitida. Pro neurologické příznaky jsme provedli MR vyšetření CNS. Interpretace MR nálezu nebyla jednoznačná, a protože jde o komplikaci, která je velmi, ale opravdu velmi výjimečná a běžný rentgenolog se s ní nesetká, konzultovali jsme MR obraz s prof. Gadnerem z AKH Vídeň. Naši lékaři z oddělení radiologické kliniky i prof. Gadner se shodli na závěru: Neurodegenerativní změny odpovídající pozdní komplikaci Langerhansovy histiocytózy.
Přesný popis MR zněl: V oblasti bazálních ganglií a dále v bílé a šedé hmotě mozkové byly nalezeny změny odpovídající neurodegenerativním změnám typickým pro LCH. Difuzní hyperintenzivní ložiska v oblasti mostu odpovídala spíše leukoencefalopatii než LCH. Infundibulum bylo zesílené.
Vzhledem k nejasnosti, zda se jedná o nádorovou infiltraci či degeneraci, bylo provedeno také PET vyšetření v Praze, kde byla popsána zvýšená kumulace v oblasti mozkového kmene. Při kontrole PET po roce již v této oblasti nebyla zvýšená kumulace značené glukózy, byl zřetelný pouze lehce asymetricky vyšší metabolizmus glukózy bez jednoznačných ložiskových změn. Ovšem ani tento zprvu pozitivní, posléze negativní PET nález neodpovídá na otázku, zda šlo o infiltraci Langerhansovými histiocyty, nebo zda šlo o zánětlivé a degenerativní změny, neboť v obou případech může být zvýšeně vychytávána 18fluorodeoxyglukóza.
Vzhledem k rychlému zhoršování stavu jsme usoudili na další, tentokráte výrazně agresivnější ataku nemoci s postižením jater a CNS. V říjnu roku 2001 byla proto zahájena léčba 2-chlordeoxyadenosinem.
Po 1. aplikaci 2-chlordeoxyadenosinu rychle poklesly hodnoty jaterních enzymů, takže jsme předpokládali postižení jater touto chorobou i bez histologického ověření.
Po 6 cyklech chemoterapie byl v únoru roku 2002 PET nález negativní, neurologické změny zůstávaly však beze změn, MR obraz CNS se nezměnil. Vyjma normalizace hodnot jaterních enzymů měla tato léčba ještě jeden pozitivní dopad: LCH infiltrace perianální krajiny vymizela a do smrti nemocného již nerecidivovala. To se předtím při lokální kortikoidní léčbě ani při předchozí chemoterapii nepodařilo.
Neurologický stav nemocného se postupně zhoršoval, pacient nebyl schopen ovládat svěrače, byl upoután na vozík a dysartrie velmi ztěžovala komunikaci. V dubnu roku 2008 pacient umřel, dle pitvy byla příčinou plicní embolie.
RTG skeletu: vyjma ložisek v oblasti kalvy nebyla na RTG snímcích dalších kostí zřetelná osteolytická ložiska. Kostní hustota Z skóre, SD od –4 do –5 SD. Těžká osteoporóza sou-visí zřejmě s insuficientní funkcí hypofýzy. Přetrvávající neurodegenerativní poškození CNS bylo následkem předchozí aktivity nemoci, takže stav jsme hodnotili jako remisi nemoci, trvající 6 let od ukončení léčby 2-chlordeoxyadenosinem.
Osudy nemocných s diagnózou LCH stanovenou v dětském věku shrnuje tab. 4.

Jednoložisková forma LCH s první manifestací v dospělém věku, bez dalších recidiv
U pacientky nar. 1978 se v roce 1996(18 let) objevila hmatná a citlivá rezistence v oblasti pravé parietální kosti s osteolytickým ložiskem pod touto rezistencí, zřetelném na RTG snímku. Byl proveden odběr materiálu pro histologické vyšetření, prokázána LCH a následně bylo toto místo lokálně ozářeno. Od té doby nedošlo k dalším recidivám.
Muž, nar. 1973. Diagnóza byla stanovena ve 25 letech (1998), kdy byla zjištěna izolovaná osteolýza postihující C4–C5. Následovala operační léčba s odstraněním měkkých nádorových hmot a fúzí obratlů C3–C6 kovovou dlahou, fixovanou 6 šrouby, s následnou radioterapií. Poslední kontrola pacienta zobrazovacími metodami byla v květnu roku 2005 v rozsahu scintigrafie skeletu, RTG snímku a MR páteře. Žádné vyšetření neprokázalo recidivu nemoci. A do roku 2008 se neobjevily nové symptomy, které by vyžadovaly další vyšetření. Doufáme, že v tomto případě se jednalo o izolované jednoložiskové postižení.
Stejně tak u ženy nar. 1980 se jednalo o jednoložiskové postižení. Diagnóza byla stanovena v 18 letech (1998), kdy bylo pro zvýšenou citlivost a hmatnou rezistenci v oblasti temporální kosti provedeno RTG vyšetření s nálezem osteolýzy. Histologie ložiska prokázala eozinofilní granulom jako jednu z forem LCH. Následovala lokální radioterapie (1998) a žena je od té doby bez recidivy.
V jednom případě jsme se setkali s uzlinovou formou LCH vzniklou v dospělosti. U pacienta nar. 1962 byla diagnóza stanovena v roce 2001, ve věku 39 let, z izolovaného zvětšené uzliny na krku. Následovalo CT media-stinálních a břišních uzlin, RTG skeletu doplněný magnetickou rezonancí zaměřenou na páteř a na femory, kde byl RTG nejasný nález. Žádné další kostní ložisko ani lymfadenopatie nebyly zjištěny. Vzhledem k tomu, že šlo o totálně extirpovanou uzlinu s nálezem histiocytózy, nebylo ani ozářeno místo po exstirpaci. Poslední kontrola u nás byla v roce 2007, kdy byl pacient bez známek relapsu nemoci.
Muž narozen 1984 měl v roce 2002(18 let) bolesti hlavy a později se na levém spánku objevila boule citlivá na tlak. V září roku 2002 byl vyšetřen na neurochirurgii, byla provedena exkochleace osteolytického ložiska kalvy a stanovena diagnóza. Další vyšetření nepotvrdila jiná ložiska, a tak byla diagnóza stanovena jako solitární eozinofilní granulom a byl ozářen 40 Gy. Od té doby bez recidivy.
Pacienty, u nichž byla stanovena diagnóza unifokální kostní formy LCH, shrnuje tab. 5.

Nemocní s první manifestací LCH v dospělém věku a s recidivami nemoci anebo s mimokostním postižením
Velmi komplikovaný průběh měla LCH u pacienta nar. 1964. Tento mladý muž neměl do svých 37 let závažnější onemocnění. V roce 1991 (v 37 letech věku) bylo nalezeno osteolytické ložisko ve femoru – řešeno ortopedicky, histologicky ověřena LCH. Po operaci následovala radioterapie. Za 2 roky, v roce 1993, byla RTG zjištěna další ložiska v oblasti pánve a obou femorů. Scintigrafie kostí popsala ještě ložiska v mandibule a pravé lopatce. Ložiska v pánvi a v L5 byla ověřena CT zobrazením.
U této víceložiskové formy jsme se rozhodli pro podání 7 cyklů chemoterapie CHOP (adriamycin, vinkristin, cyklofosfamid a prednison). První remise trvala 3 roky.
V březnu roku 1996 byla zjištěna nová ložiska v oblasti žeber a Th páteře – řešeno zářením a podáváním vinblastinu 1krát týdně po dobu 6 týdnů.
V červenci roku 1997 byla opět nová ložiska na paži v hlavici humeru a asi i další ložiska v páteři. Zároveň se nově objevil neproduktivní dráždivý kašel, námahová dušnost, bolesti na hrudníku. Proto bylo provedeno vyšetření plic metodou HRCT (high resolution CT neboli vysoce rozlišovací CT), výsledek byl kompatibilní s plicní formou histiocytózy, ale nebyl dále ověřen. Následovala chemoterapie (etoposid a prednison) a radioterapie na oblast humeru. Vzhledem k častým recidivám a neuspokojivému výsledku běžné chemoterapie byl v září roku 1997 proveden sběr kmenových krvetvorných buněk a následně podána 1. vysokodávkovaná chemoterapie (melfalan a etoposid) s podporou transplantace autologních krvetvorných buněk. Remise po této léčbě trvala jenom 2,5 roku, do června roku 2001, kdy byla scintigraficky zjištěna nová ložiska. Následovaly tedy opět 3 cykly chemoterapie (etoposid + dexametazon) a ozáření těchto ložisek. Po krátkém klidovém období se v červenci roku 2002 objevila ložiska na žebrech a páteři, zřetelná nejen na scintigrafii, ale i na RTG snímku. Pro tento relaps byla opět provedena stimulace s opakovaným sběrem kmenových krvetvorných buněk z periferní krve a následně podána 2. vysokodávkovaná chemoterapie.
Po této 2. vysokodávkované chemoterapii měla remise jen krátké trvání, 5 měsíců, a opět se objevovala další kostní ložiska, takže dále pokračovala jen paliativní perorální chemoterapie melfalanem. V únoru roku 2002 byl pacient s aktivním onemocněním naposledy na naší ambulanci a v témže roce zemřel.
V roce 1993 (21 let) vznikla u pacienta nar. 1972 první ataka LCH, která se projevila bolestí v pravé stehenní kosti a dále v obratli Th12. Osteolytická ložiska byla operována, čímž byla zjištěna diagnóza a léčena operačně alogeními štěpy. Pak byl pacient pouze sledován bez další léčby.
V roce 2003, po 10 letech, se nově objevila bolest žeber, páteře a kalvy. Na RTG snímcích bylo osteolytické ložisko v kalvě, které bylo potvrzeno CT zobrazením, průměr ložiska byl 21 mm. Na pravé straně hrudníku bylo velké osteolytické ložisko v 7. žebru a velmi suspektní nález byl také na 6., 5. a 4. žebru. Na páteři byla nápadná nerovná dolní krycí ploténka Th8 a byla zřetelná přestavba krčku pravého femoru. V těle obratle L4 CT prokázalo 3 osteolytická ložiska, největší 15 × 14 × 9 mm, a další ložiska byla i v těle obratle L5. Zobrazení páteře pomocí MR potvrdilo ložiska také v obratlech Th11, L2, L4, L5. Vzhledem k 10letému klidovému intervalu bylo resekováno 7. žebro. Histologické vyšetření prokázalo recidivu LCH.
Protože zobrazovací metody (RTG, scintigrafie skeletu, CT a MR) prokázaly mnohočetné osteolytické postižení skeletu, přistoupili jsme k systémové chemoterapii. Pacient dostal 5 cyklů chemoterapie (2-chlordeoxyadenosin 5 mg/m2 i.v. 1.–5. den). Léčba byla ukončena v listopadu roku 2003.
Poslední kontrola proběhla v prosinci roku 2005. Nález PET vyšetření byl negativní a MR páteře neprokázalo žádnou progresi, jsou však stále zřetelná původní ložiska, remise trvá déle než 2 roky. V roce 2007 při kontrole nespecifické potíže, dušnost a bolesti v oblasti hrudního koše. Zobrazovací vyšetření nepopsala progresi ve skeletu. Pro dušnost provedeno HRCT vyšetření, na němž byly popsány cystické okrsky různé velikosti, místy splývající, dále četné nodularity i fibrózní proužky. Tento nález je kompatibilní s nálezem plicní formy histiocytózy z Langerhansových buněk. Mladý muž podstoupil také bronchoskopii s bronchoavelolární laváží, v níž převažovaly makrofágy. Nebylo však provedeno imunohistochemické vyšetření přítomnosti buněk exprimujících CD1a antigen a protein S-100, bez nichž se nemůže hodnotící vyjádřit k přítomnosti či nepřítomnosti plicní formy LCH.
Mladému muži jsme doporučili nekouřit a po 6 měsících by měl mít kontrolní HCRT a funkční vyšetření, pokud bude progrese, měla by být zahájena kortikoidní léčba, jak je u plicní formy histiocytózy z Langerhanosvých buněk standardní, a při její neúčinnosti pak cytostatická léčba.
V případě pacienta nar. 1981 se první příznak objevil v 18. roce života (1999): bolest a rezistence v oblasti 8. žebra. Žebro bylo totálně resekováno a histologicky byla stanovena diagnóza unifokální LCH. Další ložisko nebylo v té době patrné. Po 5 letech, v roce 2004, přišel s bolestmi v oblasti temene, kde si nahmatal rezistenci. Na snímku kalvy měl neostré mapovité projasnění o velikosti 3 × 1 cm, odpovídající kostní formě nemoci. Následovala radioterapie, cílená na kostní ložisko. Poslední kontrola proběhla v dubnu roku 2007, mladý muž měl nejasný nález na MR zobrazení pravého kyčelního kloubu – obraz patologického signálu při okraji hlavice femoru vpravo s malým výpotkem v kloubu, PET negativní. Nemocný bude mít kontrolní zobrazení s cílem odhalit možnou recidivu LCH.
Stejně tak mladý muž nar. 1978 neměl v předchozím životě žádné vážné onemocnění, až v letech 2002/2003 (24 let) bylo nalezeno osteolytické postižení femoru vlevo. V lednu roku 2003 byla provedena v nemocnici Na Bulovce exkochleace a zpevnění hřebem. Histologie ložiska stanovila diagnózu eozinofilní granulom. Již asi od roku 2002 si povšiml rezistencí (bulek) na hlavě. Největší byla v okcipitální krajině a zvětšovala se. Způsobovala bolest hlavy a posléze i poruchy zraku. Z chirurgicky odstraněných hmot opět histologicky vyšel eozinofilní granulom (leden roku 2003). Rozsah kostního postižení: na snímku lebky osteolytické ložisko okcipitálně o průměru 2 cm. V levé lopatě kyčelní bylo ložisko 8 × 4 cm. Celotělová MIBI scintigrafie: vyšší aktivita v oblasti pánve, další ložisko v oblasti levé orbity, pokračující do os temporale.
Scintigrafie skeletu: ložiska v kalvě, fronto-parietálně a okcipitálně, další ložisko v kosti spánkové vpravo, velké ložisko v lopatě kosti kyčelní vlevo, dále ložiska v pravém femoru. Vyšší aktivita také v oblasti kolenního a talokrurálního kloubu vpravo.
CT kalvy a mozku: defekty v okcipitální oblasti oboustranně.
MR mozku: extraaxiální expanzivní proces oboustranně parasagitálně okcipitálně, šířící se defektem kalvy epidurálně až do oblasti zadní jámy lební, bez infiltrace dury. Suspektní další kostní ložiska frontoparietálně vpravo a parietálně vlevo.
Vzhledem k mnohočetným kostním ložiskům, která však u tohoto nemocného měla již i mimokostní šíření, byl proveden sběr periferních kmenových buněk po předchozí stimulační chemoterapii (etoposid a cyklofosfamid). Pak následovaly 4 cykly chemoterapie – 2-chlordeoxyadenosin 5 mg/m2 i.v. infuzně 1. až 5. den v měsíčních intervalech. Léčba byla ukončena v srpnu roku 2003.
Po ukončení léčby při kontrolním MR vyšetření bylo nalezeno jen velmi drobné reziduum měkkotkáňové expanze. V rozsahu původního defektu kalvy okcipitálně nyní jen pruh středního signálu v rozsahu diploe, bez známek sycení či expanzivních projevů, mající charakter původního infiltrátu nebo regresivních změn v místě původního infiltrátu, což nelze na MR odlišit.
Kontrolní MIBI skeletu: je patrná difuzní kumulace aktivity v páteři, náznakem v pánvi. Nejsou však patrná původní ložiska v kalvě, zřetelná při prvním MIBI vyšetření.
PET po léčbě: bez ložisek aktivity nemoci, PET vyšetření před léčbou bohužel nebylo provedeno.
Remise trvá (květen roku 2008). Stav nemoci u tohoto pacienta před a po léčbě dokumentuje obrazová dokumentace – obr. 1–4.




S primárně kožním projevem LCH přišel mladý muž narozený roku 1986. Tento muž žádné vážnější onemocnění dříve neprodělal. V roce 2004 (18 let) se objevilo nejasné kožní ložisko v oblasti metakarpofalangeálních kloubů. Morfa byla excidována a histolog morfu popsal jako dermatofibrom s příměsí Langerhansových buněk. Elektronová mikroskopie potvrdila Birbeckova granula, takže diagnóza z tohoto ložiska byla nakonec uzavřena jako kožní forma LCH. Následovalo vyšetření s cílem potvrdit či vyloučit generalizaci nemoci (zobrazení skeletu, CT břicha). Místo původního kožního defektu bylo i s bezpečnostním lemem ozářeno dávkou 30 Gy.
V roce 2005 se objevily bolesti bederní páteře. RTG snímek neprokázal patologii, zato MR bederní páteře prokázala difuzní změny kostní dřeně, které jsou jednoznačně abnormálním nálezem. Pro podezření na nepoznanou kostní formu histiocytózy provedli ortopedové cílenou biopsii kosti s odběrem materiálu na histologii. Histologie však byla nekonkluzivní, LCH nebyla potvrzena. PET byl před biopsií negativní. Vzhledem k mladému věku nemocného je pravděpodobné, že změny v oblasti páteře jsou následkem předchozí ataky kostní formy této nemoci a nekonkluzivnost histologického odběru lze mimo jiné vysvětlit tím, že se jednalo o staré ložisko již ve formě jizvení, kdy převládá zánětlivá celulizace. Další sledování ukáže, zda opravdu šlo o jedno kožní ložisko, nebo zda časem vzniknou další nová kostní ložiska.
Muž nar. 1974 byl dříve bez vážnějších nemocí, v roce 2006 se mu objevila rezistence v oblasti dásní, na linguinální straně –7 velikosti asi 8 × 2 mm. Provedena resekce a zjištěna histiocytóza z Langerhansových buněk. Exstirpace byla provedena do zdravé tkáně, takže další léčba nebyla nutná. V roce 2007 přichází s nespecifickými problémy. Jednak měl zduření a bolest pod sternomanubriálním skloubením. Scintigraficky zde byla aktivita a MR zobrazení prokázalo patologický signál, stejně tak patologický signál byl z několika dalších obratlů. Biopsie sterna a její histologické hodnocení nepřinesly objasnění, stejně jako HRCT plic nepřineslo nic patologického, pouze funkční testy prokázaly zvýšenou bronchokonstrikci. Předpokládáme, že v tomto případě teprve další průběh nemoci ukáže, zda se jednalo o jednoložiskové postižení, či zda bude docházet k recidivám, nebo zda první dechové potíže signalizovaly riziko plicní formy LCH.
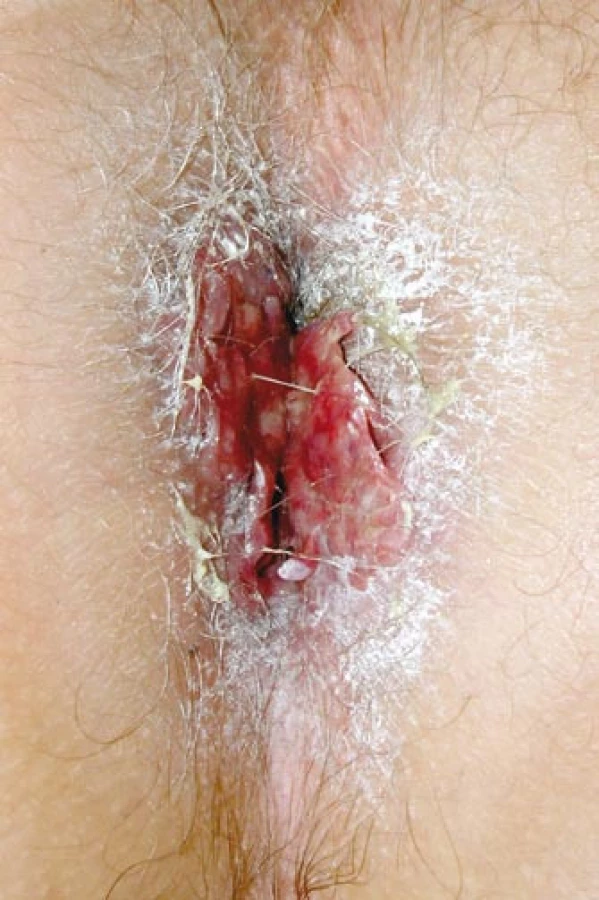
Primární postižení CNS v dospělostije velmi výjimečné. Setkali jsme se s ním u mladého muže nar. 1972, který si poprvé v létě v roce 2005 všiml, že více močí, v listopadu roku 2007 byla internisty uzavřena diagnóza diabetes insipidus. V době vzniku diabetes insipidus se začaly objevovat také bolestivé morfy perianálně. Zpočátku byly mylně hodnoceny jako anální fisury. Z původních fisur však vyrostl květákový útvar o velikosti 4 cm, zaujímající celé anální okolí, hodnoceno jako akuminátní kondylomata (obr. 5). Excize z této morfy byla provedena až po 2 letech jejich trvání, v březnu roku 2007 – histiocytóza z Langerhansových buněk – a s touto diagnózou byl poslán k nám.
První CT mozku roku 2005 nepopsalo nic patologického. MR mozku (2006) však již prokázalo rozšíření stopky hypofýzy (obr. 6 a 7). S tímto nálezem byl sledován bez dalšího zákroku. Etiologie infiltrátu v oblasti stopky hypofýzy nebyla jasná a bez poškození nemocného nebyla ověřitelná.


Na našem pracovišti jsme provedli vyšetření s cílem zjistit přesně rozsah nemoci – vyjma popsaného nebyla jiná postižení prokázána. Mimo jiné bylo provedeno FDG-PET, které v oblasti CNS nezachytilo aktivitu odpovídající maligní tkáni, zatímco v oblasti perianální bylo jasné solitární ložisko zvýšeného metabolizmu glukózy.
Pacient dostal 4 cykly chemoterapie 2-chlordeoxyadenosin, po níž se morfa v anální krajině zmenšila na 50 %. Proto po chemoterapii následovala radioterapie na perianální oblast v dávce 20 Gy. Při kontrolním vyšetření v květnu roku 2008 je perianální krajina bez patologického nálezu, mladý muž může opět vychutnávat radost z jízdy na kole. Kontrolní MR vyšetření zobrazilo již normální stopku hypofýzy. Ta byla původně rozšířena na 4,5 mm a nyní jsou její rozměry v mezích normy – 1,5 mm (obr. 8 a 9). Uvedené 4 cykly 2-chlordeoxyadenosinu vedly k vymizení infiltrátu v oblasti stopky hypofýzy, ale sekrece antidiuretického hormonu se neobnovila. Poškození této oblasti je zřejmě nevratné.


Při zahájení léčby si nemocný stěžoval na viklavost zubů, stav byl stomatologem hodnocen jako náhle vzniklá a výrazná paradentóza a nebyla odebrána žádná histologie. Není jasné, zda to byla koincidence nebo zda to byly projevy nemoci v oblasti čelisti, tak jak jsou popisovány v literatuře a jak je uvádíme v diskuzi.
Poslední pacient, muž nar. 1963, byl bez vážnějšího onemocnění až do března roku 2008. Pro bronchitidu byl přeléčen antibiotiky a pro přetrvávající kašel odeslán na plicní oddělení, kde po snímku plic provedli následně HRCT a diagnostikovali mikronodulární rozsev do 5 mm bilaterálně. Následně byla u tohoto nemocného provedena bronchoalveolární laváž. Z cytologického vyšetření získané tekutiny bylo konstatováno, že jsou přítomny s velkou převahou makrofágy a téměř všechny elementy jsou protein S-100 pozitivní, CD1 pozitivní, což svědčí pro Langerhansovy buňky.
Tímto vyšetřením byla potvrzena diagnóza. Pacient, kuřák, přestal po vysvětlení kouřit a po několika měsících by měl mít kontrolní vyšetření, po němž se dle vývoje rozhodne, zda nemoc vyžaduje či nevyžaduje medikamentózní léčbu. V běhu jsou vyšetření, která by měla potvrdit či nepotvrdit souběh s jinou formou této nemoci.
Pacienty u nás sledované s recidivující formou LCH anebo s mimokostní formou LCH shrnujeme v tab. 6.
Diskuze
Záměrem tohoto textu je jak popsat rozmanitost projevů LCH u pacientů sledovaných na našem pracovišti, tak přinést přehled publikovaných údajů o projevech této nemoci. Léčebné postupy jsme uvedli jinde [15].
Kostní projevy
Histiocytóza z Langerhansových buněk v dospělosti postihuje dominantně skelet, vytváří osteolytická ložiska podobná osteolýze při mnohočetném myelomu. Nejčastěji je uváděno postižení kalvy, následuje pak osteolytické postižení žeber. S menší frekvencí bývají postiženy další části skeletu [16–18].
U některých pacientů jsou kostní ložiska různého stáří, hojící se místa mají sklerotický lem. Ne všechna musí bolet. Zduření tkání přiléhajících ke kosti signalizuje, že choroba prorůstá do okolí a mnohdy zduření nad kostí upozorní na ložisko. Klasickou metodou průkazu ložisek je RTG snímek [19].
Kostní ložiska často způsobují zvýšený kostní obrat, a proto jsou znázornitelná metodou scintigrafie skeletu pomocí techneciumpyrofosfátu [20,21]. Novou metodou, která detekuje ložiska LCH v kostech i jinde, je FDG-PET [22]. Tato metoda vypovídá o aktivitě či neaktivitě ložiska a může pomoci při plánování radioterapie a vyhodnocování léčebné odpovědi [23,24].
Velmi citlivou metodou zobrazení patologického děje v kosti je však magnetická rezonance (MR) [25,26], která je optimální pro plánování rozsahu radioterapie [26].
Za zvláště riziková kostní ložiska jsou považovány kostní defekty v oblasti orbitální se supraorbitálními infiltráty, dále ložiska v kosti temporální. Uvedené postižení je spojené s vyšší pravděpodobností pozdějšího postižení CNS, hlavně cerebella [13].
V našem souboru 17 nemocných se postižení kalvy (primární ložisko či místo recidivy) vyskytlo celkem u 7 (41 %) nemocných. U 1 pacienta se objevilo ložisko supraorbitálně několik měsíců před 1. manifestací poškození CNS. Potvrdila se tedy v literatuře popisovaná zkušenost, že supraorbitální infiltrace často předchází pozdějšímu postižení CNS.
Z podskupiny nemocných s LCH dia-gnostikovanou v dospělém věku byly přítomny kostní projevy u 8 (62 %) nemocných ze 13, přičemž 6 ze 13 (46 %) mělo v době stanovení diagnózy přítomno jen 1 kostní ložisko a 2 ze 13 (15 %) měli v době stanovení diagnózy vícečetné kostní postižení. Kostní forma LCH vzniklá v dospělosti recidivovala u 3 (23 %) ze 13 nemocných.
Z podskupinky nemocných s LCH diagnostikovanou v dětském věku byly pozdní kostní recidivy v dospělém věku diagnostikovány u 2 ze 4 (50 %).
Kožní projevy
Infiltrace kůže buňkami Langerhansovy histiocytózy dominuje u dospělých v oblasti mediální roviny a často postihuje intertriginózní oblasti (perianální krajinu, třísla, pupek, vulvu) [28–34].
Kožní manifestace jsou u Langerhansovy histiocytózy velmi časté a mohou být vůbec prvními zachytitelnými projevy nemoci. Infiltrace kůže buňkami Langerhansovy histiocytózy převládá v oblasti mediální roviny a často postihuje intertriginózní oblasti (perianální oblast, vulva, třísla, pupek). Typickou morfou je hnědorůžová papula o velikosti 1–5 mm, při tendenci ke splývání – zvláště v oblasti kůže kštice – se objevuje i šupení. Popisovány jsou i vezikuly a pustuly. Papulózní projevy jsou často hodnoceny jako nespecifické či ekzémové. Šupící plošky jsou zaměňovány se seboroickou dermatitidou, zvláště u kojenců a malých dětí při postižení vlasaté části hlavy. Vezikulózní projevy mohou napodobovat varicelu i ekzematizovaný scabies. U nejbenignější formy choroby, Hashimoto-Pritzkerovy nemoci, se již krátce po narození objevují mnohočetné nebo solitární červenohnědé uzlíky. Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, v případě závažnějšího průběhu splývají, exulcerují a stav se může komplikovat bakteriální či mykotickou superinfekcí [35,36].
Kožní projevy se mohou sdružovats kostním či viscerálním postižením, může však jít také o izolovanou morfu, která často spontánně regreduje [37–40].
Mimo kůže vznikají někdy změny i na nehtech [41,42].
V našem souboru jsme se s kožními projevy setkali celkem u 3 (18 %) ze 17 nemocných. V prvním případě pacienta nar. 1976 trápilo svědění kůže perianálně, makroskopický nález připomínal ekzém, ale histologicky byla prokázána infiltrace LCH. U dalšího nemocného (nar. 1973) byla postižena taktéž kůže perianální oblasti. Morfy LCH zde tvořily květákovité útvary typu kondyloma acuminatum (obr. 5). U třetího nemocného, nar. 1986, se objevila morfa nad metakarpofalangeálním skloubením.
V 1. případně vymizela infiltrace a normalizoval se obraz kůže perianální krajiny po léčbě 2-chlordeoxyadenosinem, u 2. nemocného květákové útvary se sice při léčbě 2-chloradenosinem zmenšily, ale pro definitivní vymizení byla nutná radioterapie (20 Gy). Třetí muž byl léčen pouze lokální radioterapií. U všech 3 nemocných kožní formy po léčbě vymizely.
Plicní projevy
Respirační cesty jsou postiženy častěji u dospělých než u dětí, 60–100 % pacientů s plicní formou jsou kuřáci. Incidence plicního postižení mezi všemi pacienty s LCH se udává kolem 20 %. Pacienti přicházejí s anamnestickým údajem dušnosti, ale i bolestmi na hrudníku, neproduktivním kašlem, někdy udávají teploty a úbytek hmotnosti.
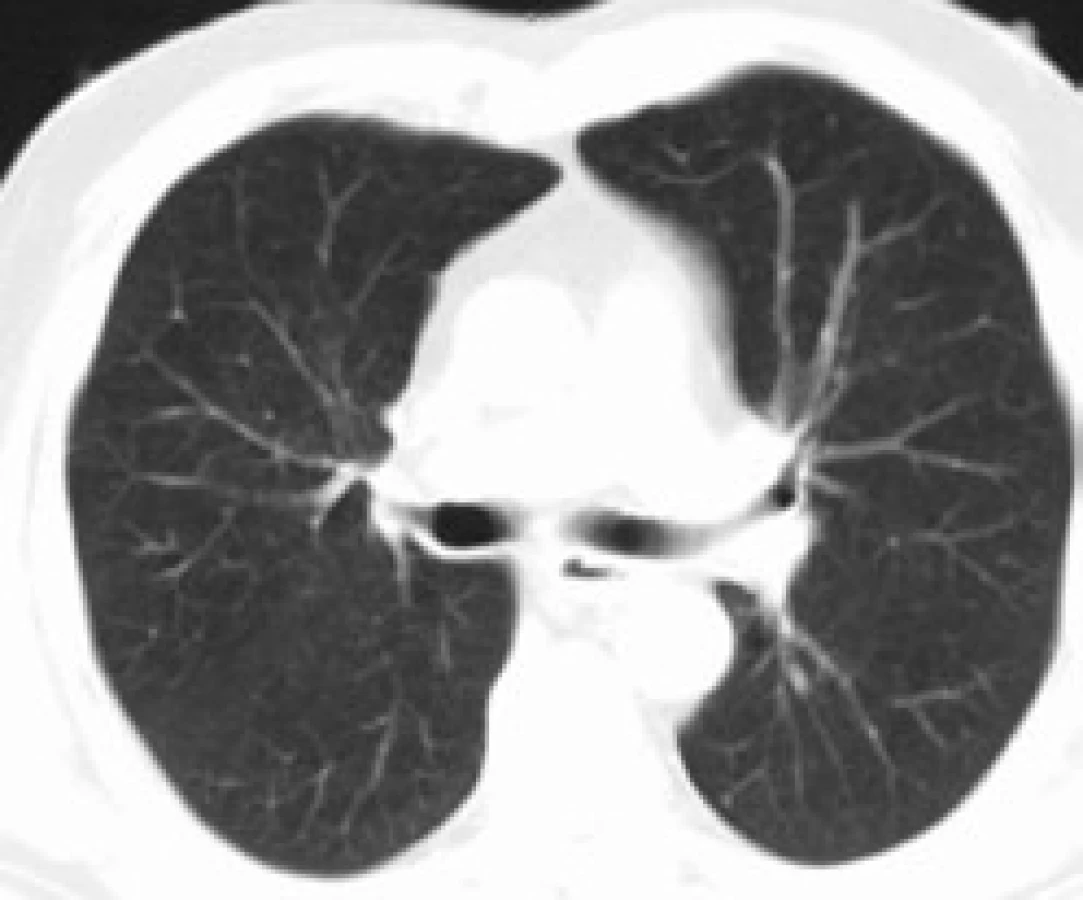
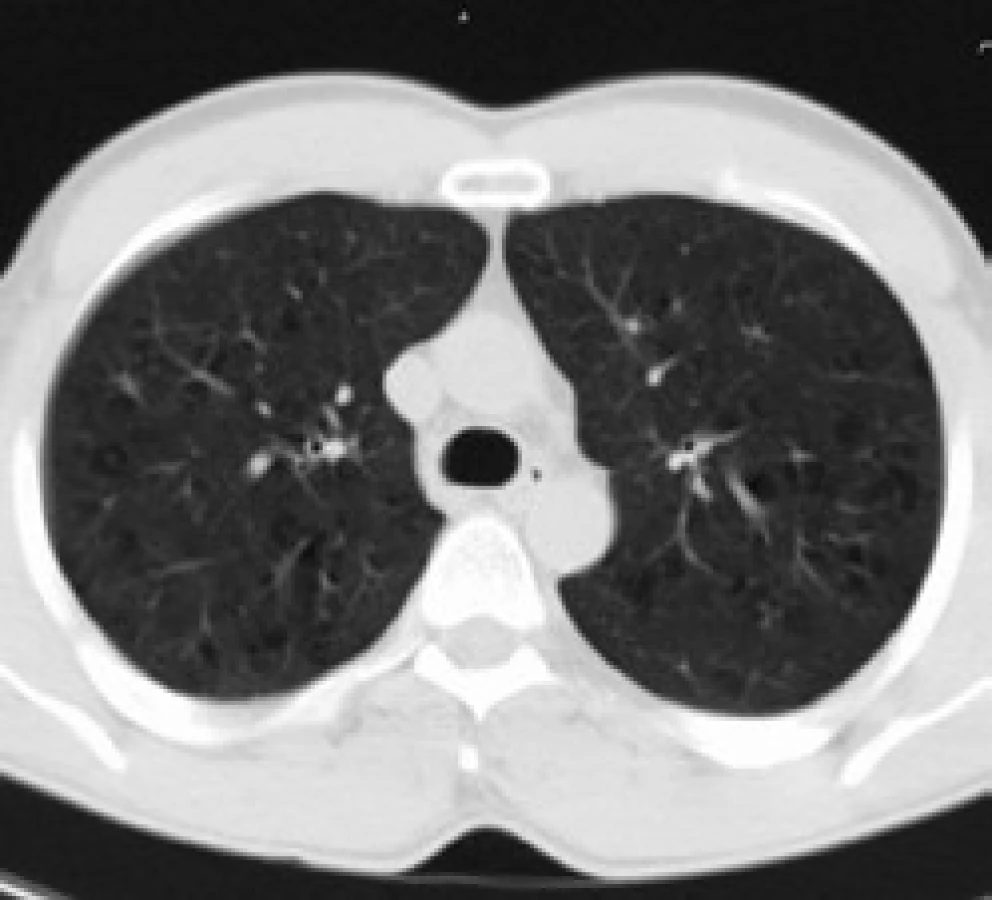
Infiltrace vyvolává restrikční změny, které předcházejí RTG změnám. Radiografický nález je tvořen cystami a intersticiálními nodulárními opacitami, obvykle blíže k hilům. Optimálním prostředkem pro diagnostiku plicní formy histiocytózy je HRCT, s jehož pomocí je možno rozpoznat tenkostěnné cysty, mikronoduly, opacity a zesílení inter-sticiálních prostorů. Cysty jsou více koncentrované v horních lalocích, méně jich je ve středních a vynechávají kostodiafragmatický úhel. Mikronoduly miliární velikosti 0,5–4,0 mm, s typickou distribucí, umožní dle ně-kte-rých autorů rozlišit pulmonální formu LCH od jiných nodulárních chorob. Zesílení intersticia je při HRCT vyšetření zřetelné hlavně bazálně [43,44]. Na obr. 10 je normální obraz plic při HRCT zobrazení a na obr. 11–13 je zobrazen intersticiální plicní proces u pacienta s původně kostní formou LCH.
Prasknutí cyst a jejich komunikace s pleurální dutinou způsobí spontánní pneumotorax [45,46]. Postižení plic histocytózou často komplikuje nasedající oportunní infekce. Odlišit ji může být problém, neboť teplota a váhový úbytek mohou být jak prvními projevy plicní histiocytózy, tak mohou mít i jiné (infekční) příčiny [48–58].
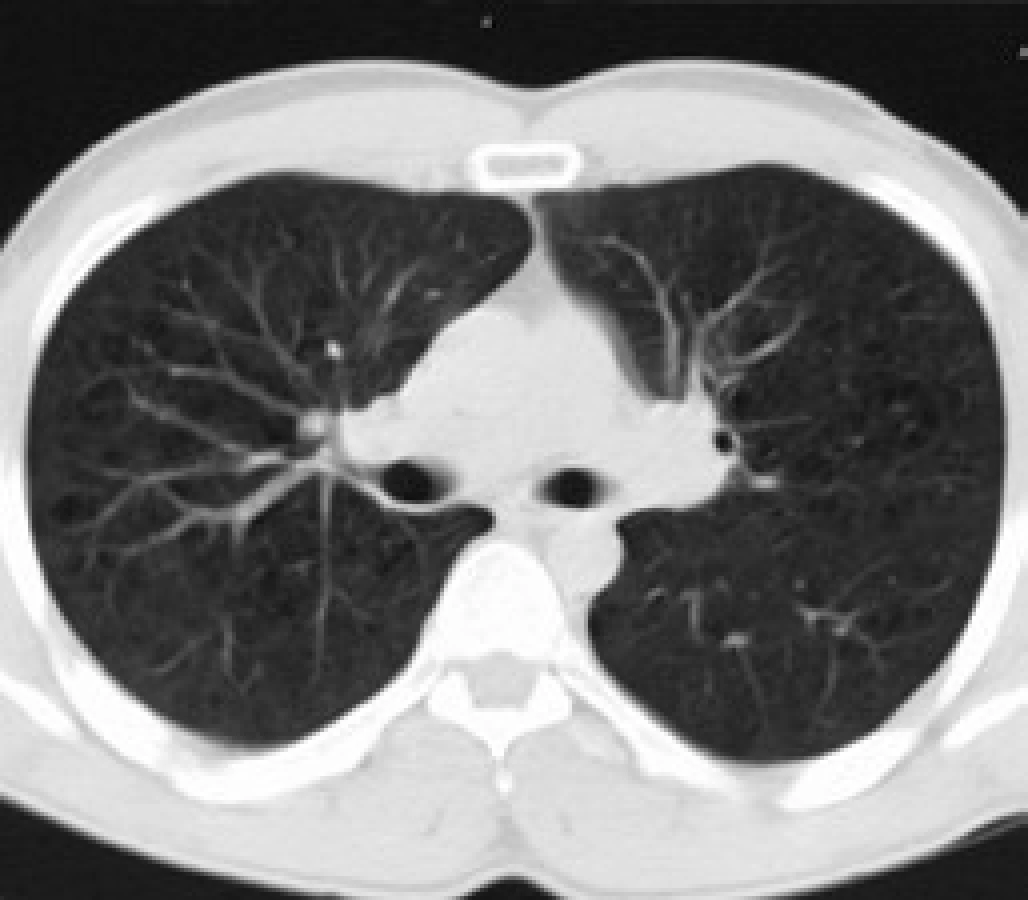
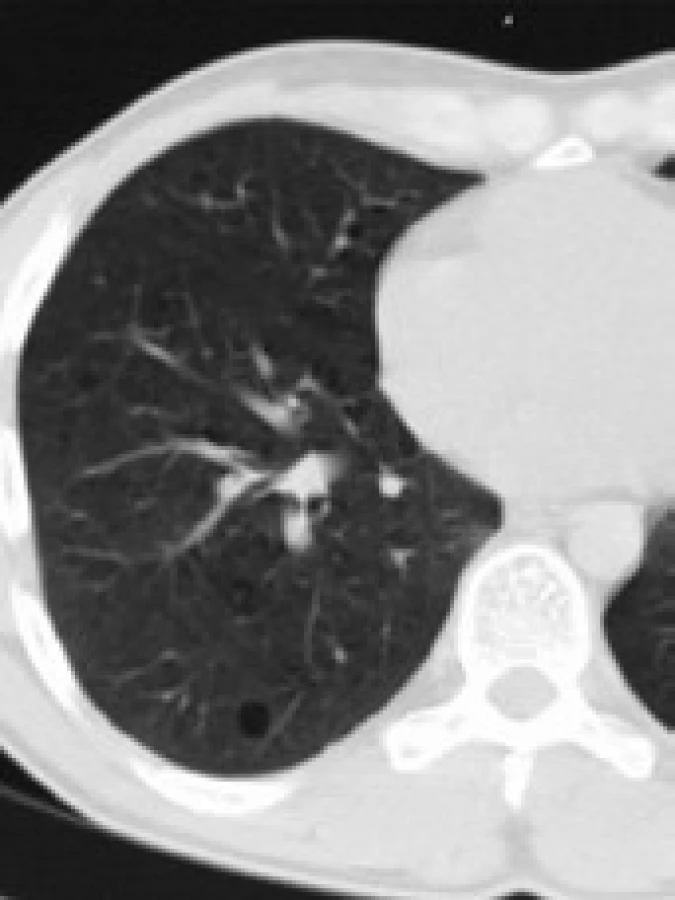
V diferenciální diagnostice pomůže buď bronchoalveolární laváž, nebo torakoskopie s excizí materiálu na histologické vyšetření.
Bronchoalveolární laváž je přínosná jedině v tom případě, že se provedou odpovídající imunohistochemické vyšetřovací metody, samotná bronchoskopie s biopsií bronchiální sliznice je pro průkaz LCH zcela nepřínosná. Langerhansovy buňky lze identifikovat flowcytometricky anebo v sedimentu z laváže imunohistochemicky průkazem makrofágů s pozitivitou znaku CD1a proteinu S-100 [59,60].
Udává se, že v případě plicní formy histiocytózy bývá v laváži více než 5 % CD1a+ buněk, zatímco u zdravých pacientů je počet CD1a+ menší než 1 %. Dále bývá zvýšen počet buněk v aspirované tekutině nad 106/ml, s prevalencí alveolárních makrofágů. Makrofágová alveolitis je totiž přítomna u kuřáků s LCH a chybí u vzácných případů plicní LCH nekuřáků [60].
Dle nálezů z konce 90. let minulého století se plicní Langerhansova histiocytóza dospělých liší od ostatních forem tím, že proliferující histiocyty jsou polyklonální, zatímco u ostatních forem jsou histiocyty spíše monoklonální. Plicní forma Langerhansovy histiocytózy se považuje za reaktivní proces na kouření či jiné stimuly. U této formy bylo také potvrzeno, že v případech pacientů se silnou vůlí, kteří dokázali přestat kouřit, dle RTG nálezů došlo k spontánní regresi nemoci. Plicní forma se považuje za relativně příznivou, pokud jde o izolovanou formu u kuřáka. Pokud však jde o plicní formu navazující na generalizované postižení skeletu a dalších orgánů nemocí, je průběh relativně nepříznivý [61–63]. V plicním parenchymu však může vzniknout i Erheimova-Chesterova choroba [64].
Pro sledování vývoje plicní formy LCH se používají jak funkční testy, tak HRCT [64]. V našem souboru nemocných byl nalezen u 3 (18 %) ze 17 pacientů HRCT obraz modularit a cyst, odpovídající plicní formě LCH. Pouze v 1 případně však bylo provedeno vyšetření tekutiny získané bronchoalveolární laváží, které potvrdilo tuto diagnózu na základně průkazu makrofágů, exprimujících znak CD1 a protein S-100. Bylo to u pacienta nar. 1963. Druhý, dle HRCT velmi suspektní případ je pacient nar. 1972, není však potvrzena imunohistochemicky, v bronchoalveolární laváži byly sice přítomny dominantně makrofágy, ale nebylo provedeno vyšetření proteinu S‑100 a CD1 antigenu. Třetí případ, kdy jsme plicní formu také předpokládali, byl pacient nar. 1964 s opakovaně recidivující kostní formou, u něhož se posléze přidružila dušnost a nález nodularit na HRCT plic, tento pacient v roce 2002 zemřel a nebyl pitván.
Proto je důležité, aby se u všech pacientů s nejasným intersticiálním plicním procesem vyšetřovaly v buňkách získaných bronchoalveolární laváží vždy také imunohistochemické znaky LCH.
Postižení endokrinního systému
Diabetes insipidus je typickým projevem dětské formy LCH, výjimečně může vzniknout i v dospělosti, v literatuře se popisuje při multifokálním kostním poškození. Zvláště v dětském věku však nepostihuje pouze sekreci adiuretinu, ale občas vzniká v důsledku nemoci deficit i ostatních hormonů, včetně poškození tvorby somatotropinu [65,66]. V našem souboru byl diabetes insipidus přítomen u 3 (75 %) ze 4 nemocných s diagnózou zjištěnou v dětském věku. U pacientů s diagnózou této nemoci v dětském věku je nutné vždy pátrat po deficitu ostatních hypofyzárních hormonů. U 1 z těchto 4 nemocných byl právě hypopituarizmus důvodem k neúspěchu in vitro fertilizace. I když pohlavní hormony a TSH byly adekvátně substituovány, tak zřejmě nedostatek somatotropinu, který nebyl substituován, způsobil neúspěch.
Ale také u dospělých může být nově vzniklý diabetes insipidus prvním projevem této nemoci [67,68]. U 1 (8 %) nemocného ze 13 s LCH diagnostikovanou v dospělém věku byl první známkou nemoci právě diabetes insipidus. Tento pacient měl zřetelnou infiltraci a rozšíření stopky hypofýzy (obr. 6–9). Tyto případy uvádí literatura formou popisů případů [69–73]. Bio-psie z infiltrátu v hypofýze by byla spojena s komplikacemi. Diagnóza LCH byla u tohoto muže stanovena histologickým vyšetřením kožních morf v oblasti anální krajiny. Zajímavé je, že tento muž s diagnózou LCH vzniklou v dospělosti neměl kostní postižení. Velmi příznivé je, že po léčbě leustatinem se šíře stopky hypofýzy upravila na normu, což signalizuje, že infiltrace v CNS příznivě reagovala na léčbu leustatinem, i když diabetes insipidus stále trvá. V literatuře je popsán případ, kdy diabetes insipidus vymizel po léčbě leustatinem [74]. V našem případě se tak nestalo a můžeme spekulovat, že je to způsobeno zahájením léčby až po 2 letech trvání diabetes insipidus, kdy se poškození hypotalamických struktur stalo nevratným.
Mozek
Velmi vzácnou manifestací LCH v dospělosti je postižení CNS. Podezření na tuto formu je nutno mít u všech nemocných s LCH, u nichž se objeví jakékoliv neurologické příznaky. Infiltráty lze znázornit pomocí MR a CT s aplikací kontrastní látky. Expanzivní ložiska jsou prokazatelná těmito metodami u 50 % pacientů s poruchou funkce hypofýzy. Disperzní infiltraci prokáže jedině autopsie. Není jasné, proč Langerhansovy buňky mají takovou afinitu k hypotalamu a jeho stopce.
Poškození CNS však může být jak přechodné [75], tak, velmi vzácně, i trvalé [76]. Vzhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií a případně analýz cerebrospinálního moku [77,78]. Dle posledních informací je možné diferencovat 3 typy postižení:
ohraničené granulomy v oblasti mozkové pojivové tkáně, zřetelné na MR zobrazení, odpovídající tumorózním ložiskům v meningách nebo v chorioideálním plexu; jejich histologie od-povídá Langerhansovým granulomům v pojivové tkáni
granulomy v oblasti pojivové mozkovétkáně s částečnou infiltrací šedé hmoty mozkové jak patologickými (CD1a+), tak i reaktivními histiocyty; tyto infiltráty byly provázené výraznou T-buněč-nou infiltrací a neurodegenerací, ztrátou neuronů, axonů a gliózou
neurodegenerativní ložiska postrádající CD1a+ buňky, nejčastěji postihující cerebellum, nucleus dentatus, cerebellární bílou hmotu a mozkový kmen, s výraznou zánětlivou infiltrací, obsahující CD8 lymfocyty. Tento pro---ces vede k degeneraci a glióze nervové tkáně. Neurodegenerativní proces se objevuje na základě T-buněčného zá--nětlivého procesu. Je provázen destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu. Tento typ postižení se objevuje až za dlouhou dobu od stanovení diagnózy, je progredující a nevratný. Projevuje se hyo--reflexií, ataxií, závratěmi, dysartrií,nystagmem, tremorem, diploopií, psy-chomotorickou retardací a neuropsy-chologickými defekty [78–84].
V našem souboru 17 nemocných se neurologické poškození vyskytlo u 2 (12 %) osob.
V jednom případě (pacient nar. 1975) byla v anamnéze ataka neurologických potíží typu ataxie netypických pro mladého muže. Tyto potíže výrazně ustoupily, byť ne zcela ad integrum. Kontrolní CT mozku bylo dle lékařských zpráv negativní, nicméně vzhledem k obtížnosti interpretací změn na CT při neurodegenerativních změnách v důsledku LCH se domníváme, že pravděpodobně šlo o nerozpoznanou mozkovou ataku LCH, podobně jak popisuje Imashuku [85].
V případě muže nar. 1976 je evidentní, jak původně dětská forma LCH přešla v dospělosti do závažného chronického onemocnění. Zpočátku jsme se domnívali, že recidivy budou omezené pouze na skelet. Výjimečné bylo, že mnohočetné kostní recidivy byly prokázány vždy pouze v oblasti kalvy. Po poslední recidivě v oblasti oka (rok 2001) následovalo za několik měsíců těžké zhoršení stavu s diplopií, skandovanou řečí a příznaky z postižení cerebela a progresivním zhoršováním funkce CNS tak, že nemocný nebyl schopen samostatné chůze, neovládal svěrače a komunikace byla velmi omezená. Tento nemocný zemřel v dubnu roku 2008. MR obraz i další průběh odpovídal neurodegenerativnímu poškození CNS, pro nějž v době stanovení diagnózy neexistovala žádná účinná léčba [85].
Lymfadenopatie
Histiocytóza obvykle nedělá výraznou lymfadenopatii, pokud ano, jde spíše o ložiskové než generalizované postižení [86]. V našem případně jsme se setkali s jedním případem izolované lymfadenopatie.
Uši
Poruchy sluchu mohou nastat jak postižením zevního sluchového kanálu, tak poruchou středního či vnitřního ucha propagací choroby z processus mastoideus. Infiltrace je nebolestivá a postupně vede k hluchotě. Časté jsou sekundární infekce, které jsou příčinou záměny za chronickou otitidu [87,88]. S tímto postižením jsme se nesetkali.
Oči
Intraokulární postižení je vzácné, zatímco infiltrace orbitálního prostoru je relativně častá. Dětští lékaři se s ní setkávají u 20–30 % nemocných Langerhansovou histiocytózou. Projevuje se ptózou víčka, edémem papily a poruchou funkce VII. nervu. Někdy může být poškozen optický nerv, což si někdy kromě systémové léčby vynutí i akutní léčbu nitroložiskovou aplikací kortikosteroidů a radioterapii [89–92]. U dospělých jsme se s těmito projevy nesetkali.
Játra a slezina
Játra i slezina mohou být touto chorobou postiženy, což se projeví jejich zvětšením. Velmi silná infiltrace jater pak může vyvolat příznaky jaterního selhání (pokles albuminu, snížení aktivity koagulačních faktorů, žloutenku bez výrazného zvýšení jaterních enzymů). V chronických formách může vzniknout periportální fibrotizace s příznaky shodnými se sklerotizující cholangoitidou a obstrukční biliární žloutenkou, kterou je nutno na základně biopsie odlišit od primární sklerotizující cholangitidy a adekvátně léčit [93–99].
V našem případně jsme na postižení jater bez histologického průkazu usuzovali u muže nar. 1976, u něhož v roce 2001 prudce vzestoupily jaterní enzymy, aniž by se našlo jiné vysvětlení než infiltrace jater LCH, proto byl tento pacient léčen leustatinem, po němž se hodnoty jaterních enzymů upravily.
Dutina ústní a trávicí trakt
Počínající infiltrace se v této oblasti projevuje zduřením dásní a sliznice patra. Může dojít k postižení kostí a uvolňování zubů či hypertrofii dásní [100]. Progrese infiltrátů pak vytváří ulcerace v ústech [101,102]. U dětí je diagnosticky významná předčasná 2. dentice.
V našem souboru je jeden nemocný (nar. 1974), u něhož byla diagnostikována infiltrace linguální části dásně a kompletně excidována. U dalšího pacienta (nar. 1973) jsme na tuto formu měli podezření. Ve stejném roce, kdy byly zjištěny hypotalamické a kožní změny perianálně, docházelo k uvolňování zubů, které bylo stomatology popsáno jako velmi rychle nastoupivší těžká paradentóza. Vzhledem k časové koincidenci se nabízí i příčinná souvislost, byť nebyla stomatology potvrzena. Žádný vzorek z dutiny ústní však nebyl k histologickému vyšetření odeslán. Pokud stomatolog nevěděl o formě LCH, postihující dásně a čelist, vedoucí k uvolňování zubů, tak ji nemohl ani diagnostikovat. Potvrdit či nepotvrdit by ji mohla jedině histologie z postižených dásní a panoramatický snímek čelisti (obr. 14).

Sliznice střevního traktu je postižena jen zřídka. Prvními příznaky je celkové neprospívání, hubnutí. Klasické projevy malabsorpce se objevují až při rozsáhlejším postižení trávicího traktu. Anální kanál a perianální oblast jsou infiltrovány často, a tvoří tak součást kožního postižení [102–105].
Sledování nemocných
Zatím nemáme v praxi dostupný žádný laboratorní parametr, takže nezbývá nic jiného než používat odpovídající zobrazovací metody a klinická vyšetření. Zatím se popsané použití sérové hladiny proteinu S-100 nedostalo do rutinní praxe [106].
Závěr
Histiocytóza z Langerhansových buněk je nemoc výjimečná svojí nízkou incidencí, ale také mnoha podobami, kterých tato nemoc může nabývat. V textu jsou uvedeny ty formy, s nimiž jsme se na našem pracovišti setkali za 18 let. V diskuzi jsme podali přehled manifestací u dospělých, jak je uvádí literatura, a srovnali je s našim pozorováním.
U dospělých by každé nové osteolytické ložisko mělo být podrobeno histologickému odběru, stejně tak jako každé patologické morfy, protože bez mikroskopického a imunohistochemického vyšetření nelze tuto nemoc rozpoznat. Pokud v dospělosti vznikne diabetes insipidus, je nutno mimo jiné hledat etiologii v LCH, stejně tak u intersticiální plicního procesu je nutno myslet na plicní formu LCH a provádět imunohistochemická vyšetření, bez nichž není možné tuto plicní formu LCH diagnostikovat.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e‑mail: z.adam@fnbrno.cz
Doručeno do redakce: 12. 5. 2008
Přijato po recenzi: 2. 7. 2008
Sources
1. Nicholson SH, Egeler M, Nesbit ME. The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998; 12 : 379–348.
2. Baumgartner I, Hochstetter A, Baumert Bet al. Langerhans cell histiocytosis in adults. Med Pediatric Oncol 1997; 28 : 9–14.
3. Bubanska E, Stančokova T, Dluholucky S. Histiocytóza z Langerhansových buněk. Čes Slov Pediat 1998; 53 : 18–19.
4. Mottl H, Koutecký J, Ganevová M. Strategieléčby histiocytózy z Langerhansových bu-něk u dětí. Čes Slov Pediat 1994; 49 : 81.
5. Mottl H, Mracek J, Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530–533.
6. Mottl H, Rob L, Stary J et al. Langerhans cell histiocytosis of vulva in adolescent. Int J Gynecol Cancer 2000; 17 : 520–524.
7. Šímová B, Mališ J, Neuwirt J. Klinické projevy histiocytózy z Langerhansových buněk. Zdrav Nov ČR Lék Listy 2003; 52 : 18.
8. Tichý M jr, Tichý M, Krč I et al. Multicentrická retikulohistiocytóza. Čes Slov Derm 1999; 74 : 168–171.
9. Toušovská K, Slavík Z. Histiocytóza z Langerhansových buněk v dětském věku. Lék Zprav lék Fak Univ Karlovy Hr Králové 1997; 42 : 127–132.
10. Gotz G, Fichter J. Langerhans’-cell histiocytosis in 58 adults. Eur J Med Res 2004; 9 : 510–514.
11. Pacovska V, Bortlova A, Homolka J et al. Granulomatóza z Langerhansových buněk. Trendy Med 2002; 4 : 59–61.
12. Malpas JS. Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Amer 1998; 12 : 259–268.
13. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol 2006; 76 : 363–368.
14. Imashuku S, Ishida S, Koike K et al. Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2004; 26 : 735–739.
15. Adam Z, Krejčí M, Vorlíček J. Maligní krevní nemoci. Praha: Grada Publishing 2008, 396 s.
16. Cheung N, Selva D, McNab AA. Orbital Langerhans cell histiocytosis in adults. Ophthalmology 2007; 114 : 1569–1573.
17. Smilek P, Krejčova B, Čada K et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994; 43 : 263–265.
18. Srikulmontree T, Massey HD, Roberts WN. Treatment of skeletal Erdheim-Chester disease with zoledronic acid: case report and proposed mechanisms of action. Rheumatol Int 2007; 27 : 303–307.
19. Hoover KB, Rosenthal DI, Mankin H. Langerhans cell histiocytosis. Skeletal Radiol 2007; 36 : 95–104.
20. Dogan S, Conway J, Miller JH et al. Detection of bone lesions in Langerhans cell histiocytosis. Complementary roles of scintigraphy and conventional radiograpy. J Pediatric Hematol/Oncol 1996; 18 : 51–58.
21. Howarth DM, Mullan BP, Wiseman GA et al. Bone scintigraphy evaluated in diagnosis and staging Langerhans’s cell histiocytosis and related disorders. J Nuclear Med 1996; 37 : 1456–1460.
22. Blum R, Seymour JF, Hicks RJ. Role of FDG-positron emission tomography scanning in the management of histiocytosis. Leukem Lymphoma 2002; 43 : 2155–2157.
23. Kaste SC, Rodriguez-Galindo C et al. PET-CT in pediatric Langerhans cell his-tiocytosis. Pediatr Radiol 2007; 37 : 615–622.
24. Weiss SE, O’Connor L, Welsh JS. Refinement of radiation therapy based on PET data in an adult with Langerhans cell histiocytosis of soft tissues. Clin Adv Hematol Oncol 2006; 4 : 290–292; discussion 292–294.
25. Furmanczyk PS, Bruckner JD, Gillespy T 3rd et al. An unusual case of Erdheim-Chester disease with features of Langerhans cell histiocytosis. Skeletal Radiol 2007; 36 : 885–889.
26. Goo HW, Yang DH, Ra YS et al. MRI of Langerhans cell histiocytosis: comparison with radiography and bone scintigraphy. Pediatr Radiol 2006; 36 : 1019–1031.
27. Olschewski T, Seegenschmiedt MH. Radiotherapy of Langerhans’ Cell Histiocytosis : Results and Implications of a National Patterns-of-Care Study. Strahlenther Onkol 2006; 182 : 629–634.
28. Fernandez Flores A, Mallo S. Langerhans cell histiocytosis of vulva. Dermatol Online J 2006; 12 : 15.
29. Ferreli C, Aste N, Pinna LA et al. Langerhans cell histiocytosis in an adult. J Eur Acad Dermatol Venereol 1997; 9 : 253–255.
30. Ferringer T, Banks PM, Metcalf JS. Langerhans cell sarcoma. Am J Dermatopathol 2006; 28 : 36–39.
31. Hagiuda J, Ueno M, Ashimine S et al. Langerhans cell histiocytosis on the penis: a case report. BMC Urol 2006; 6 : 28.
32. Mlyncek M, Uharcek P, Durcanský D. Vulvar Langerhans’ cell histiocytosis: a case report. Acta Obstet Gynecol Scand 2006; 85 : 753–755.
33. Tzung TY, Wu JC. Nonhealing perianal ulcers. Arch Dermatol 2005; 141 : 1161–1166.
34. Venizelos ID, Mandala E, Tatsiou ZA et al. Primary langerhans cell histiocytosis of the vulva. Int J Gynecol Pathol 2006; 25 : 48–51.
35. Fluri S, Nievergelt H, Kernland K et al. When atopic dermatitis doesn’t heal – an interesting case from the medical university polyclinic. Schweiz Rundsch Med Prax 2006; 95 : 945–948.
36. Punia RS, Bagai M, Mohan H et al. Langerhans cell histiocytosis of skin: a clinicopathologic analysis of five cases. Indian J Dermatol Venereol Leprol 2006; 72 : 211–214.
37. Imafuku S, Shibata S, Tashiro A. Cutaneous Langerhans cell histiocytosis in an elderly man successfully treated with narrow-band ultraviolet B. Br J Dermatol 2007; 157 : 1277–1279.
38. Lan Ma H, Metze D, Luger TA et al. Successful treatment of generalized eruptive histiocytoma with PUVA. J Dtsch Dermatol Ges 2007; 5 : 131–134.
39. Newman B, Hu W, Nigro K et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007; 56 : 302–316.
40. Munn S, Chu AC. Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Amer 1998; 12 : 269–286.
41. Ashena Z, Alavi S, Arzanian MT et al. Nail involvement in langerhans cell histiocytosis. Pediatr Hematol Oncol 2007; 24 : 45–51.
42. Mataix J, Betlloch I, Lucas-Costa A et al. Nail changes in Langerhans cell histiocytosis: a possible marker of multisystem disease. Pediatr Dermatol 2008; 25 : 247–251.
43. Canuet M, Kessler R, Jeung MY et al. Correlation between high‑resolution computed tomography findings and lung function in pulmonary Langerhans cell histiocytosis. Respiration 2007; 74 : 640–646.
44. Leatherwood DL, Heitkamp DE, Emerson RE. Best cases from the AFIP: Pulmonary Langerhans cell histiocytosis. Radiographics 2007; 27 : 265–268.
45. Kim CK, Park CB, Jin U et al. Pulmonary Langerhans’ cell histiocytosis presented with recurrent pneumothorax. Interact Cardiovasc Thorac Surg 2006; 5 : 512–513.
46. Mendez JL, Nadrous HF, Vassallo R et al. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125 : 1028–1032.
47. Adams EP, Sauceda D, Oliver J et al. Isolated pulmonary Langerhans cell histiocytosis in a 17-year-old male. J Pediatr Hematol Oncol 2007; 29 : 121–124.
48. Aerni MR, Aubry MC, Myers JL et al. Complete remission of nodular pulmonary Langerhans cell histiocytosis lesions induced by 2-chlorodeoxyadenosine in a non‑smoker. Respir Med 2008; 102 : 316–319.
49. Bernstrand C, Cederlund K, Sandstedt B et al. Pulmonary abnormalities at long term follow up of patients with Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 459–468.
50. Braier J, Latella A, Balancini B et al. Isolated pulmonary Langerhans cell histiocytosis presenting with recurrent pneumothorax. Pediatr Blood Cancer 2007; 48 : 241–244.
51. Bittenglova R, Pešek M, Mukenšnabl P et al. Granulomatóza z Langerhansových buněk. Stud pneumol phtiseol 2002; 62 : 196–202.
52. Brown RE. Bisphosphonates as antialveolar macrophage therapy in pulmonary Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 641–643.
53. Green MB, Allen JN. Cough, dyspnea,and reticulonodular opacities in a 58-year-old smoker. Chest 2007; 132 : 700–703.
54. Jülg BD, Weidner S, Mayr D. Pulmonary manifestation of a Langerhans cell sarcoma: case report and review of the literature. Virchows Arch 2006; 448 : 369–374.
55. Rožánek P, Molnar V, Rešl M. Tři případy plicní granulomatózy z Langerhansových buněk. Lék zpr lék Fak Univ Karlovy Hr Králové 1998; 43 : 127–132.
56. Sundar KM, Gosselin MV, Chung HL et al. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology and clinical evolution of the disease. Chest 2003; 123 : 1673–1683.
57. Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J. 2006; 27 : 1272–1285.
58. Skacel Z, Marel M, Vraštilova P et al. Histiocytóza z Langerhansových buněk. Přehled literatury a vlastní pozorování. Stud pneumol phtiseol 2000; 60 : 150–156.
59. Zeppa P, Cozzolino I, Russo M et al. Pulmonary langerhans cell histiocytosis (histiocytosis X) on bronchoalveolar lavage: a report of 2 cases. Acta Cytol 2007; 51 : 480–482.
60. Tötsch M, Guzman J, Theegarten D et al. Bronchoalveolar lavage. Pathologe 2007; 28 : 346–353.
61. Hidalgo A, Franquet T, Giménez A. Smoking‑related interstitial lung diseases: radiologic-pathologic correlation. Eur Radiol 2006; 16 : 2463–2470.
62. Liu YH, Fan XH, Fang K. Langerhans’ cell histiocytosis with multisystem involvement in an adult. Clin Exp Dermatol 2007; 32 : 765–768.
63. Marten K. Smoking‑related interstitial lung diseases. Rofo 2007; 179 : 68–75.
64. Negrin‑Dastis S, Butenda D, Dorzee J et al. Complete disappearance of lung abnormalities on high‑resolution computed tomography: a case of histiocytosis X. Respir J 2007; 14 : 235–237.
65. Amato MC, Elias LL, Elias J et al. Endocrine disorders in pediatric – onset Langerhans Cell Histiocytosis. Horm Metab Res 2006; 38 : 746–751.
66. Donadieu J, Rolon MA, Pion I et al. Incidence of growth hormone deficiency in pediatric onset Langerhans cell histiocytosis: Efficacy and safety of Growth hormone treatment. J Clin Endocrinol Metabolism 2004; 89 : 604–609.
67. Makras P, Alexandraki KI, Chrousos GP et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 2007; 18 : 252–257.
68. Neji S, Ben Slama C, Zidi B. Hypo-tha-lamic-pituitary Langerhans cell histiocytosis in adults. Presse Med 2006; 35 : 1263–1266.
69. Halefoglu AM. Magnetic resonance imaging of thickened pituitary stalk proceeding to Langerhans cell histiocytosis in a child. Australas Radiol 2006; 50 : 175–178.
70. Horn EM, Coons SW, Spetzler RF et al. Isolated Langerhans cell histiocytosis of the infundibulum presenting with fulminant diabetes insipidus. Childs Nerv Syst 2006; 22 : 542–544.
71. Kandpal H, Subramanian S, Hari S. Langerhans cell histiocytosis of pituitary stalk. Neurol India 2007; 55 : 91–92.
72. Ouyang DL, Roberts BK, Gibbs IC et al. Isolated Langerhans cell histiocytosis in an adult with central diabetes insipidus: case report and review of literature. Endocr Pract 2006; 12 : 660–663.
73. Ottaviano F, Finlay JL. Diabetes insipidus and Langerhans cell histiocytosis: a case report of reversibility with 2-chlorodeoxyadenosine. J Pediatr Hematol Oncol 2003; 25 : 575–577.
74. Prosch H, Grois N, Prayer D et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 594–599.
75. Ryan P, Walterfang M, Scholes A et al. Recovery of cognitive function in neuropsychiatric Langerhan’s cell histiocytosis. Psychiatry Clin Neurosci 2006; 60 : 629–632.
76. Nanduri VR, Lillywhite L, Chapman Cet al. Cognitive outcome of long term survivors of multisystem Langerhans cell histiocytosis. J Clin Oncol 2003; 21 : 2961–2967.
77. Prosch H, Feldges A, Grois N et al. Demonstration of CD1a positive cells in the cerebrospinal fluid-A clue to diagnosis of isolated Langerhans cell histiocytosis of the hypothalamic-pituitary axis? Med Pediatr Oncol 2003; 41 : 474–476.
78. Prosch H, Grois N, Wnorowski M et al. Long‑term MR imaging course of neurodegenerative Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2007; 28 : 1022–1028.
79. Bös M, Grothe C, Urbach H et al. Cerebellar syndromes in Langerhans’ cell histiocytosis. Nervenarzt 2007; 78 : 437–440.
80. Dhall G, Finlay JL, Dunkel IJ et al. Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer 2008; 50 : 72–79.
81. Grois N, Prayer D, Prosch H et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829–838.
82. Martin‑Duverneuil N, Idbaih A, Hoang-Xuan K et al. French Langerhans Cell Histiocytosis Study Group. MRI features of neurodegenerative Langerhans cell histiocytosis. Eur Radiol 2006; 16 : 2074–2082.
83. Mittheisz E, Seidl R, Prayer D et al. Central nervous system‑related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2007; 48 : 50–56.
84. Steiner M, Prayer D, Asenbaum S et al. Modern imaging methods for the assessment of Langerhans’ cell histiocytosis‑associated neurodegenerative syndrome: case report. J Child Neurol 2005; 20 : 253–257.
85. Imashuku S, Okazaki NA, Nakayama M et al. Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 2008; 50 : 308–311.
86. Edelweiss M, Medeiros LJ, Suster S et al. Lymph node involvement by Langerhans cell histiocytosis: a clinicopathologic and immunohistochemical study of 20 cases. Hum Pathol 2007; 38 : 1463–1469.
87. Baumann C, Reschke K, Jungehülsing M et al. Destruction of the vestibular organ by Langerhans’ cell histiocytosis. Eur Radiol 2006; 16 : 1177–1178.
88. Kürten T, Groeger M, Angerstein W. Frequency of hearing disorders in children with langerhans’ cell histiocytosis. Laryngorhinootologie 2008; 87 : 96–99.
89. Anton M, Holoušova M, Řehůřek J et al. Histiocytoza X a dětská očnice. Čs Ophthal 1992; 48 : 176–180.
90. Bermingham N, Townley D, Fenton S et al. Ocular langerhans cell histiocytosis. Eye 2007; 21 : 1127–1128.
91. D’Angio GJ. Langerhans cell histiocytosis affecting the eyes. Pediatr Blood Cancer 2006; 47 : 639.
92. Tsai JH, Galaydh F, Ching SS. Anterior uveitis and iris nodules that are associated with angerhans cell histiocytosis. Am J Ophthalmol 2005; 140 : 1143–1145.
93. Dina I, Copaescu C, Herlea V et al. Liver involvement in Langerhans’ cell histiocytosis. Case report. J Gastrointestin Liver Dis 2006; 15 : 57–59.
94. Griffiths W, Davies S, Gibbs P et al. Liver transplantation in an adult with sclerosing cholangitis due to Langerhans cell histiocytosis. J Hepatol 2006; 44 : 829–831.
95. Guthery SL, Heubi JE. Liver involvement in childhood histiocytic syndromes. Curr Opin Gastroenterol 2001; 17 : 474–478.
96. Choi SW, Bangaru BS, Wu CD et al. Gastrointestinal involvement in disseminated Langerhans cell histiocytosis (LCH) with durable complete response to 2-chlorodeoxyadenosine and high‑dose cytarabine. J Pediatr Hematol Oncol 2003; 25 : 503–506.
97. Hait E, Liang M, Degar B et al. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 2006; 118 : 1593–1599.
98. Hait E, Liang M, Degar B et al. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and lite-rature review. Pediatrics 2006; 118: e1593–e1599.
99. Konno S, Hizawa N, Betsuyaku T et al. Adult Langerhans cell histiocytosis with independently relapsing lung and liver lesions that was successfully treated with etoposide. Intern Med 2007; 46 : 1231–1235.
100. Klein F, Krigar D, Petzoldt D et al. Periodontal manifestation of Langerhans’ cell histiocytosis in a young man: case report with a 24-month follow‑up. Quintessence Int 2006; 37 : 175–182.
101. Silvestros SS, Mamalis AA, Sklavounou AD et al. Eosinophilic granuloma masquerading as aggressive periodontitis. J Periodontol 2006; 77 : 917–921.
102. Querings K, Starz H, Balda BR. Clinical spectrum of cutaneous Langerhans’ cell histiocytosis mimicking various diseases. Acta Derm Venereol 2006; 86 : 39–43.
103. Arizo M, Egeler M. Clinical aspectsof Langerhans cell histiocytosis. Hema-tology/Oncology Clinics North Amer 1998; 12 : 247–267.
104. Chang KL, Snyder DS. Langerhans cell histiocytosis. Cancer Treat Res 2008; 142 : 383–398.
105. Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol 2004; 50 : 157–174.
106. Ugurel S, Pröhler G, Tilgen W et al. S100-β serum protein – a new marker in the diagnosis and monitoring of Langerhans cell histiocytosis. Brit J Dermatol 2000; 143 : 201–202.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2008 Issue 11
Most read in this issue
- Hypokalemická periodická paralýza u pacientů s hypertyreózou
-
Histiocytóza z Langerhansových buněk u osob dospělého věku – nemoc s mnoha tvářemi.
Zkušenosti jednoho pracoviště a přehled projevů nemoci - Bezpečnost dlouhodobého podávání losartanu v běžné klinické praxi: neintervenční studie NCT-CZ 14/04/LOZ
- Význam stanovování inhibinu B v klinické andrologické praxi