
Dvacet let péče o dospělé nemocné s cystickou fibrózou v České republice
Twenty years of care for cystic fibrosis adults in Czech Republic
Introduction:
Most of cystic fibrosis (CF) patients survive now into adulthood and they are transferred to pulmonologist care. Aims: An overview of progress in care for CF adults in Czech Republic and evaluation of relationship of pulmonary function, nutritional status and airway colonization.
Methods:
All adult CF patients followed in pulmonary departments from December 1987 to December 2007 were included into study. Data about survival status, pulmonary function, nutritional status, airway colonization and other pulmonary and extrapulmonary manifestations of CF were collected from patients’ records.
Results:
Total of 206 patients (96 females) were followed. Pancreatic insufficiency was present in 175 (85.0%), liver disease in 61 (29.6%) and insulin treatment in 58 (28.2%) patients. Bone disease was found in 70 (46.7%) from 150 examined patients. Sixty-two patients (23 females) died at mean age 25.4 ± 5.5 years (median 24.3 years). Worse survival was recorded in patients with Burkholderia cepacia complex (BCC) airway colonization (24.4 ± 4.0 vs 28.5 ± 7.0 years, p = 0.012). One hundred forty-four living patients were followed to date of the 31st December 2007 with mean age 27.5 ± 6.5 years (median 26.5 years), FEV1 64.4 ± 28.5% pred. and BMI 20.9 ± 3.1 kg/m2. Worse pulmonary function was present in patients with BCC colonization (FEV1 58.8 ± 21.9 vs 67.8 ± 27.3% pred., p = 0.041) and in malnourished patients (FEV1 49.5 ± 18.5 vs 69.7 ± 25.9% pred., p < 0.0001). BCC colonization was found in 54 (37.5%), Pseudomonas aeruginosa (PA) colonization in 92 (63.9%) and colonization without BCC or PA in 40 (27.8%) patients, respectively. Malnutrition (BMI < 19.0 kg/m2) was recorded in 38 (26.4%) patients.
Conclusion:
This study confirms growing number of CF adults in Czech Republic, close relationship of pulmonary function and nutritional status and also unfavourable influence of BCC colonization.
Key words:
cystic fibrosis – adults
Authors:
L. Fila 1; V. Sedlák 2; I. Binková 3; P. Jakubec 4; R. Bittenglová 5; J. Musil 1
Authors‘ workplace:
Pneumologická klinika 2. lékařské fakulty UK a FN Motol Praha, přednosta doc. MUDr. Jaromír Musil, Ph. D.
1; Plicní klinika Lékařské fakulty UK a FN Hradec Králové, přednosta doc. MUDr. František Salajka, CSc.
2; Klinika nemocí plicních a tuberkulózy Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednostka prof. MUDr. Jana Skřičková, CSc.
3; Klinika plicních nemocí a tuberkulózy Lékařské fakulty UP a FN Olomouc, přednosta prof. MUDr. Vítězslav Kolek, DrSc.
4; Klinika tuberkulózy a respiračních nemocí Lékařské fakulty UK a FN Plzeň, přednosta prof. MUDr. Miloš Pešek, CSc.
5
Published in:
Vnitř Lék 2009; 55(6): 542-548
Category:
Original Contributions
Overview
Úvod:
Díky pokrokům v péči o nemocné s cystickou fibrózou (CF) se většina těchto pacientů nyní dožívá dospělosti a je předávána do péče pneumologa. Cíl: Přehled vývoje a současného stavu péče o dospělé nemocné s CF v České republice a zhodnocení vztahu stavu plicních funkcí, stavu výživy a kolonizace dýchacích cest.
Metody:
Soubor je tvořen dospělými s CF sledovanými na pneumologických pracovištích od prosince roku 1987 do prosince roku 2007. Ze zdravotní dokumentace byly zaznamenány údaje o přežití, stavu plicních funkcí a stavu výživy, kolonizaci dýchacích cest a dalších plicních i mimoplicních projevech nemoci.
Výsledky:
Celkem bylo sledováno 206 pacientů (96 žen). Pankreatická insuficience byla zjištěna u 175 (85,0 %), hepatopatie u 61 (29,6 %) a léčba inzulinem u 58 (28,2 %) nemocných. Metabolická kostní nemoc byla zachycena u 70 (46,7 %) ze 150 vyšetřených nemocných. Ze sledovaných nemocných 62 zemřelo (23 žen) v průměrném věku 25,4 ± 5,5 roku (medián 24,3 roku). Přežití bylo horší u nemocných s kolonizací dýchacích cest komplexem Burkholderia cepacia (BCC) (24,4 ± 4,0 vs 28,5 ± 7,0 let, p = 0,012). K 31. 12. 2007 bylo sledováno 144 žijících pacientů (73 žen) průměrného věku 27,5 ± 6,5 roku (medián 26,5 roku), FEV1 64,4 ± 28,5 % náležité hodnoty a BMI 20,9 ± 3,1 kg/m2. Horší plicní funkce měli nemocní s kolonizací BCC (FEV1 58,8 ± 21,9 vs 67,8 ± 27,3 % náležité hodnoty, p = 0,041) a nemocní v malnutrici (FEV1 49,5 ± 18,5 vs 69,7 ± 25,9 % náležité hodnoty, p < 0,0001). BCC bylo kolonizováno 54 (37,5 %), Pseudomonas aeruginosa (PA) 92 (63,9 %) a bez kolonizace BCC či PA bylo 40 (27,8 %) pacientů. Malnutrice (BMI < 19,0 kg/m2) byla přítomna u 38 (26,4 %) pacientů.
Závěr:
Práce dokumentuje rostoucí počet dospělých pacientů s CF, těsný vztah stavu plicních funkcí a stavu výživy a nepříznivý vliv kolonizace BCC.
Klíčová slova:
cystická fibróza – dospělí
Úvod
Za první vědecký popis cystické fibrózy (CF) je pokládána publikace americké patoložky Andersenové z roku 1938 [1]. V České republice byl první nemocný s touto diagnózou popsán v květnu roku 1946 a malý soubor pacientů byl publikován v roce 1948 [2]. O dr. Dagmar Benešové, spoluautorce tohoto sdělení, se dodnes mluví jako o „české Dorothy Andersen“.
Systematická péče o tyto nemocné je pak spojena se jménem doc. MUDr. Věry Vávrové a začíná zavedením vyšetření chloridů v potu s využitím pilokarpinové iontoforézy v roce 1960 a komplexní léčby v roce 1965 [3,4].
Nemocní s touto chorobou ve 40. a 50. letech minulého století umírali obvykle již v předškolním věku. V následujících letech se s přibývajícími poznatky o tomto onemocnění zlepšovala dostupná péče a s ní i prognóza pacientů tak, že v současnosti činí predikované přežití ve vyspělých zemích 30–40 let. CF tedy již řadu let není doménou pouze pediatrů [5].
O prvním dospělém pacientovi s CF bylo ve světě referováno již v roce 1946 [6]. Shwachman a Kulczycki ve známé práci [7] uvádějí mezi 105 nemocnými sledovanými alespoň 5 let 3 pacienty ve věku 18 a více let, přičemž nejstaršímu z nich bylo 22,9 roku. Další dospělí nebyli pro dobu sledování < 5 let do studie zahrnuti (nejstaršímu z nich bylo 33 let).
V českých zemích o CF u dospělých referoval poprvé Netoušek [8] a malý soubor pacientů publikoval Ledeč v roce 1966 [9]. Pro pneumology však byla tato diagnóza dlouho exotická, o čemž svědčí např. článek Marela z roku 1986 [10].
Motolské plicní oddělení bylo díky návaznosti na zdejší pediatrii logicky prvním v českých zemích, kde se o dospělé s CF začali pneumologové starat. První pacient s CF zde byl hospitalizován pro pneumotorax v květnu roku 1984 a nemocní začali být systematicky sledováni v poradně pro dospělé s CF od prosince roku 1987. Péče o ně je v těchto letech spojena se jménem doc. MUDr. Jaromíra Musila, který publikoval desetileté zkušenosti s péčí o tyto pacienty v roce 1997 [11].
S narůstajícím počtem dospělých nemocných s CF vyvstala potřeba vzniku poraden i při dalších pneumologických pracovištích fakultních nemocnic, konkrétně v roce 2000 v Hradci Králové a v Brně a v roce 2001 v Olomouci a v Plzni. Těchto 5 poraden je dodnes základem péče o dospělé s CF v České republice. Současná péče se opírá o konsenzus Evropské společnosti pro CF (ECFS) z roku 2005 [12], dobrou orientaci v této problematice poskytuje monografie doc. Vávrové et al, vydaná v lednu roku 2006 [13].
V předkládaném sdělení se autoři pokusili shrnout vývoj a současný stav péče o dospělé nemocné s CF za posledních 20 let a zhodnotit vztah stavu plicních funkcí, stavu výživy a kolonizace dýchacích cest.
Metody
Soubor pacientů je tvořen všemi nemocnými, kteří byli sledováni od prosince roku 1987 na 5 výše uvedených pracovištích s diagnózou CF ověřenou přítomností klinických příznaků a dysfunkce CFTR [14].
Ze zdravotní dokumentace nemocných byla s využitím jednotného dotazníku retrospektivně zaznamenána tato data: pohlaví, věk (u žijících k 31. 12. 2007, u zemřelých při úmrtí), koncentrace chloridů v potu, mutace transmembránového regulátoru vodivosti (CFTR), doba sledování, výskyt vybraných komplikací v oblasti dýchacích ústrojí, a to nosní polypózy, pneumotoraxu (PNO), hemoptýzy a alergické bronchopulmonální aspergilózy (ABPA), dále výskyt pankreatické insuficience, hepatopatie (fibróza a cirhóza jater), diabetes mellitus (DM), metabolické kostní nemoci (osteopenie a osteoporóza) a rovněž reprodukční problematika. U současného souboru nemocných pak stav výživy (hodnocení pomocí body mass indexu – BMI) a plicních funkcí (hodnoceno pomocí objemu usilovně vydechnutého vzduchu za první sekundu – FEV1), kolonizace dýchacích cest vybranými patogeny (Pseudomonas aeruginosa, komplex Burkholderia cepacia a netuberkulózní mykobakteria) a vybrané terapeutické postupy (dlouhodobá domácí oxygenoterapie – DDOT, plicní transplantace – LuTx, nutriční podpora, inhalace alfadornázy, dlouhodobá inhalační léčba antibiotiky tobramycinem a kolistinem a dlouhodobá protizánětlivá léčba perorálním azitromycinem). Údaje o kolonizaci, resp. genomovaru komplexu Burkholderia cepacia byly zaznamenány u všech nemocných sledovaných od roku 1994, resp. od roku 2001.
Při statistickém hodnocení byl použit software Primer of Biostatistics, 6th ed.,McGraw-Hill, Blacklick, OH, USA, 2005.Ze statistických testů byly k hodnocení použity t‑test, χ2 test a Pearsonův korelační koeficient. Pokud není uvedeno jinak, jsou data vyjádřena jako průměr ± směrodatná odchylka (SD).
Výsledky
Soubor nemocných
Celkem bylo sledováno 206 pacientů, 110 mužů a 96 žen. Průměrná hodnota koncentrace chloridů v potu byla 92,9 ± 18,2 mmol/l. Výsledky vyšetření mutací CFTR a genotypů nemocných jsou uvedeny v tab. 1, 2. Vývoj počtu sledovaných pacientů v plné péči v jednotlivých centrech je uveden na obr. 1.


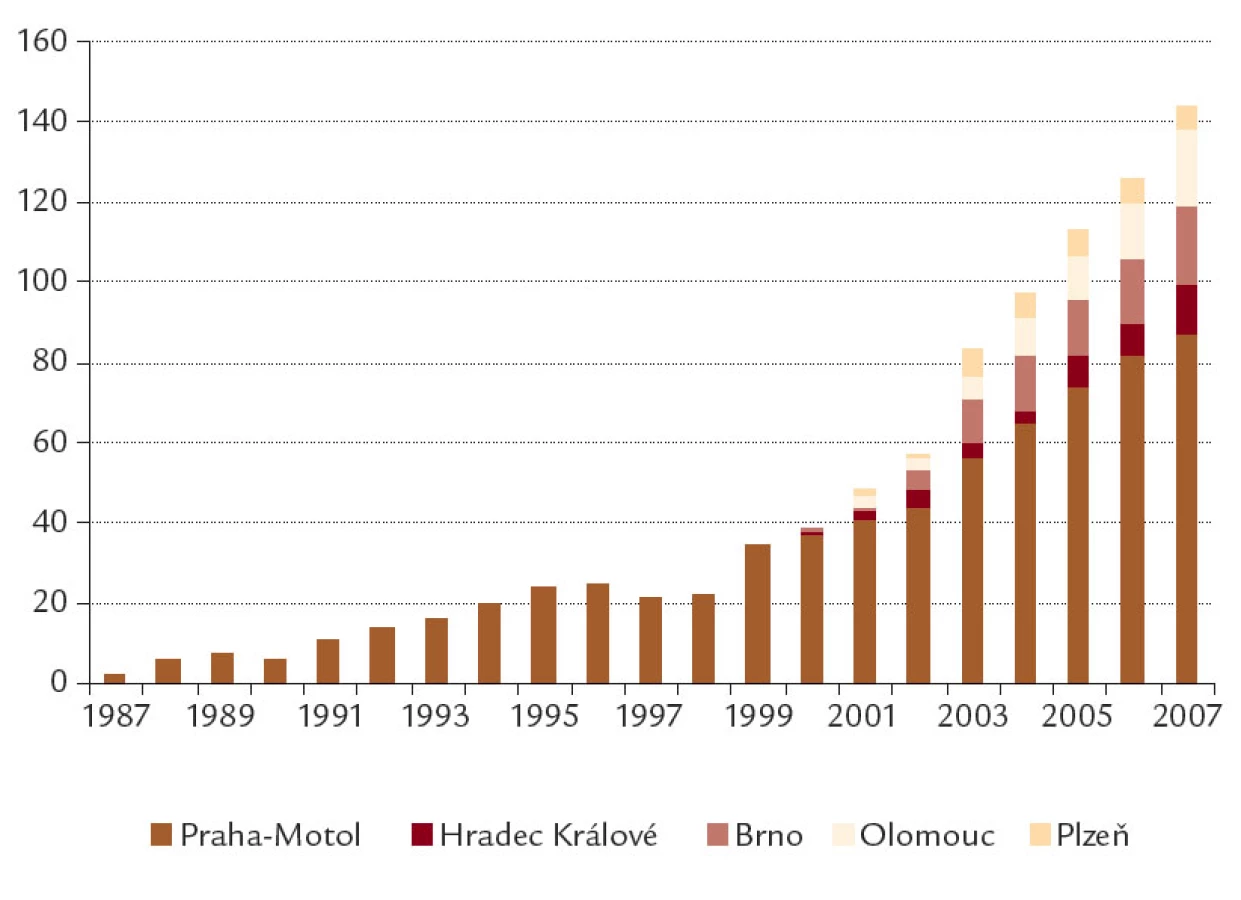
Pankreatická insuficience byla zjištěna u 175 nemocných (85,0 %), nosní polypóza u 60 (29,1 %), PNO u 20 (9,7 %), závažná hemoptýza vyžadující hospitalizaci u 18 (8,7 %) a ABPA u 9 (4,4 %) nemocných. DDOT bylo léčeno 6 žijících a 25 zemřelých nemocných. LuTx prodělalo 10 nemocných, z nich 6 dosud žije. Na čekací listině LuTx je v současnosti 6 nemocných; dalších 5 pacientů zařazených na čekací listinu zemřelo.
Hepatopatie byla zjištěna u 61 (29,6 %) nemocných a porušená glukózová tolerance nebo DM s nutností léčby inzulinem u 58 (28,2 %) nemocných. Metabolická kostní nemoc byla zachycena u 70 (46,7 %) ze 150 vyšetřených nemocných.
Šestnáct žen (16,7 %) a 8 mužů (7,3 %) má děti. U mužů šlo ve 2 případech o plodnou mutaci 3849+10kbC>T, v jednom o asistovanou reprodukci, ve 3 o oplodnění semenem dárce a ve 2 případech již tyto údaje nebyly k dispozici.
Podsoubor zemřelých nemocných
Ze sledovaných nemocných 62 (30,1 %) zemřelo, z toho 39 mužů a 23 žen. Průměrný věk při úmrtí byl 25,4 ± 5,5 roku, medián 24,3 roku a rozmezí 18,4 až41,8 roku. Analýza podskupin zemřelých podle pohlaví, roku úmrtí a kolonizace komplexem Burkholderia cepacia (BC) je uvedena v tab. 3.

Podsoubor žijících nemocných
Soubor žijících nemocných k 31. prosinci 2007 byl tvořen 144 pacienty, 71 mužem a 73 ženami. Průměrný věk u nich byl 27,5 ± 6,5 roku, medián 26,5 a rozmezí 19,3–50,4 roku. Demografická data tohoto souboru jsou uvedena v tab. 4.

Kolonizace dýchacích cest zahrnovala komplex BC u 54 (37,5 %) pacientů, Pseudomonas aeruginosa (PA) u 92 (63,9 %) a bez kolonizace komplexem BC a PA bylo 40 (27,8 %) pacientů. Dále byla v respiračních sekretech zjištěna netuberkulózní mykobakteria u 6 (4,2 %) pacientů, z toho šlo ve 4 případech o Mycobacterium avium a ve 2 o Mycobacterium fortuitum.
Normální hodnoty plicních funkcí (FEV1 ≥ 80 % náležitých hodnot – NH) mělo 41 (28,5 %) nemocných. Ventilační porucha byla v pásmu lehké (FEV1 60,0–79,9 % NH), středně těžké (FEV1 45,0–59,9 % NH), resp. těžké (FEV1 < 45,0 % NH) obstrukce u 37 (25,7 %), 31 (21,5 %), resp. 35 (24,3 %) pacientů.
Normální stav výživy (BMI ≥ 19,0 kg/m2) byl přítomen u 106 (73,6 %) nemocných, v malnutrici (BMI < 19,0 kg/m2) bylo 38 (26,4 %) pacientů.
Závislost stavu plicních funkcí a stavu výživy na věku a kolonizaci dýchacích cest komplexem BC je uvedena v tab. 5; korelace hodnot FEV1 a BMI (r = 0,424, p < 0,0001) je uvedena na obr. 2.


V léčbě se uplatňovala alfadornáza u 109 (75,7 %), inhalační tobramycin u 25 (17,3 %), inhalační kolistin u 21 (14,6 %), dlouhodobě podávaný azitromycin u 22 (15,3 %) a nutriční podpora u 58 (40,3 %) pacientů.
Diskuze
Vývoj přežívání nemocných s CF byl v českých zemích obdobný jako ve světě. Se zlepšováním péče se přežívání prodlužovalo a zprvu pediatričtí nemocní začali dosahovat dospělosti a byli předáváni na pneumologická pracoviště. Tento jinak příznivý vývoj však byl v našich podmínkách negativně ovlivněn přítomností epidemického kmene komplexu BC (genomovar 3A, nově Burkholderia cenocepacia), dle typizace pomocí metody MLST (multilocus sequence typing) šlo o kmen ST‑32 (původně označovaný CZ1) [15,16].
V roce 1960, odkdy se datuje systematická péče o CF nemocné v tomto státě, přežíval z období 1946–1959 jediný diagnostikovaný nemocný [4]. 30. 6. 1981 bylo ze 135 žijících pacientů sledovaných doc. Vávrovou 14 (10,4 %) ve věku 15–20 let a pouze 5 (3,7 %) bylo ve věku 21 a více let [3]. K 31. 12. 1988 bylo v ČR známo 271 nemocných s CF, z nich však pouze 22 (8,1 %) dosáhlo věku ≥ 18 let [17]. V období roku 1985 až 1. 7. 1998 bylo v ČR evidováno 349 nemocných s CF, 254 žijících a 95 zemřelých. Z nich se 126 (36,1 %) dožilo ≥ 18 let. Tento soubor dospělých s CF byl tvořen 85 žijícími a 41 zemřelými nemocnými (průměrný věk při úmrtí 22,7 ± 5,0 roku, rozmezí 18,1–37,5 roku). Dospělí na konci uvedeného období tvořili 33,5 % souboru žijících nemocných. Žijící dospělí nemocní sledovaní ve FN Motol (n = 57) měli průměrný věk 23,1 ± 6,5 roku a průměrnou hodnotu FEV1 60 ± 28 % NH [5]. K 1. 7. 1998 bylo ze 192 nemocných pravidelně sledovaných ve FN Motol 47 (24,5 %) kolonizováno komplexem BC, průměrný věk v době kolonizace byl 13,6 roku. U osob starších 15 a více let byla prevalence kolonizace BC dokonce 44 % [18]. Zemřelí nemocní s CF v ČR měli v období 1985–1990 průměrný věk při úmrtí 12,9 roku (medián 12,2 roku) a v období 1991–1999 18,4 roku (medián 19,0 roku) [19]. Medián věku dožití CF pacientů narozených v letech 1976–1985 byl 25,3 roku, u osob narozených v období 1960–1975 byl pouze 16,4 roku [20]. K 1. 7. 2007 žilo v ČR 471 nemocných s CF, z nich 180 (38,2 %) ve věku ≥ 18 let. Medián věku tehdy činil 14,5 roku a nejstaršímu nemocnému bylo 53,1 roku [21].
Další data se týkají zastoupení dospělých mezi pacienty s CF ve světě. V Německu v roce 2005 tvořili dospělí 43,4 % populace nemocných s CF, přitom v celé populaci pacientů s CF byl tehdy průměrný věk 17,7 roku a medián věku při úmrtí 23,7 roku [22]. Ve Francii představovali dospělí v roce 2003 37,9 % celé populace pacientů s CF, medián věku byl 14,0 roku a medián věku při úmrtí 22,0 let [23]. V USA v roce 2006 tvořili dospělí 44,6 % ze všech pacientů s CF, nejstaršímu nemocnému bylo tehdy 78 let [24]. Ve Velké Británii v roce 2004 představovali pacienti ve věku ≥ 16 let 51,4 % populace nemocných s CF. Průměrný věk byl tehdy 18,0 roku, medián věku 16,4 roku a medián věku zemřelých 25,6 roku [25]. Nemocní narození v roce 2003 ve Velké Británii měli očekávané průměrné přežití 42,6 roku (muži), resp. 36,9 roku (ženy), a nemocní, kteří v roce 2003 dosáhli 20 let, měli očekávané průměrné přežití 45,4 roku (muži), resp. 40,4 roku (ženy) [26]. Predikované střední přežití v USA činilo v roce 2006 36,9 roku [24]. Mortalita nemocných s CF v USA v období 1985–1999 ve věkové skupině 2–15 let u obou pohlaví klesla o 45–70 %. Ve věkové skupině > 15 let však bylo zlepšení méně výrazné, navíc dívky ve věkové skupině 2–20 let měly horší přežití než chlapci [27].
Obdobné výsledky poskytují i námi zjištěná data. Přežití nemocných zemřelých v letech 2001–2007 se proti období 1987–2000 zlepšilo a bylo horší u nemocných kolonizovaných komplexem BC. Rovněž jsme zjistili nesignifikantně častější úmrtí u mužů, a to ve vyšším věku než u žen. To je v souladu s uváděnou horší prognózou dívek ve věku 2–20 let, které z důvodu časnějšího úmrtí (před dosažením dospělosti) na pneumologická pracoviště předány být vůbec nemohly.
Co se týče bronchopulmonálního onemocnění a stavu výživy nemocných s CF, je i zde k dispozici srovnání se světem. V Německu v roce 2005 v centrech s 50 a více pacienty byla u osob ve věku ≥ 18 let průměrná hodnota FEV1 58,6 ± 24,9 % NH a průměrná hodnota BMI 20,5 ± 2,8 kg/m2 (70,1 % nemocných mělo BMI ≥ 19,0 kg/m2) [22]. Ve Velké Británii v roce 2004 měli nemocní ve věku ≥ 16 let v 78,7 % BMI ≥ 19,0 kg/m2 a v 49,7 % FEV1 ≥ 60 % NH. Během roku 2004 pak měli v 58,5 % chronickou infekci PA, v 7,0 % nosní polypy, v 6,3 % ABPA, v 1,2 % PNO, v 0,9 % masivní hemoptýzu a v 5,3 % alespoň jeden záchyt komplexu BC [25]. V USA v roce 2006 bylo léčeno alfadornázou 67,4 % nemocných ve věku ≥ 6 let. Kolonizaci PA mělo v celé populaci CF nemocných 55,0 % a komplexem BC 2,9 % [24]. V roce 2000 byla průměrná hodnota FEV1 u dospělých s CF v USA 60,8 % NH. 36 % pacientů mělo FEV1 ≥ 70 % NH, 39 % FEV1 40–69 % NH a 25 % pacientů FEV1 < 40 % NH. Častější u dospělých oproti dětem v tomto roce byla kolonizace PA (79 vs 47 %) a komplexem BC (6 vs 2 %) a výskyt masivních hemoptýz (1,8 vs 0,1 %) a PNO (1,4 vs 0,2 %) [28]. Výskyt nosní polypózy u pacientů s CF je udáván v 26 % [29], což odpovídá i námi zjištěné prevalenci. Diagnostická kritéria ABPA u nemocných s CF byla publikována v roce 2003 [30]. Výskyt ABPA v evropských zemích u nemocných ve věku ≥ 6 let byl 10 %, rozdíly mezi jednotlivými zeměmi i centry však byly značné [31]. PNO je závažnou komplikací, obvykle u starších osob s pokročilejším plicním onemocněním. Během života (analýza dat z USA 1990–1999) postihne 3,4 % CF populace, medián věku prvního výskytu je 21,0 roku. K 72,4 % případů PNO dojde u osob ve věku ≥ 18 let a k 75 % případů u osob s FEV1 < 40 % NH [32]. Masivní hemoptýza (definovaná jako > 240 ml/den nebo > 100 ml/den několik dnů) je rovněž závažnou komplikací a postihuje starší nemocné s pokročilejším plicním onemocněním. Během života (analýza dat z USA 1990–1999) k ní dojde u 4,1 % nemocných s CF, medián věku prvního výskytu je 23,0 roku. K 75 % případům masivní hemoptýzy dojde u osob ve věku ≥ 18 let a k 61,2 % případů u osob s FEV1 < 40 % NH [33].
Námi zjištěná data týkající se plicních funkcí a stavu výživy jsou s výše uvedenými údaji srovnatelná. U dalších charakteristik týkajících se bronchopulmonálního postižení je srovnatelný výskyt kolonizace PA a nosní polypózy. Zásadní rozdíl je v prevalenci kolonizace komplexem BC, a to vzhledem k výskytu epidemického kmene, jak již bylo uvedeno výše.
Z mimoplicních manifestací je sledován především výskyt tzv. velkých komplikací, a to DM, hepatopatie a osteoporózy. Ve věkové kategorii 20–29 let, resp. ≥ 30 let, mělo DM 35 %, resp. 43 %, dospělých s CF [34]. Hepatopatie se rozvíjí obvykle do 10 let života a postihuje 27–35 % nemocných s CF, přičemž v asi 5 % případů progreduje do end‑stage multilobulární biliární cirhózy [35]. Nízká kostní denzita je zjišťována u 50–75 % dospělých s CF [36]. Námi zjištěné prevalence jsou s těmito daty srovnatelné. V uvedených publikacích je rovněž podrobně rozebrána problematika těchto komplikací CF.
Dospělá populace nemocných s CF se od dětské pochopitelně liší i reprodukční problematikou. Od 90. let minulého století ročně otěhotní 3–4 % žen s CF ve věku ≥ 17 let [37]. Plodných je pouze 1–2 % mužů s CF, v ostatních případech jde o obstruktivní azoospermii [28].
Principy léčby dospělých s CF se i nás v současnosti opírají o konsenzuální dokumenty podporované americkou Cystic Fibrosis Foundation [28,38].
Závěr
Uvedený článek demonstruje několik důležitých skutečností. Jde především o narůstající počet dospělých pacientů s CF a jejich prodlužující se přežití, dále o těsný vztah stavu plicních funkcí a stavu výživy a v neposlední řadě o nepříznivý vliv kolonizace komplexem BC. K prevenci kolonizace epidemickými kmeny jsou dnes podrobně rozpracované režimy [39]. Upozornit je třeba i na možnost stanovení diagnózy CF i v dospělosti, především u pankreaticky suficientních forem onemocnění [40].
Poděkování
Tento článek by nemohl vzniknout bez přispění pediatrů, kteří se o pacienty v dětském věku starali a do naší péče je po dosažení dospělosti předávali (za všechny jmenujeme doc. MUDr. Věru Vávrovou); pracovníků chloridových laboratoří a klinických genetiků (za všechny jmenujeme prof. MUDr. Milana Macka), bez nichž by dysfunkce CFTR nemohla být ověřena; dále pracovníků gastroenterologických laboratoří, bez nichž by nemohla být ověřena pankreatická insuficience vyšetřením elastázy 1 ve stolici; lékařských mikrobiologů, bez nichž by nebyly dostupné údaje o kolonizaci dýchacích cest; pneumologů v uvedených fakultních nemocnicích i v terénních pracovištích, kteří se na péči o nemocné s CF podíleli; kolektivu transplantačního oddělení III. chirurgické kliniky 1. LF UK a FN Motol, které je jediným pracovištěm, do jehož péče můžeme nemocné po vyčerpání našich možností léčby předat (za všechny jmenujeme doc. MUDr. Roberta Lischkeho) a v neposlední řadě lékařů dalších odborností (otorinolaryngologů, gastroenterologů, diabetologů, osteologů a řady dalších), kteří se podíleli na péči o mimoplicní komplikace u našich nemocných a bez nichž by multidisciplinární přístup nebyl možný.
Zkratky
ABPA alergická bronchopulmonální aspergilóza
BC Burkholderia cepacia
BMI body mass index
CF cystická fibróza
CFTR transmembránový regulátor vodivosti
DDOT dlouhodobá domácí oxygenoterapie
DM diabetes mellitus
ECFS European Cystic Fibrosis Society
FEV1 usilovně vydechnutý objem za první vteřinu
LuTx transplantace plic
MLST multilocus sequence typing
NH náležitá hodnota
PA Pseudomonas aeruginosa
PNO pneumotorax
Doručeno do redakce: 10. 11. 2008
Přijato po recenzi: 20. 1. 2009
MUDr. Libor Fila
www.fnmotol.cz
e‑mail: libor_fila@seznam.cz
Sources
1. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. A clinical and pathological study. Amer J Dis Child 1938; 56 : 344–399.
2. Švejcar J, Benešová D, Houštěk J. Cystická fibróza pankreatu. Čas Lék Česk 1948; 87 : 1116–1122.
3. Houštěk J, Vávrová V. Cystická fibróza. In: Houštěk J, Syrovátka A (eds). Pokroky v pediatrii 7. Praha: Avicenum 1983 : 139–180.
4. Vávrová V, Zemková D, Macek M Jr. Pohled na cystickou fibrózu v roce 2002. Trendy v medicíně 2002; 4 : 24–34.
5. Vávrová V, Zemková D, Bartošová J et al. Cystická fibróza – nemoc dospívajících a dospělých? Čas Lék Česk 1999; 138 : 654–659.
6. Hellerstein H. Cystic fibrosis of the pancreas in an adult. Ohio Med J 1946; 42 : 616–617. Citace in: Widerman E, Millner L, Sexauer W et al. Health status and sociodemographic characteristics of adults receiving a cystic fibrosis diagnosis after age 18 years. Chest 2000; 118 : 427–433.
7. Shwachman H, Kulczycki LL. Long‑term study of one hundred five patients with cystic fibrosis. Studies made over a five - to fourteen-year period. AMA J Dis Child 1958; 96 : 6–15.
8. Netoušek M. Mucoviscidosis adultorum hereditaria. Čas Lék Česk 1960; 99 : 1525–1527.
9. Ledeč J, Šimonová J. Syndrom mukoviscidózy u dospělých. Vnitř Lék 1966; 12 : 525–530.
10. Marel M. Neobvyklý případ mukoviscidózy u mladého muže. Prakt Lék 1986; 66 : 794–795.
11. Musil J, Rozehnalová H, Kvapil M. Deset let péče o dospělé nemocné s cystickou fibrózou. Vnitř Lék 1997; 43 : 728–732.
12. Kerem E, Conway S, Elborn S et al. Consensus Committee. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 2005; 4 : 7–26.
13. Vávrová V a kol. Cystická fibróza. Praha: Grada Publishing 2006.
14. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr 1998; 132 : 589–595.
15. Baldwin A, Mahenthiralingam E, Thickett KM et al. Multilocus sequence typing scheme that provides both species and strain differentiation for the Burkholderia cepacia complex. J Clin Microbiol 2005; 43 : 4665–4673.
16. Drevinek P, Vosahlikova S, Cinek O et al. Widespread clone of Burkholderia cenocepacia in cystic fibrosis patients in the Czech Republic. J Med Microbiol 2005; 54 : 655–659.
17. Houstek J, Hruskovic I, Vyhnálek M et al. Care for cystic fibrosis patients in Czechoslovakia. Acta Univ Carol Med (Praha) 1990; 36 : 217–219.
18. Zemková D, Vávrová V, Bartošová J B. Cepacia v ČR – pro Zpravodaj CF. Zpravodaj Klubu rodičů a přátel dětí nemocných cystickou fibrózou 1998 : 28–30.
19. Vávrová V, Zemková D, Bartošová J et al. Cystic fibrosis in the Czech Republic from 1960–2000. Alergie 2001; 3 (Suppl 1):S46.
20. Zemková D, Skalická V, Bartošová J et al. Moderní management cystické fibrózy a jeho vliv na zdravotní stav a přežívání českých nemocných. Čes Slov Pediatr 2008; 63 : 76–82.
21. Vávrová V, Zemková D, Skalická V et al. Cystic fibrosis in Czech Republic. ECFS Newsletter 2008; 25 : 4–9.
22. Stern M, Wiedemann B, Wenzlaff P. German Cystic Fibrosis Quality Assessment Group. From registry to quality management: the German Cystic Fibrosis Quality Assessment project 1995–2006. Eur Respir J 2008; 31 : 29–35.
23. Bellis G, Cazes MH, Parant A et al. Cystic fibrosis mortality trends in France. J Cyst Fibros 2007; 6 : 179–186.
24. Cystic Fibrosis Foundation. Patient Registry 2006 Annual Report, Bethesda, Maryland USA.
25. Cystic Fibrosis Trust. Annual Data Report 2004. Dundee: UK CF Database, University of Dundee Dundee 2006.
26. Dodge JA, Lewis PA, Stanton M et al. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007; 29 : 522–526.
27. Kulich M, Rosenfeld M, Goss CH et al. Improved survival among young patients with cystic fibrosis. J Pediatr 2003; 142 : 631–636.
28. Yankaskas JR, Marshall BC, Sufian B et al. Cystic fibrosis adult care: consensus conference report. Chest 2004; 125 (Suppl 1): 1S–39S.
29. Orenstein DM, Winnie GB, Altman H. Cystic fibrosis: a 2002 update. J Pediatr 2002; 140 : 156–164.
30. Stevens DA, Moss RB, Kurup VP et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. State of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis 2003; 37 (Suppl 3): S225–S264.
31. Mastella G, Rainisio M, Harms HK et al.Allergic bronchopulmonary aspergillosis in cystic fibrosis. A European epidemiological study. Epidemiologic Registry of Cystic Fibrosis. Eur Respir J 2000; 16 : 464–471.
32. Flume PA, Strange C, Ye X et al. Pneumothorax in cystic fibrosis. Chest 2005; 128 : 720–728.
33. Flume PA, Yankaskas JR, Ebeling M et al. Massive hemoptysis in cystic fibrosis. Chest 2005; 128 : 729–738.
34. Moran A, Hardin D, Rodman D et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus. A consensus conference report. Diabetes Res Clin Pract 1999; 45 : 61–73.
35. Colombo C, Russo MC, Zazzeron L et al. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr 2006; 43 (Suppl 1): S49–S55.
36. Aris RM, Merkel PA, Bachrach LK et al. Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab 2005; 90 : 1888–1896.
37. Edenborough FP, Borgo G, Knoop C et al. Guidelines for the management of pregnancy in women with cystic fibrosis. J Cyst Fibros 2008; 7 (Suppl 1): S2–S32.
38. Flume PA, O’Sullivan BP, Robinson KA et al. Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176 : 957–969.
39. Saiman L, Siegel J. Infection control in cystic fibrosis. Clin Microbiol Rev 2004; 17 : 57–71.
40. Fila L. Diagnostika cystické fibrózy v dospělosti. Lék Listy 2008; 57 : 22–23.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue 6
Most read in this issue
- Léčba selhání ledvin u mnohočetného myelomu
- Je průkaz adenomu nadledviny u pacientů s primárním hyperaldosteronizmem dostatečný pro indikaci adrenalektomie?
- Využití farmakogenetiky při léčbě warfarinem
- Případ familiární adenomatózní polypózy a návrh systému dispenzarizace