
Pacientka s AL‑amyloidózou a závažným deficitem faktoru X je po vysokodávkované chemoterapii již 7 let v kompletní hematologické remisi s normální aktivitou faktoru X. Popis případu a přehled literatury
A patient with AL amyloidosis and severe factor X deficiency has been in complete haematological remission with normal factor X activity for 7 years following high‑dose chemotherapy. A case study and literature review
Disturbance of haemostasis and bleeding are rather frequent complications of AL amyloidosis. These are frequently caused by increased fragility of capillaries, thrombocyte function disorders and coagulation cascade defects. The most frequent coagulation disorder is decreased factor X activity. We describe a 34‑year old female after hysterectomy for myomatous uterus and metrorhagia. Before the surgery, the attending physicians did not identify any pathological changes suggesting a need for further investigations or presence of AL amyloidosis. Post‑surgery development was complicated by life-threatening diffuse haemorrhage. Extended investigations of coagulation cascade revealed reduction of factor X activity to 16%. Targeted histological examination of the resected uterus confirmed AL amyloid deposits consisting of κ chains. The patient’s bone marrow contained certain small level of multiplied κ chains-expressing plasma cells (< 10%); monoclonal immunoglobulins IgG κ and free κ chains were identified in serum. At that time, the patient did not satisfy the then valid Durie - Salmon criteria for multiple myeloma and thus the patient was diagnosed with primary systemic AL amyloidosis. The patient’s condition gradually improved following substitution therapy (Prothromplex, fresh frozen plasma and erythrocyte transfusion) and bleeding slowly ceased so that chemotherapy with VAD (vincristine, adriamycin and dexamethasone) was initiated 6 weeks after the surgery. A total of 8 chemotherapy cycles were administered and complete haematological remission was achieved after the 5th cycle. Administration of the 8 VAD chemotherapy cycles resulted in increased factor X activity; bleeding complications subsided completely, thereby decreasing the risk of life - threatening mucositis‑associated haemorrhage. Consequently, tandem high‑dose chemotherapy (melphalan 100 mg/ m2) with autologous haematopoietic stem cells transplantation was added to the treatment plan. Treatment was completed at the beginning of 2003 and, from that time, the patient is on continuous maintenance therapy with interferon α. Seven years from the diagnosis and 6 years from the completion of treatment the patient is in complete haematological remission, with no signs of organic damage caused by AL amyloid and with normal factor X activity. Factor X activity increased at the time when complete haematological remission was achieved after 8 cycles of VAD chemotherapy to 42%, it reached 68% the second year following high‑dose chemotherapy, 77% after 5 years and 85% after 7 years. We had considered administration of high‑dose chemotherapy in the standard regimen, i.e. following 4 cycles of VAD chemotherapy, as too high risk in the described young female patient. Therefore, we administered 8 cycles of conventional chemotherapy and only after complete haematological remission and partial organ response (factor X activity increased to 42%) were achieved, we added tandem high‑dose chemotherapy to the treatment. We thus achieved long‑term (7‑years so far) complete haematological and organ remission. Increase in factor X activity is explicit over the entire 7‑year observational period. We recommend starting treatment of high‑risk transplant patients with AL amyloidosis with traditional chemotherapy regimen and, in case of positive haematological and organ treatment response, we recommend re‑examination of potential benefits and risks of high‑dose chemotherapy with autologous transplantation.
Key words:
factor X – AL amyloidosis – multiple myeloma – monoclonal gammopathy – high‑dose chemotherapy with autologous transplantation – coagulopath
Authors:
Z. Adam 1; M. Matýšková 2; M. Krejčí 1; L. Pour 1; J. Kissová 2; M. Šlechtová 2; G. Chlupová 2; Y. Stavařová 3; J. Simonides 4; M. Penka 2; J. Mayer 1; R. Hájek 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
1; Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
2; Hematologicko transfuzní oddělení Baťovy krajské nemocnice, a. s., Zlín, přednostka prim. MUDr. Yveta Stavařová
3; Hematologicko‑transfuzní oddělení Nemocnice Znojmo, přednosta prim. MUDr. Jan Simonides
4
Published in:
Vnitř Lék 2010; 56(1): 67-78
Category:
Case Reports
Overview
Poměrně častou komplikací při AL‑amyloidóze je narušení hemostázy a krvácivé příznaky. Bývají způsobeny zvýšenou fragilitou kapilár a malých cévek, poruchou funkce trombocytů a defekty v koagulační kaskádě. Nejčastější koagulační poruchou je snížení aktivity faktoru X. Popisujeme 34letou ženu, která podstoupila hysterektomii pro myomatózní uterus a metroragii. Před operačním zákrokem ošetřující lékaři na nemocné neshledali žádné patologické změny, které by vedly k dalšímu vyšetřování či k podezření na AL‑amyloidózu. Pooperační průběh byl komplikován život ohrožujícím difuzním krvácením. Rozšířené koagulační vyšetření odhalilo sníženou aktivitu faktoru X na 16 %. Cílené histologické vyšetření resekované dělohy prokázalo AL‑amyloidová depozita, tvořená κ řetězci. Pacientka měla v kostní dřeni nepatrně zmnožené plazmatické buňky (< 10 %), exprimující pouze řetězce κ a v séru prokázaný monoklonální imunoglobulin IgG κ a volné κ řetězce. Pacientka nesplňovala v té době platná Durieho a Salmonova kritéria pro mnohočetný myelom, a tak byla diagnóza uzavřena jako primární systémová AL‑amyloidóza. Při substituční léčbě (Prothromplex, čerstvě zmražená plazma a erytrocytární transfuze) se stav nemocné velmi pozvolna zlepšoval a difuzní krvácení se pomalu zmírňovalo natolik, že po 6 týdnech od operace byla zahájena chemoterapie VAD (vinkristin, adriamycin a dexametazon). Celkem bylo aplikováno osm cyklů chemoterapie a bylo dosaženo kompletní hematologické remise po 5. cyklu. Po osmi cyklech chemoterapie VAD se zvýšila aktivita faktoru X a zcela vymizely krvácivé projevy, čímž se snížilo riziko život ohrožujícího krvácení při mukozitidě. Proto byl doplněn léčebný plán o tandemovou vysokodávkovanou chemoterapii (melfalan 100 mg/m2) s autologní transplantací krvetvorných kmenových buněk. Léčba byla ukončena počátkem roku 2003, od té doby je pacientka trvale na udržovací léčbě interferonem α. Sedm let od stanovení diagnózy a šest let od ukončení léčby je v kompletní hematologické remisi bez známek orgánového poškození AL‑amyloidem, s normální aktivitou faktoru X. Aktivita faktoru X se v době dosažení kompletní hematologické remise, po osmi cyklech chemoterapie VAD, zvýšila na 42 %, druhý rok po vysokodávkované chemoterapii dosáhla 68 %, po pěti letech 77 %, po sedmi letech 85 %. U popisované mladé nemocné jsme považovali ze vysoce rizikové podat vysokodávkovanou chemoterapii standardně, tedy po čtyřech cyklech chemoterapie VAD. Proto jsme podali osm cyklů konvenční chemoterapie a teprve po dosažení kompletní hematologické remise a parciální orgánové léčebné odpovědi (zvýšení aktivity faktoru X na 42 %) jsme léčbu doplnili o tandemovou vysokodávkovanou chemoterapii. Dosáhli jsme tak dvoudobé (již sedm let trvající) kompletní hematologické a orgánové remise. Zvyšování aktivity faktoru X je zřetelné po celý sedmiletý interval sledování. V případě vysokého transplantačního rizika u pacientů s AL‑amyidózou považujeme za vhodné začít léčbu klasickým chemoterapeutickým režimem a v případě dosažení hematologické a orgánové léčebné odpovědi znovu přehodnotit přínos a rizika vysokodávkované chemoterapie s autologní transplantací.
Klíčová slova:
faktor X – AL‑amyloidóza – mnohočetný myelom – monoklonální gamapatie – vysokodávkovaná chemoterapie s autologní transplantací – koagulopatie
Úvod
AL‑amyloidóza je popisný termín pro patologické stavy způsobené ukládáním lehkých řetězců (většinou typu l) ve tkáních a orgánech ve formě fibrilární hmoty, která vykazuje typické histochemické reakce [1,2]. U každého zdravého člověka koluje určité množství volných lehkých řetězců k i l v séru, aniž by docházelo k jejich ukládání a tvorbě amyloidu. V patologickém klonu plazmocytů může dojít ke změně genetické informace pro tvorbu lehkých řetězců. Tato změna způsobí tvorbu nefyziologických lehkých řetězců, které mají tendenci k vytváření tkáňových fibrilárních depozit ve formě AL‑amyloidu.
Pokud proliferace těchto patologických klonálních plazmatických buněk má maligní charakter, pak se jedná o sekundární AL‑amyloidózu při mnohočetném myelomu či Waldenströmově makroglobulinemii.
Jestliže má proliferace těchto patologických klonálních plazmatických buněk benigní charakter, podobně jako plazmocyty u monoklonální gamapatie nejistého významu, pak se jedná o primární systémovou AL‑amyloidózu.
Pro úplnost musíme uvést, že zcela vzácně se vyskytne ložisková AL‑amyloidóza, třeba v bronchu či ORL oblasti, u níž se předpokládá v ložisku existující patologický klon plazmocytů, který secernuje do svého okolí volné lehké řetězce a z nich se pak tvoří solitární ložisko amyloidu.
Problém primární systémové AL‑amyloidózy je v tom, že klon plazmocytů je sice benigní, ale jeho produkt – amyloidogenní lehké řetězce – vytvářejí depozita takového rozsahu, že mohou vést ke smrti nemocného.
To, v kterém orgánu či tkáni bude docházet k ukládání amyloidu, je důsledkem tropizmu těchto lehkých řetězců. Orgánový tropizmus amyloidogenních lehkých řetězců je závislý na tom, který z genů pro variabilní část molekuly lehkého řetězce byl začleněn do DNA, kódující právě tento lehký řetězec [3].
Příznaky AL‑amyloidózy se odvíjejí od toho, který orgán je zásadně postižen a jsou přehledně popsány v jiných publikacích [1,4–7].
V následujícím popisu případu se zabýváme vlivem AL‑amyloidu na koagulační vlastnosti krve.
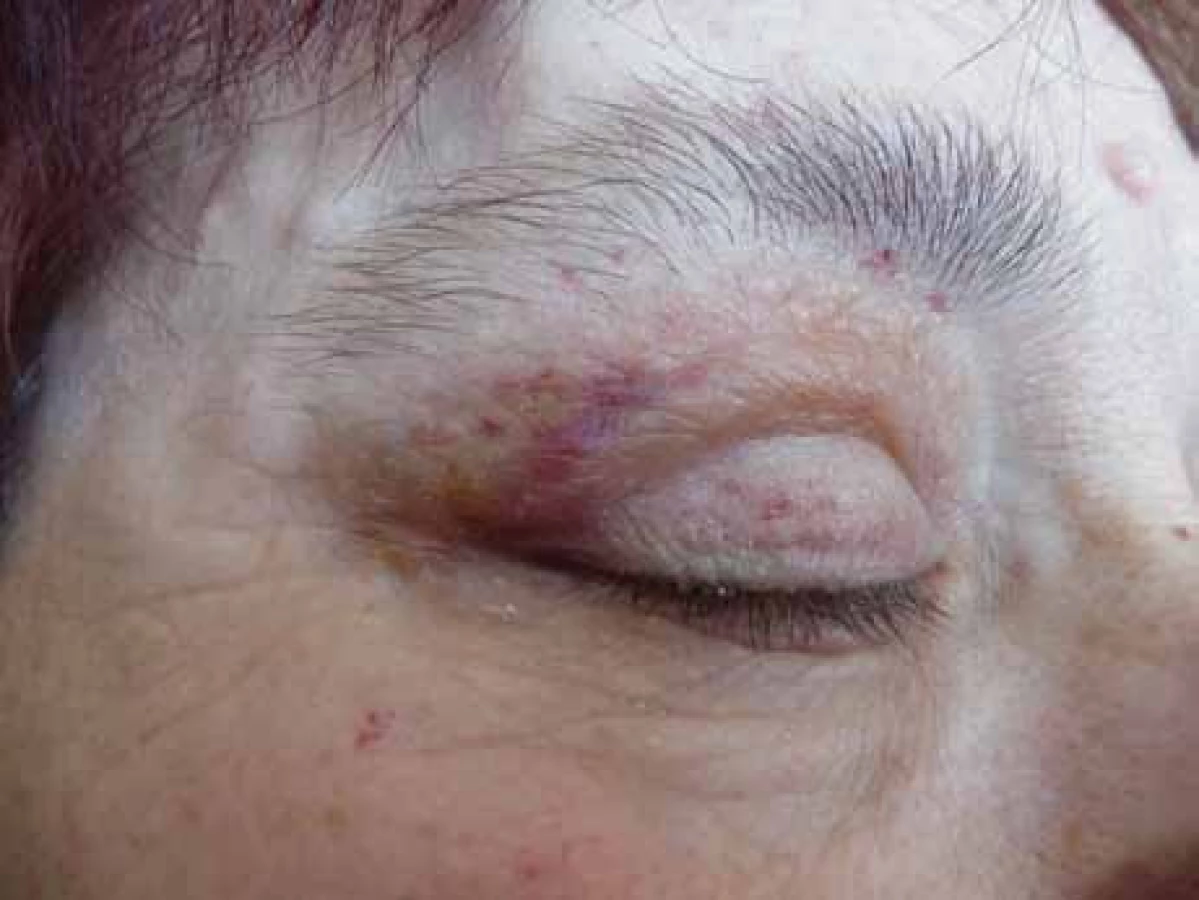
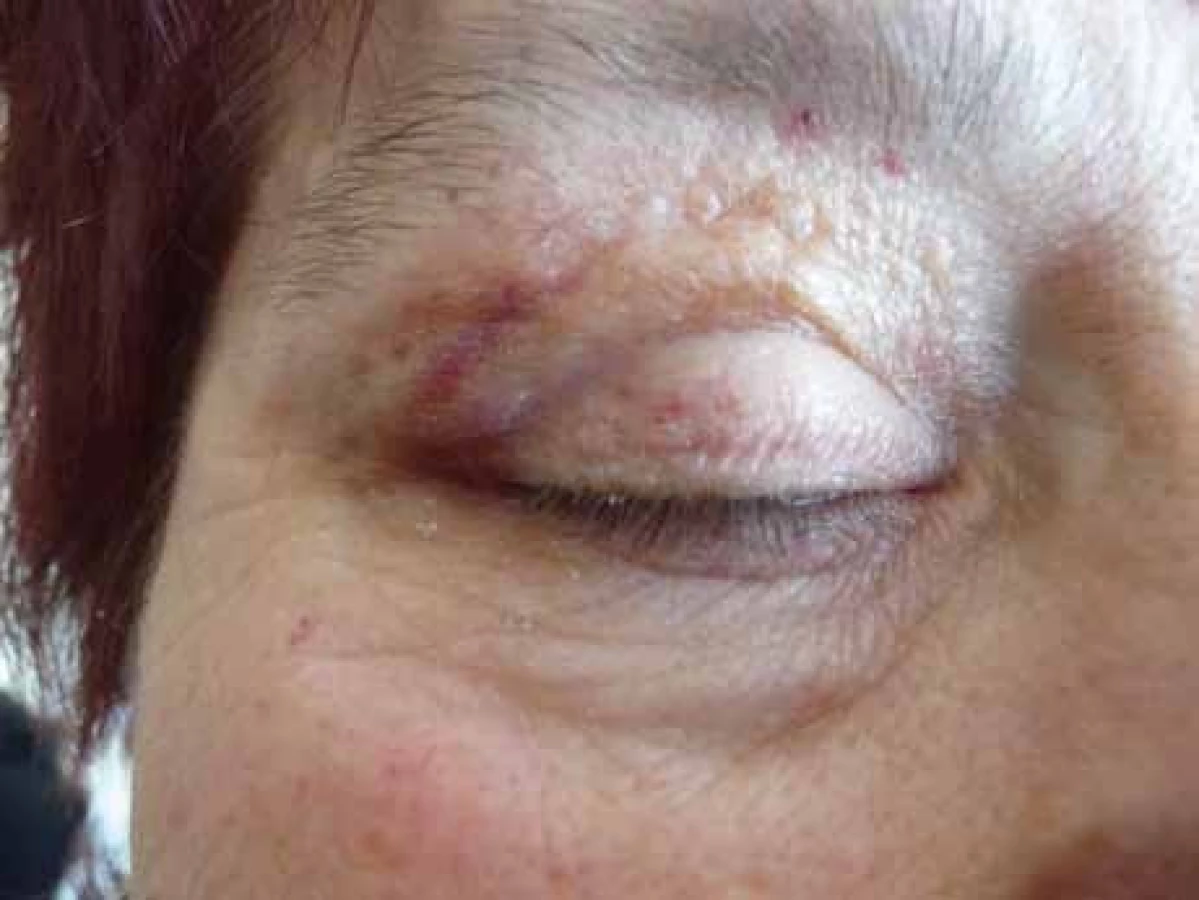
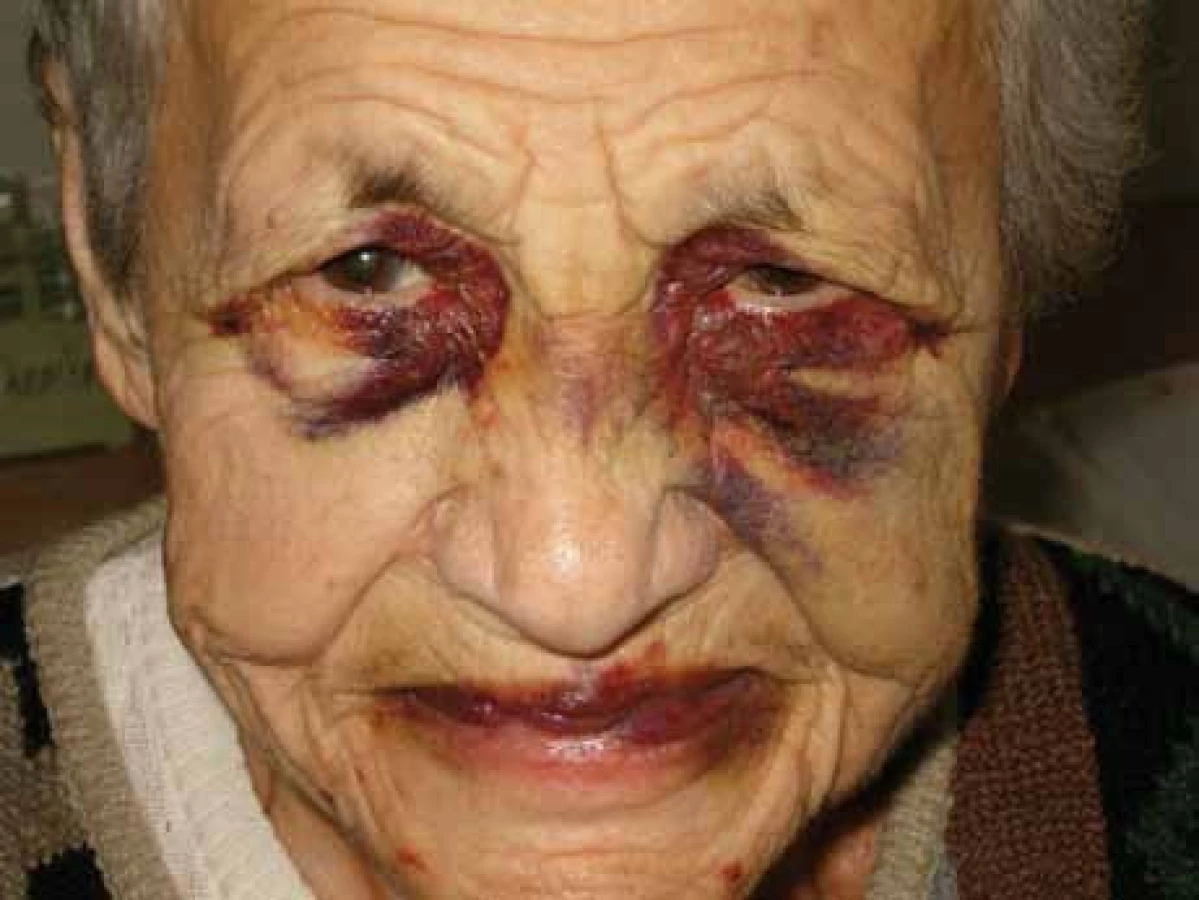
Poruch hemostázy a krvácivé projevy různé intenzity jsou relativně častým projevem AL‑amyloidózy [8–14]. U nemocných osob s AL‑amyloidózou je zvýšená fragilita drobných cév, která je způsobena depozity amyloidu v jejich stěnách. Dosud není zcela jasné, proč se spontánní krvácení, způsobené zvýšenou fragilitou kapilár, nejčastěji manifestuje v oblasti víček [15,16]. Drobné hematomy v oblasti víček ilustruje obr. 1 a pokročilé obr. 2 a 3.
Ke zvýšené cévní fragilitě přistupují ještě další faktory. Deficit specifických koagulačních faktorů byl popsán poprvé v roce 1962 [17]. Tento jev byl posléze potvrzen dalšími popisy případů [18–22]. Nejvíce popsaných deficitů se týkalo faktoru X [23–29]. Jen zcela výjimečně byly popsány deficity dalších faktorů:
- deficit faktoru IX [30,31]
- deficit faktoru II [30,31]
- deficit faktoru VII [30]
- deficit faktoru V [32,33]
- deficit faktoru X a XII [34]
- získaný von Willebrandův syndrom u pacienta s AL‑amyloidózou [35]
- získaný deficit faktor faktoru X a sou-časně i antitrombinu [36]
- současný deficit více koagulačních faktorů včetně snížené koncentrace fibrinogenu byl popsána u AL‑amyloidózy v souvislosti s diseminovanou intravaskulární koagulací [37,38] a také nerovnováhou v systému trom-bin‑antitrombin [39,40]
Průkazy koagulačních faktorů navázaných na AL‑amyloidové hmoty [41,42] a ojedinělé zprávy o tom, že koagulační abnormality ustoupily po odstranění amyloidem napěchované sleziny [43], vedly k vytvoření teorie, že u AL‑amyloidózy dochází k sekvestraci koagulačních faktorů na amyloidu.
Teprve v novějších pracích bylo prokázáno, že funkční deficit faktoru X u AL‑amyloidózy souvisí jak s jeho sekvestrací na amyloidu, tak se snížením aktivity faktoru X. V následujícím popisu chceme upozornit na závažné koagulační problémy, které mohou ohrozit život, přičemž v pozadí může být nepoznaná AL amyloidóza.
Popis případu
V době diagnózy se jednalo o 34letou ženu (narozená 1968). V rodinné anamnéze nebyly žádné informace o výskytu krvácivých či trombotických projevů. V osobní anamnéze byly častější záněty žil bez bližší specifikace, 2krát porod císařským řezem, bez trombotických či krvácivých komplikací. Mladá žena byla bez dietního omezení, bez užívání antibiotik a bez antikoagulační léčby. Neměla žádné příznaky z oblasti gastrointestinálního traktu. Do roku 2002 byla bez výskytu krvácivých projevů. Od začátku roku 2002 popisovala častější epistaxe, od poloviny dubna roku 2002 menometroragie. Proto byla hospitalizována na spádovém gynekologickém pracovišti a provedena 2krát separovaná abraze pro opakované gynekologické krvácení. Tím se však krvácení nevyřešilo. Gynekologové stanovili diagnózu endometrióza a myomatózní uterus a indikovali hysterektomii.
Před plánovanou operací však vznikl otok pravé dolní končetiny, dle ultrazvukového vyšetření bylo vysloveno podezření na hlubokou žilní trombózu lýtkové žíly vpravo. Z důvodu této komplikace před plánovaným operačním zákrokem byla přeložena na Gynekologickou kliniku FN Brno.
V květnu roku 2002 byla provedena hysterektomie. Samotný výkon proběhl bez komplikací. První pooperační den poklesly však hodnoty krevního obrazu, změnil se palpační nález na břiše a objevila se hypotenze a tachykardie. Byla provedena operační revize s nálezem hemoperitonea – asi 2 000 ml jen částečně sražené krve. Krvácení bylo difuzní, nebyl nalezen chirurgicky řešitelný zdroj krvácení. Gynekologové odstranili hematom z Retziova prostoru, asi 100 ml.
Při řešení komplikace bylo provedeno první rozsáhlejší hematologické vyšetření.
V krevním obraze byla lehká leukocytóza, 10,5 ×109/l, anémie, koncentrace hemoglobinu 83,4 g/l, lehká trombocytopenie 117 × 109/l. V základním koagulogramu byly prodloužené hodnoty PT – R 1,56 aPTT – 47,4 s, normální hladina fibrinogenu – 2,73 g/l, lehké snížení hladiny antitrombinu (AT) na 77 %, zvýšené D‑dimery – 1,95 μg/l, prodloužení trombinového času (TČ) – 23 s.
Gynekologové začali při pokračujícím krvácení podávat čerstvě zmraženou plazmu. Po výše uvedeném koagulačním vyšetření byla zahájena pravidelná aplikace koncentrátu protrombinového komplexu – Prothromplex – v intervalu 6–8 hod a pravidelné podávání antifibrinolytik (Pamba).
Leč ne vždy se v životě ihned zdaří to, o co usilujeme. I přes výše popsanou substituční léčbu se pacientce po další dva dny zhoršovaly bolesti břicha a pokračovalo krvácení. Stav nemocné se prostě nelepšil. Pacientka byla přeložena na Kliniku anesteziologie, resuscitace a intenzivní medicíny (KARIM) FN Brno, mimo jiné pro snadnější dostupnost hematologické péče.
Vstupně při překladu do FN Brno: v krevním obrazu opět lehká leukocytóza, anémie a lehká trombocytopenie. V koagulaci prodloužené PT (R 1,61), hraničně vyšší aPTT (42,6 s/R 1,3), normální hladina fibrinogenu (2,26 g/l), TČ (19,7 s) v normě. Hladina AT lehce pod dolní hranicí normy (78 %), elevace D‑dimerů (3,48 μg/ml). Dále byla výrazná hyperbilirubinemie, 59 μmol/l, elevace aktivity enzymů AST, GMT, ale hodnota ALT byla v normě. Provedeno kompletní hematologické vyšetření s následujícími výsledky:
- retrakce koagula – 77,3 %, snížená
- euglobulinová lýza – 180 min
- vyšetření funkční aktivity koagulačních faktorů: faktor II – 75 %, faktor V – 83 %, faktor IX – 103 %, lehké snížení faktoru VII – 50 %, velmi závažné snížení faktoru X – 16 %, při opakování 20 %
- ostatní vyšetření: vWF – Ag (LIA) nad 150 %, inhibitor faktoru X nepřítomen, screening na lupus antikoagulans opakovaně negativní
Byla provedena následující zobrazovací vyšetření: RTG snímek plicní – normální nález, ultrazvukové vyšetření břicha – normální obraz parenchymatózních orgánů, tekutina kolem jater, duplexní vyšetření hlavního žilního systému dolních končetin – bez průkazu trombotické okluze.
HIV negativní, sérologie hepatitidy negativní, jen anti‑HAV pozitivní.
Klinický nález: při vědomí, kardio-pulmonálně kompenzována, břicho vzedmuté, difuzně bolestivé.
Dále opakované krevní ztráty do drénů až 1 700 ml denně, 9. den po provedené hysterektomii evakuace hematomu ve stěně břišní subfasciálně, následující den revize stěny břišní.
17. pooperační den zjištěn pomocí sonografie a CT rozsáhlý hematom stěny břišní, peritonea, různých denzit a stáří – opět operační revize. V dalším průběhu vaginální krvácení z poševního pahýlu.
Měsíc od provedené hysterektomie slábnutí krvácivých projevů, ale objevily se známky postižení jater – hepatomegalie, elevace jaterních enzymů, zejména GMT.
Diferenciální diagnostika defektu faktoru X u pacientky:
- hereditární defekt faktoru X – nesvědčila pro to rodinná anamnéza a osobní anamnéza (do roku 2002bez krvácivých komplikací včetně obou císařských řezů)
- toxonutritivní postižení jater – nepodpořeno anamnestickými údaji
- specifický inhibitor proti faktoru X – vyšetřen, ale nebyl prokázán
- AL‑amyloidóza
Proto na požádání hematologa bylo provedeno dovyšetření histologického preparátu dělohy a doplněno barvení na amyloid.
Histologie resekované dělohy: stěna dělohy obvyklého uspořádání, některé cévy mají ztluštěnou medii a při cytochemickém barvení kongo-červení se jednoznačně barví cihlově červeně. Po obarvení thioflavinem se v některých cévách objevují fluoreskující ložiska, nikoliv však souvislá depozita. Imunohistochemické barvení prokázalo depozita tvořená lehkými k řetězci. Nález připouští interpretaci jako depozita AL‑amyloidu z k řetězců.
Laboratorní vyšetření z doby stanovení diagnózy AL‑amyloidóza: běžná biochemická vyšetření, urea, kreatinin, ionty v normě. Pouze aktivita jaterních enzymů byla mírně zvýšená, ALT 3,19 μkat/l, AST 1,07 μkat/l, GMT 7,76 μkat/l, ALP 0,68 μkat/l. Imunoglobuliny kvantitativně: IgG 18,3 g/l, IgM 0,68 g/l, IgA 1,02 g/l. Imunofixační elektroforéza prokázala v séru monoklonální imunoglobulin typu IgG κ a volné k řetězce. V moči byla nízká koncentrace bílkoviny 0,09 g/l a byly přítomny volné řetězce κ.
Histologie válečku kostní dřeně: Krevní koagulum s ojedinělými drobnými částečkami kostní dřeně s krvetvornými buňkami. Imunohistochemicky jsou prokazatelné nehojné plazmocyty, reagující pouze s protilátkou proti k lehkým řetězcům, exprese l řetězců nebyla prokázána na žádných plazmocytech.
Hodnocení myelogramu: V nátěru jsou patrné v krajích zmnožené plazmocelulární buňky, místy až 10 %, ve středu nátěru prakticky nejsou přítomny vůbec. Plazmatické buňky nejsou normální morfologie, mají charakter plamenných buněk. Celkové zastoupení plazmocytů v preparátu bylo 0,8 %.
Další vyšetření: Spirální CT břicha: hepatomegalie, játra zvětšená doleva ke slezině, pravý lalok zvětšený kaudálně, dosahuje pod úroveň aortální bifurkace. Slezina hraniční velikosti. Ultrazvuk střev: zesílení řas v oblasti jejuna může souviset s AL‑amyloidózou. ECHO srdce: bez patologického nálezu, ejekční frakce 63 %. Funkční plicní vyšetření také zcela v normě.
Stanovení diagnózy: Počet plazmatických buněk, které sice byly klonální, nepřevyšovaly 10 %, přítomnost kompletní molekuly monoklonálního imunoglobulinu IgG κ a volných řetězců κ, nepřítomnost osteolytických kostních defektů, nesnížené koncentrace polyklonálních imunoglobulinů typu IgM a IgA a histologický průkaz AL‑amyloidu nesplňoval kritéria mnohočetného myelomu dle Durieho a Salmona, takže diagnóza byla uzavřena jako primární systémová AL‑amyloidóza. Dle nových kritérií by však bylo možné diagnózu uzavřít jako symptomatický mnohočetný myelom, jak rozvedeme v diskuzi.
Terapie: Po 6 týdnech od operace byla 16. 6. 2002 zahájena chemoterapie VAD (vinkristin 0,5 mg v celkové dávce a adriamycin 9 mg/m2 i.v. infuze 1.–4. den, dexametazon 40 mg p.o. 1.–4., 10.–13. a 20.–23. den ve 28denních cyklech. Celkem bylo aplikováno 8 VAD cyklů, poslední byl podán v lednu roku 2003.
Koncentrace monoklonálního imunoglobulinu v séru již v průběhu léčby poklesla ze vstupní hodnoty 15,2 g/l na nekvantifikovatelnou hodnotu před aplikací 5. cyklu chemoterapie VAD a v prosinci roku 2002, tedy před podáním 7. cyklu, již byla imunofixace negativní. Bylo tedy dosaženo hematologické kompletní remise nemoci.
Vzhledem k tomu, že u pacientky odezněly postupně veškeré krvácivé projevy, jsme po 8 cyklech změnili původní rozhodnutí (pro riziko krvácení nepoužít vysokodávkovanou chemoterapii s autologní transplantací) a provedli jsme sběr periferních kmenových buněk s cílem provést vysokodávkovanou chemoterapii s autologní transplantací. Sběr periferních kmenových krvetvorných buněk byl proveden po stimulačním režimu cyklofosfamid 2,5 g/m2 a G‑CSF (Neupogen) v dávce 5 μg/kg s výbornou výtěžností 22,73 × 106/l CD34+ buněk/kg.
Následně byla provedena v březnu a červenci roku 2003 tandemová vysokodávkovaná chemoterapie (melfalan 100 mg/m2) s transplantací kmenových krvetvorných buněk, získaných z periferní krve. Vysokodávkovanou chemoterapii snesla nemocná bez komplikací, bez mukozitidy. Od té doby je pacientka na udržovací léčbě interferonem a. Pro patologickou únavu – fatigue v posledních dvou letech snížena dávka interferonu a z původní dávky 3krát 3 miliony jednotek na 2krát týdně 3 miliony jednotek. V této dávce interferon snáší velmi dobře.
V průběhu sedmi let od transplantace je pacientka v kompletní hematologické remisi bez známek orgánového poškození a bez průkazu tvorby monoklonálního imunoglobulinu, s negativními výsledky imunofixační elektroforézy. Teprve od roku 2008 máme u pacientky stanovené hodnoty volných lehkých řetězců l a k, a můžete tedy jejich hladiny používat k monitorování aktivity nemoci. Vývoj monoklonálního imunoglobulinu při léčbě a aktivitu faktoru X uvádí tab. 1.

Diskuze
Primární systémová AL‑amyloidóza nebo sekundární amyloidóza při mnohočetném myelomu? Nomenklaturní zařazení má terapeutické důsledky!
Klinické projevy AL‑amyloidózy odvisí od orgánového tropizmu amyloidogenních lehkých řetězů, a tedy orgánů a tkání, v nichž postupně narůstají amyloidová depozita, poškozují je a způsobují jejich selhávání. Depozita AL‑amyloidu často poškozují srdce, ledviny játra, střevo, nervový systém a další tkáně [1].
Jak je v úvodu uvedeno, AL‑amyloidóza může být jak sekundární při mnohočetném myelomu, tak primární v případech, kdy nejsou splněna kritéria mnohočetného myelomu. V našem případě jsme se drželi v roce 2002 stále používaných kritérií dle Durieho a Salomona (tab. 2). O rok později byla přijata nová kritéria, která uvádíme v tab. 3. V nich není pro symptomatický mnohočetný myelom početní (10 %) hranice plazmocytů v kostní dřeni, ale je zde nově zaveden průkaz klonality plazmocytů v kostní dřeni, a pokud je v kostní dřeni prokázána klonální proliferace plazmocytů a je přítomno orgánové poškození, mezi něž patří i amyloidóza, lze nemoc klasifikovat jako mnohočetný myelom. Dle nových kritérií by tedy tato pacientka již splnila kritéria symptomatického mnohočetného myelomu.


Je to formalita, ale dnes důležitá formalita, protože nové účinné léky jsou standardně používány pro léčbu mnohočetného myelomu, zatímco pro léčbu nemocných s primární AL‑amyloidózou nejsou v ČR hrazené, ač mezi primární systémovou AL‑amyloidózou a sekundární AL‑amyloidózou při myelomu je kontinuální přechod. Současné publikace popisují vysokou efektivitu nových léků nejen u myelomu, ale také u primární systémové AL‑amyloidózy. Chceme proto upozornit, že histologické hodnocení kostní dřeně s průkazem klonality plazmocytů může posunout diagnózu z primární systémové AL‑amyloidózy do sekundární AL‑amyloidózy při mnohočetném myelomu a umožnit těmto pacientům přístup k bortezomibu, thalidomidu a lenalidomidu. Investovat peníze do registrace těchto léků pro velmi vzácně se vyskytující choroby, mezi něž jistě patří i primární systémová AL‑amyloidóza, považují obvykle farmaceutické firmy ze svého ekonomického úhlu pohledu za nepřínosné a neinvestují peníze do registračních studií pro vzácné diagnózy.
Příčiny snížení aktivity a antigenní koncentrace faktoru X
Relativně častým projevem primární i sekundární AL‑amyloidózy bývá narušená hemostáza. Nejčastěji jsou krvácivé projevy způsobené zvýšenou fragilitou kapilár, typické jsou hematomy v oblasti víček, ale také difuzní ekchymózy (obr. 1–3).
Prodloužení alespoň jednoho koagulačního parametru se popisuje jako časté, Mumfort uvádí, že 51 % vyšetřených pacientů s AL‑amyloidózou mělo prodlouženo alespoň jeden ze tří základních parametrů (PT, aPTTT a TT).
První popis snížené hodnoty aktivity faktoru X je z roku 1962 [45]. Při pozdějším zjišťování kinetiky faktoru X bylo popsáno značné zkrácení poločasu tohoto faktoru a zvýšené vychytávání v játrech a ve slezině, kde se vázal na amyloidová depozita [41,42]. Citované pozorování spolu s popisy ojedinělých případů, kdy po odstranění sleziny s velkým depozitem amyloidu se zlepšily parametry hemostázy [43,46,47], vedlo k vytvoření hypotézy, že defekt faktoru X je u AL‑amyloidózy způsoben sekvestrací tohoto koagulačního faktoru na amyloidových masách [19].
Frekvence tohoto jevu byla předmětem analýzy dvou studií. Greipp (1981) popsal tento defekt u 7 % pacientů s AL ‑ amyloidózou [31] a zatím největší studie Mumford z roku 2000 jej popisuje u 14 % vyšetřených nemocných [48]. Dominujícím zdrojem informací o této poruše u AL‑amyloidózy jsou popisy jednotlivých případů [47], protože výskyt AL‑amyloidózy je nízký.
Mechanizmus, jak dochází k deficitu aktivity faktoru X u AL‑amyloidózy, bude zřejmě složitější než původní představa o absorpci faktoru X na amyloid [19]. Mumfordova skupina prokázala, že u pacientů s krvácivým stavem na podkladě nedostatečné aktivity faktoru X není pokles aktivity faktoru X úměrný poklesu antigenní koncentrace faktoru X. Také míra splenického postižení nesouvisela s aktivitou faktoru X.
Autoři, kteří hodnotili deficit faktoru X u AL‑amyloidózy, používali jeho funkční průkaz.
Pouze v práci z roku 1985 a následně z roku 2000 [48] byla analyzována jak aktivita, tak i antigenní kvantita a obě práce konstatují, že pokles aktivity není provázen úměrným poklesem koncentrace antigenu faktoru X. Uvedené autorské kolektivy uzavírají: funkční deficit faktoru X u AL‑amyloidózy souvisí:
- zčásti s vazbou faktoru X na amyloidová depozita
- a z části je důsledkem poškození funkce faktoru X.
Přítomnost inhibitoru neprokázali, a tak se domnívají, že snížená aktivita je způsobena buď defektem potranslační modifikace (glykosylace) [49], nebo inaktivace aktivní části faktoru X navázáním ligandu, kterým může být monoklonální imunoglobulin nebo jeho fragment [48]. Starší práce připouštějí i existenci inhibitor faktoru X [50] a popisují interferenci monoklonálního imunoglobulinu či jeho fragmentů s polymerizací fibrinu [51].
Jak z námi popsaného případu vyplývá, neměla pacientka před operačním zákrokem klinické známky AL‑amyloidózy, depozita amyloidu v těle byla ještě malá na to, aby interferovala s funkcí orgánů a tkání, a proto nebyla tato diagnóza rozpoznána v rámci interního předoperačního vyšetření, v rámci něhož není standardně prováděna ani imunofixační elektroforéza, ani stanovení kvantity lehkých řetězců. Nicméně do té doby nepoznaná AL‑amyloidóza způsobila závažný deficit faktoru X, který způsobil život ohrožující krvácení. Klinicky se tato zatím latentní porucha objevila až při větších nárocích na koagulační systém při operačním zákroku.
Léčba AL amyloidózy se sníženou aktivitou faktoru X, způsobující závažné krvácivé projevy
Kauzální léčba deficitu faktoru X vyžaduje odstranění produkce monoklonálního imunoglobulinu a depozit amyloidu [52]. Infuze faktoru X měly jen velmi krátkou účinnost (krátký poločas) [53,54]. U několika pacientů s AL‑amyloidózou a extrémně velkou slezinou s depozity amyloidu vedlo její odstranění k eliminaci absorpčního mechanizmu deficitu faktoru X a ke zlepšení koagulace [43,46]. Léčebný úspěch splenektomie však byl popsán pouze v několika případech. Jsou popsány případy, kdy opakovaná plazmaferéza vedla k úpravě aktivity faktoru X [44,55,56].
Kauzální možností léčby deficitu faktoru X je potlačit produkci monoklonálních lehkých řetězců odpovídající léčbou [1] neboli zničit patologický plazmocelulární klon, produkující toxické volné lehké řetězce.
Obecně prvním cílem při léčbě AL‑amyloidózy je dosažení kompletní hematologické remise – zastavení produkce klonálních lehkých řetězců (dosažení negativní imunofixace) a normalizace koncentrace volných lehkých řetězců. Zásadní snížení koncentrace či nepřítomnost těchto lehkých řetězců pak umožní reparačním pochodům v orgánech poškozených depozity amyloidu upravit jejich funkci, neboli dosáhnut orgánové léčebné odpovědi [57]. Orgánová léčebná odpověď však nastupuje s několikaměsíčním opožděním za hematologickou léčebnou odpovědí, pokud se reparačním mechanismům podaří obnovit funkci amyloidem poškozeného orgánu.
Pomalý nástup orgánové léčebné odpovědi – postupné zvyšování aktivity faktoru X – je zřetelný z tab. 1.
Pro léčbu AL amyloidózy se používají podobné režimy jako pro léčbu mnohočetného myelomu.
V případě AL‑amyloidózy s deficitem faktoru X byly použity jak klasické léčebné postupy a v některých případech bylo dosaženo léčebné odpovědi [58–61], tak i vysokodávkovaná chemoterapie s následnou normalizací aktivity faktoru X [62,63].

Přínos vysokodávkované chemoterapie pro nemocné s AL‑amyloidózou
Vysokodávkovaná chemoterapie s následující autologní transplantací krvetvorné tkáně je velmi účinnou volbu pro vhodné nemocné s touto nemocí. Nízký výskyt AL‑amyloidózy limituje tvorbu randomizovaných studií, ale srovnání typu case control study prokázalo lepší výsledky vysokodávkované chemoterapie ve srovnání s konvenční chemoterapií [64].
Prospektivně randomizovaně tento problém analyzovala francouzská studie [65], která prokázala profit vysokodávkované chemoterapie pouze pro pacienty s nevelkým orgánovým poškozením. Kritici této francouzské studie uvádějí, že měla nezvykle vysokou mortalitu ve skupině s vysokodávkovanou chemoterapií – 24 %, způsobenou zařazením pacientů se značným orgánovým poškozením do transplantačního programu. Domníváme, že i tato studie prokazuje, že vysokodávkovaná chemoterapie s autologní transplantací je optimálním režimem pro nemocné s nevelkým orgánovým poškozením.
Důležitost dosažení kompletní remise pro prognózu nemocných zdůrazňuje analýza 80 pacientů, kteří podstoupili vysokodávkovanou chemoterapii s autologní transplantací před 10 lety. Sice 22 % léčených umřelo v průběhu 1. roku (14 % důsledkem toxicity léčby a 8 % důsledkem progrese). Z 63 pacientů, kteří přežili 1. rok po transplantaci, se 51 dostalo do kompletní remise nemoci. Pro celou skupinu 80 pacientů byl medián přežití 4,75 roku. Medián přežití pacientů, kteří se dostali do kompletní remise, přesahoval 10 roků, zatímco medián přežití těch, kteří se nedostali do kompletní remise, byl pouze 50 měsíců. Tato studie prokázala, že dosažení kompletní remise navodí u AL‑amyloidózy dlouhodobé přežití [66,67].
Rizika vysokodávkované chemoterapie u pacientů s AL‑amyloidózou
Problémem vysokodávkované chemoterapie u AL‑amyloidózy je kumulace toxicit, a proto je spojena s mnohem vyšším rizikem úmrtí než u mnohočetného myelomu. Již sběr periferních kmenových buněk byl zcela ojediněle komplikován nekardiálním plicním edémem s hypoxií. U tohoto pacienta byla při sekci rozpoznána depozita AL‑amyloidu v plicích [68]. Také ruptura sleziny v průběhu aplikace leukocytárních růstových faktorů byla popsána [69].
V případě postižení zažívacího traktu depozity amyloidu může následná mukozitida po chemoterapii způsobit značné krvácení a ohrozit život (toxické megakolon) [69–71].
Další závažnou komplikací vysokodávkované chemoterapie je zhoršení funkce ledvin. Vzestup kreatininu nejméně o 44 μmol/l (0,5 mg/dl) byl v jedné studii zaznamenán u 19 % [72,73]. V další studii byl vzestup kreatininu nejméně o 88 μmol/l v potransplantačním období (melfalan 100–200 mg/m2) zjištěn dokonce u 23 % nemocných [74].
Vysokodávkovaná chemoterapie má u pacientů s mnohočetným myelomem nízkou mortalitu (2–3 %), pokud se ale stejná léčba použila u nemocných s AL‑amyloidózou, je mortalita podstatně vyšší. Analýza výsledků ze 48 transplantačních center (107 pacientů prokázala) transplantační mortalitu 18 % do 30. dne od provedení vysokodávkované chemoterapie s autologní transplantací, při mediánu přežití čtyři roky. Výsledná časná transplantační mortalita (18 %) je vyšší, než je obvykle publikováno v analýzách větších center, která uvádějí 10–15% mortalitu spojenou s vysokodávkovanou chemoterapií s autologní transplantací [75]. Vyšší peritransplantační mortalita je způsobená součtem poškození organizmu depozity amyloidu a toxického vlivu vysokodávkované chemoterapie.
Kteří pacienti s AL‑amyloidózou jsou vhodní pro vysokodávkovanou chemoterapii?
Vysokodávkovaná chemoterapie s au-tologní transplantací se doporučuje pro osoby s amyloidovým postižením jednoho nebo maximálně dvou orgánů. Nesmí ale předcházet krvácení do zažívacího traktu způsobené depozity amyloidu [76], nesmí být přítomno klinicky závažné postižení srdce a ledvin [77,78].
Bostonské centrum např. podmínky pro podání vysokodávkované chemoterapie uvádí následovně: pro pacienty mladší 65 let s ejekční frakcí levé komory nad 45 % použili dávku 200 mg/m2. Starším pacientům nad 65 let nebo se sníženou ejekční frakcí na 40–45 % podávali 140 mg/m2. Aplikaci melfalanu rozdělili vždy do dvou dávek a aplikovali je –2 a –1 den před transplantací. U takto vybrané skupiny nemocných měli v kategorii do 65 let 10,4% mortalitu a kategorii nad 65 let 12,3%. Počet kompletních remisí byl u mladší skupiny 43 % a u starší 32 % a medián přežití byl čtyři roky [75]. Kritéria pro volbu dávky Italské myelomové skupiny uvádí tab. 5 [79].

Úvahy o optimální léčbě naší nemocné a začlenění vysokodávkované chemoterapie do léčebného plánu
U naší pacientky se jednalo o relativně časně diagnostikové onemocnění, které se projevilo až při chirurgickém zákroku krvácivým stavem [80–83]. V rámci interního předoperačního vyšetření nebylo vysloveno žádné podezření na koagulopatii či na AL‑amyloidózu.
Zobrazovací vyšetření, která byla provedena před zahájením chemoterapie, připouštěla depozita amyloidu v játrech a ve slezině a také ve sliznici duodena. Invazivní ověřování rozsahu nemoci a prokazování amyloidu v dalších orgánech nepřicházelo pro riziko krvácení v úvahu. Proto riziko krvácení při mukozitidě jsme v době zahájení chemoterapie neuvažovali o vysokodávkované chemoterapii a naplánovali jsme 8 cyklů chemoterapie obsahující vinkristin, adriamycin a dexametazon. Tato léčba vedla ke kompletní remisi – vymizení monoklonálního imunoglobulinu a negativní imunofixaci a vymizení krvácivých projevů. Při úspěchu klasické chemoterapie a postupném zvyšování aktivity faktoru X jsme nakonec přehodnotili původní léčebný plán a doplnili jej v odstupu 9 měsíců od zahájení chemoterapie o tandemovou vysokodávkovanou chemoterapii s redukovanou dávkou melfalanu (melfalan 100 mg/m2). Cílem bylo prohloubit léčebnou odpověď. Tuto dávku melfalanu pacientka tolerovala velmi dobře, bez známek mukozitidy. Důvodem pro nepodání klasické dávky 200 mg/m2 byly obavy z obnovení neztišitelného krvácení při mukozitidě, kterou by vyšší dávka mohla vyvolat.
Normalizace aktivity faktoru X při chemoterapii bylo již dříve popisováno a v literatuře jsme našli dvě publikace popisující přínos vysokodávkované chemoterapie pro tyto nemocné [62,63].
Léčba byla ukončena v roce 2003 a od té doby má pacientka udržovací léčbu interferonem a. Nyní je již 7 let od stanovení diagnózy v kompletní remisi nemoci, bez známek krvácení a známek AL‑amyloidózy. Monitorování choroby jsme v posledních dvou letech doplnili o vyšetřování koncentrace volných lehkých řetězců. Toto vyšetření nebylo v době stanovení diagnózy dostupné [84].
Závěr
Jedním z projevů AL‑amyloidózy je krvácivý stav na podkladě snížení aktivity i antigenní koncentrace faktoru X. K uvedené poruše dochází jednak vychytáváním faktoru X na amyloidové hmoty a jednak snížením aktivity cirkulujícího faktoru X zatím neznámým mechanizmem. Vznikne‑li krvácení z nejasné příčiny, je vždy vhodné zapátrat, zda není přítomen AL‑amyloid.
Za nejúčinnější léčebný postup u AL‑amyloidózy je stále považována vysokodávkovaná chemoterapie s autologní transplantací. Je sice spojena s vyšší peritransplantační mortalitou, ale pokud se podaří dosáhnout kompletní remise nemoci, přináší pacientům dlouhodobé přežití, v našem případě je tato pacientka již 7 let od stanovení diagnózy a 6 let od ukončení léčby v kompletní remisi.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
Doručeno do redakce: 25. 5. 2009
Přijato po recenzi: 30. 6. 2009
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e‑mail: z.adam@fnbrno.cz
Sources
1. Ščudla V, Pika T. Současné možnosti diagnostiky a léčby systémové AL‑amyloidózy. Vnitř Lék 2009; 55 (Suppl 1): S77–S87.
2. Tichý M. Primární amyloidóza. Lék Zpr. LF UK Hradec Králové 1999; 44 : 5–6.
3. Abraham RS, Geyer SM, Price-Troska TL et al. Immunoglobulin light chain variable (V) region influence clinical presentation and outcome in light chain associated amyloidosis(AL). Blood 2003; 101 : 3801–3808.
4. Kroupa R, Dastych M, Šenkyřík M et al. Systémové amyloidóza s dominující klinickou manifestací v trávicím traktu. Vnitř Lék 2005; 51 : 588–592.
5. Linhartová K, Daum O. Srdeční amyloidóza. Cor Vasa 2005; 47 : 328.
6. Ryšavá R. Amyloidóza ledvin. Postgrad Med 2006; 8 : 207–212.
7. Ryšavá R. Léčba paraproteinemické nefropatie a primární amyloidózy ledvin. Aktual v Nefrol 2005; 11 : 62–65.
8. Butler WM, Baldwin PE. Prolongation of thrombin and reptilase times in patients with amyloidosis and acquired factor X deficiency. South Med J 1984; 77 : 648–651.
9. Dray X, Treton X, Joly F et al. Hemorrhagic bullous colitis as a primary manifestation of AL amyloidosis. Endoscopy 2006; 38 (Suppl 2): E15–E16.
10. James DG, Zuckerman GR, Sayuk GS et al. Clinical recognition of Al type amyloidosis of the luminal gastrointestinal tract. Clin Gastroenterol Hepatol 2007; 5 : 582–588.
11. Stammler F. Haemorrhagic diathesis as an early symptom of systemic amyloidosis. Dtsch Med Wochenschr 2006; 131 : 17–21.
12. Sucker C, Hetzel GR, Grabensee B et al. Amyloidosis and bleeding: pathophysiology, diagnosis, and therapy. Am J Kidney Dis 2006; 47 : 947–955.
13. Takahashi T, Suzukawa M, Akiyama M et al. Systemic AL amyloidosis with disseminated intravascular coagulation associated with hyperfibrinolysis. Int J Hematol 2008; 87 : 371–374.
14. Yamamoto T, Maeda N, Kawasaki H. Hepatic failure in a case of multiple myeloma‑associated amyloidosis (kappa-AL). J Gastroenterol 1995; 30 : 393–397.
15. Higgins GT, Olujohungbe A, Kyle G. Recurrent subconjunctival and periorbital haemorrhage as the first presentation of systemic AL amyloidosis secondary to myeloma. Eye 2006; 20 : 512–515.
16. Hoshino Y, Hatake K, Muroi K et al. Bleeding tendency caused by the deposit of amyloid substance in the perivascular region. Intern Med 1993; 32 : 879–881.
17. Korsan–Bengtsen K, Hjort PF, Ygge J. Acquired factor X deficiency in a patient with amyloidosis. Tromb Diath Haemorrh 1962; 7 : 558–566.
18. Galbraith PA, Sharma N, Parker WL et al. Acquired factor X deficiency. Altered plasma antithrombin activity and association with amyloidosis. JAMA 1974; 230 : 1658–1660.
19. Glenner GG. Factor X deficiency and systemic amyloidosis. N Engl J Med 1977; 297 : 108–109.
20. Fogdall RP, Fischbach DP. Factor X deficiency in primary amyloidosis. N Engl J Med 1980; 302 : 923.
21. Girmann G, Wilker D, Stadie H et al. Acquired isolated factor X deficiency associated with systemic amyloidosis. Case report and review of literature. Klin Wochenschr 1980; 58 : 859–862.
22. Gloy J, Böhler J, Schollmeyer P et al. Primary amyloidosis with severe nephrotic syndrome and acquired factor X deficiency. Nephrol Dial Transplant 1997; 12 : 588–590.
23. Van Dam FE, de Pauw BE, Schutjes CP et al. Isolated factor X deficiency in amyloidosis. Case report and review of the literature. Neth J Med 1975; 18 : 129–134.
24. Wick A, Sulser H, Pflugshaupt R. Acquired isolated factor X deficiency in generalized amyloidosis. Schweiz Med Wochenschr 1972; 102 : 1799–1803.
25. Krause JR. Acquired factor X deficiency and amyloidosis. Am J Clin Pathol 1977; 67 : 170–173.
26. Lucas FV, Fishleder AJ, Becker RC et al. Acquired factor X deficiency in systemic amyloidosis. Cleve Clin J Med 1987; 54 : 399–406.
27. Rapoport M, Yona R, Kaufman S et al. Unusual bleeding manifestations of amyloidosis in patients with multiple myeloma. Clin Lab Haematol 1994; 16 : 349–353.
28. Yood RA, Skinner M, Rubinow A et al. Bleeding manifestations in 100 patients with amyloidosis. JAMA 1983; 249 : 1322–1324.
29. Zeitler KD, Blatt PM. Amyloidosis and factor X deficiency. South Med J 1982; 75 : 306–308.
30. McPherson RA, Onstad JW, Ugoretz RJ et al. Coagulopathy in amyloidosis: combined deficiency of factors IX and X. Am J Hematol 1977; 3 : 225–235.
31. Greipp PR, Kyle RA, Bowie EJ. Factor-X deficiency in amyloidosis: a critical review. Am J Hematol 1981; 11 : 443–450.
32. Gatel A, Cacoub P, Piette JC. AL amyloidosis combined with acquired factor V deficiency. Ann Intern Med 1998; 128 : 604–605.
33. Emori Y, Sakugawa M, Niiya K et al. Life–threatening bleeding and acquired factor V deficiency associated with primary systemic amyloidosis. Blood Coagul Fibrinolysis 2002; 13 : 555–559.
34. Wiest R, Klouche M, Härle P et al. Acquired combined factor X and XII deficiency with isolated bleeding complications in a patient with AL amyloidosis. Ann Hematol 2005; 84 : 196–199.
35. Kos CA, Ward JE, Malek K et al. Association of acquired von Willebrand syndrome with AL amyloidosis. Am J Hematol 2007; 82 : 363–367.
36. Quitt M, Aghai E, David M et al. Acquired factor X and antithrombin III deficiency in a patient with primary amyloidosis and nephrotic syndrome. Scand J Haematol 1985; 35 : 155–157.
37. Perlin E, Brankman P, Berg HS et al. Enhanced blood coagulation and fibrinolysis in a patient with primary amylidos. Tromb Diath Haemorrh 1971; 26 : 9–14.
38. Liebman H, Chinowsky M, Valdin J et al. Increased fibrinolysis and amyloidosis. Arch Intern Med 1983 : 143 : 678–682.
39. Gastineau DA, Gertz MA, Daniels TM et al. Inhibitor of thrombin in the systemic amyloidosis. A common coagulation abnormality. Blood 1991; 77 : 2637–2640.
40. Gamba G, Montani N, Anesi E et al. Abnormalities in thrombin‑antithrombin pathway in AL amyloidosis. Amyloid 1999; 6 : 273–277.
41. Furie B, Greene E, Furie BC. Syndrome of acquired factor X deficiency and systemic amyloidosis in vivo studies of the metabolic fate of factor X. N Engl J Med 1977; 297 : 81–85.
42. Furie B, Voo L, McAdam KP et al. Mechanism of factor X deficiency in systemic amyloidosis. N Engl J Med 1981; 304 : 827–830.
43. Greipp PR, Kyle RA, Bowie EJ. Factor X deficiency in primary amyloidosis: resolution after splenectomy. N Engl J Med 1979; 301 : 1050–1051.
44. Beardell FV, Varma M, Martinez J. Normalization of plasma factor X levels in amyloidosis after plasma exchange. Am J Hematol 1997; 54 : 68–71.
45. Ménmaché D, Boivin P. Deficit acquis en facteur X chez un malade atteint d’amyloisoe primitive. Nouv Rev Fr Hematol 1962; 71 : 764–667.
46. Rosenstein ED, Itzkowitz SH, Penziner AS et al. Resolution of factor X deficiency in primary amyloidosis following splenectomy. Arch Intern Med 1983; 143 : 597–599.
47. Bohler A, Lämmle B. Decreased Quick percentage, acquired factor X deficiency, hemarthrosis and ecchymosis: amyloidosis. Ther Umsch 1999; 56 : 523–525.
48. Mumford AD, O‘Donnell J, Gillmore JD et al. Bleeding symptoms and coagulation abnormalities in 337 patients with AL‑amyloidosis. Br J Haematol 2000; 110 : 454–460.
49. Sinha U, Wolf DL. Carbohydrate residues modulate the activation of coagulation factor X. J Biol Chem 1993; 268 : 3048–3051.
50. Mulhare PE, Tracy PB, Golden EA et al. A case of acquired factor X deficiency with in vivo and in vitro evidence of inhibitor activity directed against factor X. Am J Clin Pathol 1991; 96 : 196–200.
51. Wissløff F, Michaelsen TE, Godal HC. Inhibition or acceleration of fibrin polymerisation by monoclonal immunoglobulin fragments. Tromb Res 1984; 35 : 81–90.
52. le Quellec A, Sotto A, Ciurana AJ. Spontaneous resolution of acquired factor X deficiency in amyloidosis. J Intern Med 1993; 234 : 329–330.
53. Duncan EM, Cole J, Clarkson AR et al. Poor recovery and short survival of infused factor X in a case of acquired factor X deficiency and amyloidosis. Thromb Haemost 1999; 82 : 1375–1376.
54. Takabe K, Holman PR, Herbst KD et al. Successful perioperative management of factor X deficiency associated with primary amyloidosis. J Gastrointest Surg 2004; 8 : 358–362.
55. Smith SV, Liles DK, White GC 2nd et al. Successful treatment of transient acquired factor X deficiency by plasmapheresis with concomitant intravenous immunoglobulin and steroid therapy. Am J Hematol 1998; 57 : 245–252.
56. Spero JA, Lewis JH, Hasiba U et al. Treatment of amyloidosis associated factor X deficiency. Thromb Haemost 1976; 35 : 377–381.
57. Gertz MA, Comenzo R, Falk RA. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis. A consensus opinion from the 10th international symposium on amyloid and amyloidosis. Am J Hematol 2005; 79 : 319–328.
58. Camoriano JK, Greipp PR, Bayer GK et al. Resolution of acquired factor X deficiency and amyloidosis with melphalan and prednisone therapy. N Engl J Med 1987; 316 : 1133–1135.
59. Levin M, Chokas W. Acquired factor X deficiency and amyloidosis treated with melphalan and prednisone. N Engl J Med 1987; 317 : 1155–1156.
60. Mohri H. Resolution of acquired factor X deficiency with amyloidosis secondary to plasma cell dyscrasia. Am J Hematol 2004; 77 : 421–422.
61. Schwarzinger I, Stain‑Kos M, Bettelheim P et al. Recurrent, isolated factor X deficiency in myeloma: repeated normalization of factor X levels after cytostatic chemotherapy followed by late treatment failure associated with the development of systemic amyloidosis. Thromb Haemost 1992; 68 : 648–651.
62. Breems DA, Sonneveld P, de Man RA et al. Successful treatment of systemic amyloidosis with hepatic involvement and factor X deficiency by high dose melphalan chemotherapy and autologous stem cell reinfusion. Eur J Haematol 2004; 72 : 181–185.
63. Choufani EB, Sanchorawala V, Ernst T et al. Acquired factor X deficiency in patients with amyloid light‑chain amyloidosis: incidence, bleeding manifestations, and response to high‑dose chemotherapy. Blood 2001; 97 : 1885–1887.
64. Dispenzieri A, Kyle RA, Lacy MQ et al. Superior survival in primary systemic amyloidosis patients udergoing peripheral blood stem cell transplantation. A case control study. Blood 2004; 103 : 3960–3963.
65. Jaccard A, Moreau P, Leblond V et al. High dose melphalan versus melphalan plus dexamethasone for AL‑amyloidosis. N Engl J Med 2007; 357 : 1083–1093.
66. Sanchorawala V, Seldin DC, Magnani B et al. Serum free light‑chain responses after high‑dose intravenous melphalan and autologous stem cell transplantation for AL (primary) amyloidosis. Bone Marrow Transplant 2005; 36 : 597–600.
67. Sanchorawala V, Skinner M, Quillen K et al. Long‑term outcome of patients with AL amyloidosis treated with melphalan and stem cell transplantation. Blood 2007; 110 : 3561–3563.
68. Gertz MA, Lacy MQ, Bjornsson J et al. Fatal pulmonary toxicity related to the administration of granulocyte colony stimulation factor in amyloidosis. A report and review of growth factor pulmonary toxicity. J Hematother Stem Cell Res 2000; 9 : 635–643.
69. Hayes–Lattin BM, Curtin PT, Fleming WH et al. Toxic megacolon: a life threatening complication of high dose therapy and autologous transplantation among patients with AL‑amyloidosis. Bone Marrow Transplant 2002; 30 : 279–285.
70. Kumar S, Dispenzieri A, Lacy MQ et al. High incidence of gastrointestinal tract bleeding after autologous stem cell transplant for primary systemic amyloidosis. Bone Marrow Transplant 2001; 28 : 381–385.
71. Kyle RA, Gertz MA, Lacy MQ et al. Localized AL amyloidosis of the colon: an unrecognized entity. Amyloid 2003; 10 : 36–41.
72. Gertz MA, Lacy MQ, Dispenzieri A et al. Amyloidosis. Best Pract Res Clin Haematol 2005; 18 : 709–727.
73. Leung N, Slezak JM, Bergstralh EJ. Acute renal insufficiency after high dose melphalan in patients with primary systemic amyloidosis during stem cell transplantation. Am J Kidney Dis 2005; 45 : 102–111.
74. Fadia A, Casserly LF, Sanchorawala V et al. Incidence and outcome of acute renal failure complicating autologous stem cell transplantation for AL‑amyloidosis. Kidney Int 2003; 63 : 1868–1873.
75. Vesole DH, Akasheh M, Peréz WS et al. High dose therapy and autologous hematopoietic stem cell transplantation for patients with primary systemic amyloidosis. A center for International blood and marrow transplant research study. Mayo Clin Proc 2006; 81 : 880–888.
76. Spier BJ, Einstein M, Johnson EA et al. Amyloidosis presenting as lower gastrointestinal hemorrhage. WMJ 2008; 107 : 40–43.
77. Röcken C, Ernst J, Hund E et al. Interdisciplinary guidelines on the diagnosis and therapy of extracerebral amyloidosis: issued by the German Society of Amyloid Disease e.V. (www.amyloid.de). Med Klin (Munich) 2006; 101 : 825–829.
78. United kingdom myeloma forum: Guidelines on the diagnosis and management of AL‑amyloidosis. Brit J Hematology 2004; 125 : 681–700.
79. Perfetti V, Siena S, Palladini G et al. Long term results of a risk adapted approach to melphalan conditioning in autologous peripheral blood stem cell transplantation for primary AL amyloidosis. Haematologica 2006; 91 : 1635–1643.
80. Böhrer H, Waldherr R, Martin E et al. Splenectomy in an uraemic patient with acquired factor X deficiency due to AL amyloidosis. Nephrol Dial Transplant 1998; 13 : 190–193.
81. Enjeti AK, Walsh M, Seldon M. Spontaneous major bleeding in acquired factor X deficiency secondary to AL‑amyloidosis. Haemophilia 2005; 11 : 535–538.
82. Adam Z, Ščudla V, Tomíška M. Léčba AL‑amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 46–52.
83. Adam Z, Ščudla V. Klinické projevy a diagnostika AL‑amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 36–45.
84. Ščudla V, Minařík J, Schneiderka P et al. Význam sérových hladin volných lehkých řetězců imunoglobulinu v diagnostice a hodnocení aktivity mnohočetného myelomu a vybraných monoklonálních gamapatií. Vnitř Lék 2005; 51 : 1249–1259.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 1
Most read in this issue
- Nemocní starší 80 let s de novo akutními myeloidními leukemiemi bez dysplazie v erytroblastické a/nebo megakaryocytární řadě dosahují kompletní remise a delšího přežití po klasické chemoterapii 3+7
- Paraneoplastický tromboembolický syndróm ako prvý príznak zhubného ochorenia
- Dysfunkce pravé komory po implantaci levostranné mechanické srdeční podpory
- Epidemiológia cievnych chorôb