
Prekancerózní stavy v gastroenterologii a jejich molekulární genetika z pohledu klinické praxe
Clinical perspective of precancerotic states in gastroenterology and their molecular genetics
Gastrointestinal tract tumours represent an important cause of death in the Czech Republic. Diagnosis of the disease at its advanced stage with progression precluding radical surgical solution is a frequent common denominator. Detection of precancerous states, including congenital genetic defects or premalignant syndromes and malignant lesions may serve as a useful tool for early detection of disease risk or disease onset. Considering the incidence of individual types of tumours, this paper focuses mainly on precancerotic states leading to malignant alterations of oesophagus, colorectum and pancreas. Apart from a concise morphological description of each premalignancy and its malignant transformation, a description of traditional molecular models of development and progression is also provided. Furthermore, we include a brief list of the most important genes contributing to the mechanism of malignant transformation in these three most important tumour diseases.
Key words:
gastrointestinal tract – tumour diseases – genetics – oesophagus – colon – rectum – pancreas – DNA – mutation
Authors:
P. Mináriková 1,2; M. Zavoral 1,2
Authors‘ workplace:
Interní klinika 1. lékařské fakulty UK a ÚVN Praha, přednosta prof. MUDr. Miroslav Zavoral, Ph. D.
1; Subkatedra gastroenterologie IPVZ Praha, přednosta prof. MUDr. Miroslav Zavoral, Ph. D.
2
Published in:
Vnitř Lék 2010; 56(1): 44-48
Category:
Reviews
Overview
Nádory gastrointestinálního traktu zapříčiňují významnou část úmrtí u nás. Častým jmenovatelem je diagnóza v pokročilé fázi s progresí vylučující radikální chirurgické řešení. Detekce prekancerózních stavů, zahrnující vrozené genetické postižení či premaligní syndromy a premaligní léze, je vhodným nástrojem pro časné rozpoznání rizika, či přímo nastupujícího onemocnění. Vzhledem k zastoupení jednotlivých typů nádorů je tato práce věnována především prekancerózám vedoucím k maligním postižením jícnu, kolorekta a slinivky břišní. Kromě stručného morfologického popisu dané premalignity a její maligní transformace je vždy připojen popis klasických molekulárních modelů vzniku a progrese. Součástí je i stručný výčet nejvýznamnějších genů podílejících se na mechanizmech maligní transformace u těchto tří nejvýznamnějších nádorových postižení.
Klíčová slova:
gastrointestinální trakt – nádorová onemocnění – genetika – jícen – kolon – rektum – slinivka – DNA – mutace
Úvod
Prekancerózní stav je definován jako onemocnění, syndrom nebo klinické zjištění, které, pokud není léčeno, vede ke vzniku nádoru [1]. Z uvedené definice vyplývá, že snahou lékařů by mělo být zachytit tento stav co nejdříve, nejlépe v asymptomatickém, tj. bezpříznakovém stadiu, a zabránit tak vývoji v karcinom. Odhady odborníků jsou alarmující. V současné době v České republice ročně onemocní gastrointestinálními nádory přibližně 13 500 osob a téměř 8 800 jich zemře [2]. Při pokračování současného trendu může v příštích 20 letech dojít až k 3násobnému nárůstu incidence a mortality. To je jeden z hlavních důvodů, proč se výzkum zaměřuje na stále nové možnosti diagnostiky premaligních stavů a jejich časné léčby.
V gastroenterologii se setkáváme s prekancerózami velmi často, bohužel, nejsme však schopni v rámci dostupných screeningových či diagnostických postupů je odhalit vždy včas, abychom vývoji karcinomu skutečně zabránili. V posledních letech se pozornost proto soustřeďuje na nové možnosti časného záchytu, a to především pomocí metod molekulární biologie a genetiky. Výzkum genetické podstaty nádorů se zdá být klíčový u několika forem karcinomů. U nádorů gastrointestinálního traktu došlo k největším pokrokům na poli molekulárně‑biologickém v oblasti výzkumu karcinomu jícnu, tlustého střeva a pankreatu. Následující souhrn se proto zabývá právě těmito malignitami. Je zaměřen na základní principy, které se již staly součástí diagnostiky či jejichž využití lze předpokládat v blízké budoucnosti.
Důvodem vzniku karcinomu je nekontrolované buněčné dělení způsobené mutací genetické informace uložené v DNA. Vrozené formy mutací vedou ke vzniku hereditárních forem nádorů a ty představují pouze malou část nádorů (5 %) z celkového počtu nádorů. Ostatní, tzv. sporadické formy nádorů jsou způsobené somatickými mutacemi, které vznikají působením vnějších faktorů, jako jsou kouření, konzumace alkoholu, přepálených živočišných tuků, působení slunečního záření a některých chemických látek. Mutace nejčastěji postihují geny, které spouštějí, nebo naopak potlačují růst a dělení buněk.
Genetickou podstatu zhoubného bujení lze charakterizovat 3 hlavními skupinami genů:
- onkogeny – tzv. spouštěcí geny (např. geny KRAS, EGFR), u kterých dochází v důsledku mutace k permanentní aktivaci jejich funkce stimulující buněčný růst a dělení
- tumor supresorové geny (např. geny APC, DCC, TP53), u kterých dochází v důsledku mutace k vyřazení z funkce, což má za následek buněčné dělení
- geny zajišťující opravy DNA (např. geny MSH2, MLH1), jejichž ztráta funkce v důsledku genové mutace způsobí nekontrolované hromadění dalších mutací (chyb) v ostatních genech.
Schéma jednotlivých stadií vývoje zhoubného nádoru je uvedeno v grafu 1. Z normální epiteliální buňky se v důsledku nahromadění genetických poruch stává buňka maligní. Přítomnost spouštěcí mutace v genu A (např. gen pro kontrolu a opravy chyb DNA – MSH2) způsobí zvýšení četnosti náhodných mutací dalších genů. Následné mutace v dalších genech (gen B, např. onkogen KRAS) mají za následek zvýšení frekvence buněčného dělení mimo kontrolní mechanizmy. Během tohoto nekontrolovaného dělení dochází kromě mutací i k dalším typům poškození, např. ke ztrátám části genů (nukleotidové delece), ke ztrátě chromozomálních úseků obsahujících geny (alelické delece) nebo v neposlední řadě i ke ztrátě celých chromozomů (buněčná aneuploidie). Na morfologické úrovni dochází k dysplastickým změnám a progredující neoplazie vede nejprve ke vzniku maligního karcinomu in situ. Jeho další proliferace vede ke generalizaci onemocnění v důsledku lokální progrese onemocnění do okolních orgánů a zakládání vzdálených metastáz [3].

Časné rozpoznání morfologických změn provázejících nádorové bujení je klíčem pro časný záchyt premaligních či časně maligních stavů, a tedy naprosto zásadní pro další prognózu onemocnění, možnosti léčby a přežití [4].
Prekancerózy jícnu – sekvence Barrettův jícen – adenokarcinom jícnu
Barrettův jícen (BJ) je onemocnění definované jako metaplastická přeměna dlaždicového epitelu jícnu na specializovaný cylindrický epitel střevního typu, tzv. intestinální metaplazie [5]. Tento typ epitelu byl nazván specializovaná intestinální metaplazie (SIM). Již v 70. letech minulého století byla popsána výrazná souvislost BJ a adenokarcinomu jícnu [6]. Vzhledem k nárůstu adenokarcinomu jícnu především u pacientů s BJ na rozdíl od karcinomu spinocelulárního se začalo uvažovat o možných vazbách a souvislostech a BJ začal být považován za prekancerózu. Adenokarcinom jícnu se vyvine asi u 0,5 % nemocných s BJ za rok. Prognóza adenokarcinomu je špatná, 5leté přežití je pouze 11 %. Dalšími rizikovými faktory karcinomu jícnu jsou:
- refluxní choroba jícnu (gastroesophageal reflux disease – GERD) trvající déle než 5 let
- abdominální obezita
- rasa (časté onemocnění u kavkazské populace)
- pohlaví (postihuje častěji muže)
Vývoj adenokarcinomu jícnu na podkladě BJ lze charakterizovat jako třífázový proces, na jehož počátku dochází k tzv. iniciaci, kdy rozhodující úlohu hrají uvedené rizikové faktory. V další fázi dochází k tzv. formaci nového fenotypu a následuje závěrečná fáze, tzv. progrese, tj. vývoj genetických abnormalit, nekontrolovaná proliferace a vývoj časné a progredující neoplazie.
Dosavadní používané diagnostické metody jsou především metody endoskopické, kdy makroskopický nález BJ (obr. 1) je potvrzen histologickým vyšetřením.V posledních letech došlo k obrovskému pokroku ve zpřesnění endoskopické diagnostiky použitím metod, jako jsou endoskopie s vysokou rozlišovací schopností (HRE – high resolution endoscopy), kterou lze doplnit chromodiagnostikou za použití barviva indigokarmínu. Jinými metodami bez užití barviva je NBI (narrow band imaging) či autofluorescenční endoskopie (AFE). Všechny uvedené metody si kladou za cíl odhalit časná stadia maligní transformace, resp. různé stupně dysplastických změn. Dysplazie je histologický termín zahrnující strukturální a cytologické změny, které vznikají u klonu epiteliálních buněk v důsledku genetických alterací. Proto je v poslední době dysplazie nahrazena termínem intraepiteliální neoplazie (IEN).
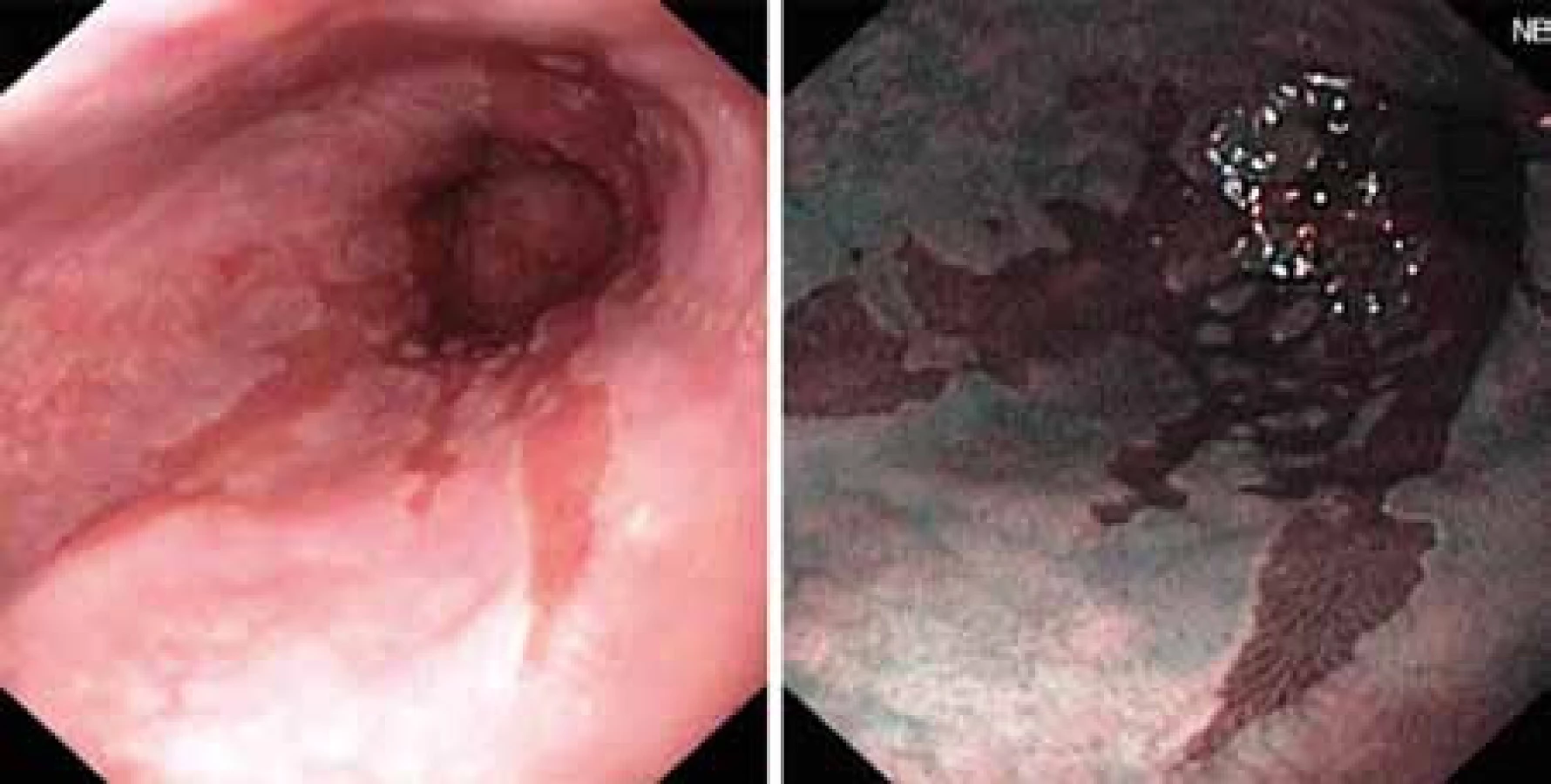
Histologicky lze dysplazii hodnotit 4 stupni, tj. nepřítomna, přítomna, a to buď vysokého stupně – tzv. high grade dysplasia (HGD), nebo nízkého stupně (low grade dysplasia) nebo dysplazie neurčitá (indeterminate dysplasia) [7]. Vztah rizika vzniku adenokarcinomu jícnu v závislosti na stupni dysplazie je uveden v grafu 2.

Nové způsoby vyhledávání rizikových osob, tj. osob s BJ, u nichž je vysoké riziko vzniku karcinomu jícnu, by mohly být založeny na poznatcích molekulárně biologických a na sledování tzv. biomarkerů. Těmito biomarkery jsou především geny KRAS, TP53, KRT1 (cytokeratin 1), COX2 (cyklooxygenáza 2)a další. Mutace TP53 tumor supresoru je prokazována u 95 %, mutace KRAS onkogenu u 5 %, zatímco pozitivní exprese COX2 u 70–90 % adenokarcinomů jícnu. Na základě znalostí o úloze genu COX2 v proliferaci a angioinvazi lze inhibitory cyklooxygenáz užít v chemoprevenci karcinomu jícnu [8].
Prekancerózy tlustého střeva a konečníku
Kolorektální karcinom je nejčastější malignitou gastrointestinálního traktu a Česká republika zaujímá světový primát v incidenci tohoto onemocnění. Právě u tohoto nádoru dospěl výzkum molekulárních mechanizmů a biologické podstaty nejdále [9]. Kolorektální tumory typicky začínají na základě DNA mutace, případně více mutací, postihující jedinou epiteliální buňku. Ve smyslu pochodů uvedených v grafu 1 dochází ke genetické nestabilitě a nádorové proliferaci. Dle původu prvotní mutace lze rozlišit vrozené a sporadické formy.
Vrozené formy kolorektálního karcinomu
Přibližně 15% kolorektálních nádorů má hereditární charakter, neboli je způsobeno vrozenou predispozicí ve formě dědičně přenášené genové poruchy. Nejznámější typy vrozených forem kolorektálního nádoru jsou familiální adenatomatózní polypóza a hereditární nepolypózní kolorektální karcinom. Familiární adenomatózní polypóza (FAP) je autozomálně dominantní onemocnění s prevalencí 1/8 000, které se projevuje výskytem stovek polypů, především v tlustém střevě a rektu. Toto onemocnění je nejčastěji způsobeno zárodečnou mutací v genu APC, v menším počtu případů (asi 15 %) také vrozenou mutací genu MYH. Na rozdíl od FAP u hereditárního nepolypózního kolorektálního karcinomu (HNPCC) ke vzniku žádného polypu nedochází. Toto autozomálně dominantní postižení, často označované jako tzv. Lynchův syndrom [10], vzniká v důsledku mutace v některém z genů ze skupiny tzv. „mismatch‑repair genů“ (MMR). Tyto geny, z nichž nejznámější jsou hMSH2 a hMLH1, zajišťují systém oprav náhodných chyb vzniklých při standardní DNA replikaci.
Genetické testování obou nejčastějších hereditárních forem kolorektálního karcinomu je principiálně založené na detekci mutací v genech APC a MYH u familiální adenomatózní polypózy a genů hMSH2, hMLH1 (a případně dalších) u Lynchova syndromu. Vzhledem k vysokým nákladům na DNA sekvenování bývá při diagnostice FAP nejprve použito tzv. PTT testu (protein truncation test), který odhalí mutaci v genu APC způsobující předčasné ukončení APC proteinu. Teprve při negativním výsledku se provádí kompletní APC sekvenace. Obdobně u diagnostiky Lynchova syndromu se před nákladnou sekvenací mnoha potenciálních genů provádí jako předkrok test na přítomnost tzv. mikrosatelitní nestability (MSI – microsatellite instability). Přítomnost MSI je detekovatelná ve formě variace délky DNA úseků obsahující dinukleotidová opakování. Tato variace nastává jako projev nedokonalosti systému DNA oprav zajišťujícího správnost syntetizované DNA. Tumory vykazující MSI jsou následně vyšetřeny sekvenací.
Sporadické formy kolorektálního karcinomu
Více než 85 % případů kolorektálních nádorů je sporadických, vznikajících primárně z adenomových polypů na základě působení vnějších faktorů, nikoli dědičné predispozice. V roce 1990, Fearon a Vogelstein prezentovali důkazy o několikastupňovém modelu formace kolorektálního karcinomu na základě postupného postižení několika onkogenů a tumor supresorových genů [11]. Mutované geny na počátku této dráhy jsou tumor supresorový gen APC (jde však o mutace v jiných lokacích než u FAP) a onkogen KRAS [12]. V pozdní fázi jsou pozorovány delece na chromozómu 18q (oblast genu DCC – Deleted in Colorectal Carcinoma) a mutace genu TP53. V poslední době se v souvislosti s kolorektálním karcinomem také často zmiňuje jeho vysoký stupeň biologické heterogenity, která je přičítána existenci dalších molekulárních mechanizmů při jeho vzniku [13]. Mezi další potenciální mechanizmy je tak zařazována tzv. epigenetická inaktivace některých tumor‑supresorových genů nebo opravných genů způsobená patologickou metylací jejich promotorových DNA oblastí [14]. Uvádí se také, že primární poruchy systému řízené buněčné smrti (apoptózy) mohou zapříčinit vznik kolorektálních nádorů [15].
Za kvalitativně odlišný typ prekancerózy tlustého střeva mohou být považovány nespecifické střevní záněty, které mohou v malém procentu případů (asi 1 %) vést ke vzniku sporadického kolorektálního karcinomu. Hlavními rizikovými faktory v těchto případech jsou trvání nemoci a rozsah zánětu. Významným faktorem je i primární sklerózující cholangitida (PSC), autoimunitní onemocnění extra ‑ a intrahepatálních žlučovodů, postihující 3–5 % nemocných s ulcerózní kolitidou. Rizikem kolorektálního nádoru je i pozitivní rodinná anamnéza kolorektálního karcinomu, zvyšující riziko dvojnásobně. V neposlední řadě je rizikovým faktorem aktivita zánětlivých změn. Genetický základ a karcinogeneze u případů vznikajících jako pozdní komplikace nespecifických střevních zánětů jsou obdobné jako u sporadického karcinomu. Zásadní rozdíl je v časové posloupnosti výskytu mutací u jednotlivých genů s tím, že gen APC bývá postižen v závěrečné fázi maligní transformace, zatímco mutace genu TP53 nebo ztráty alel a mikrosatelitní nestabilita se objevují i v časných fázích, kdy ještě nelze dysplazii morfologicky verifikovat.
Genetické testování sporadických forem kolorektálních nádorů je založeno na detekci výše popsaných genetických defektů. Nejčastěji je sledován panel mutací genů APC, KRAS a TP53, případně metylace genu VIM (vimentin). Kromě již klasických vyšetření tkáňových vzorků adenomů získaných při kolonoskopickém vyšetření se začínají uplatňovat i neinvazivní postupy genetického testování ze stolice [16]. Zcela novým fenoménem je také možnost záchytu cirkulujících nádorových buněk či volné nádorové DNA, u kterých byla zjištěna korelace se stupněm onemocnění [17,18].
Prekancerózy pankreatu
Nádory pankreatu představují i přes významné pokroky molekulárně biologických metod nejobávanější zhoubné malignity nejen v oblasti gastrointestinálního traktu. Hodnoty mortality dosahují téměř úroveň incidence s ročním počtem dosahujícím 19 diagnostikovaných a 17 zemřelých pacientů na 100 000 obyvatel. Hlavním exogenním rizikovým faktorem je kouření (u kuřáků je popisováno 2–3krát vyšší riziko karcinomu pankreatu) a alkohol [19]. Z endogenních faktorů lze uvést chronickou pankreatitidu a především rodinnou historii onemocnění [20].
Morfologický přerod normální pankreatické tkáně v karcinom je charakterizován přechodem tzv. pankreatické intraepiteliální neoplazie (PanIN) charakterizující prekurzorové intraduktální léze podle stupně cytologické a architektonické atypie od lehké (stupeň 1) po těžkou (stupeň 3) [21]. Jednotlivé stupně jsou též charakterizovány řadou genetických alterací, jak je zobrazeno na obr. 2. Vstupní genetickou poruchou je mutace onkogenu KRAS, detekována již ve stupni PanIN‑1A (v invazivních karcinomech dosahuje frekvence 80–90 %). Ten je následován dalšími defekty, jako např. hypermetylace promotorové oblasti tumor‑supresoru p16(INK4) ve stupni PanIN‑2 (v invazivních karcinomech bývá aktivován až v 98 %) nebo mutacemi genu TP53 ve stupni Pan IN‑3 (v invazivních karcinomech aktivován v 50–75 %, nejčastěji mechanizmem mutace a následné alelické delece). V pozdních fázích PanIN‑3 dochází ke ztrátě lokusu 18q (lokus genu DPC – Deleted in Pancreatic Cancer), jehož kompletní absenci lze v invazivních karcinomech detekovat v asi 55 %. Vyšetřením sady těchto molekulárních variací lze zpřesnit diagnostiku ložiskových lézí, především v kombinaci s endosonografickým vyšetřením a současně provedenou tenkojehlovou biopsií pankreatu (EUS/FNA) především v případech inkonkluzivního výsledku cytologického vyšetření [22]. Kombinace těchto metod je tedy výhodná pro výběr pacientů – kandidátů chirurgické resekce pankreatu z důvodu ložiskového procesu, avšak záchyt žádného z uvedených genetických markerů nemá význam pro odhad postupu a prognózy onemocnění [23].
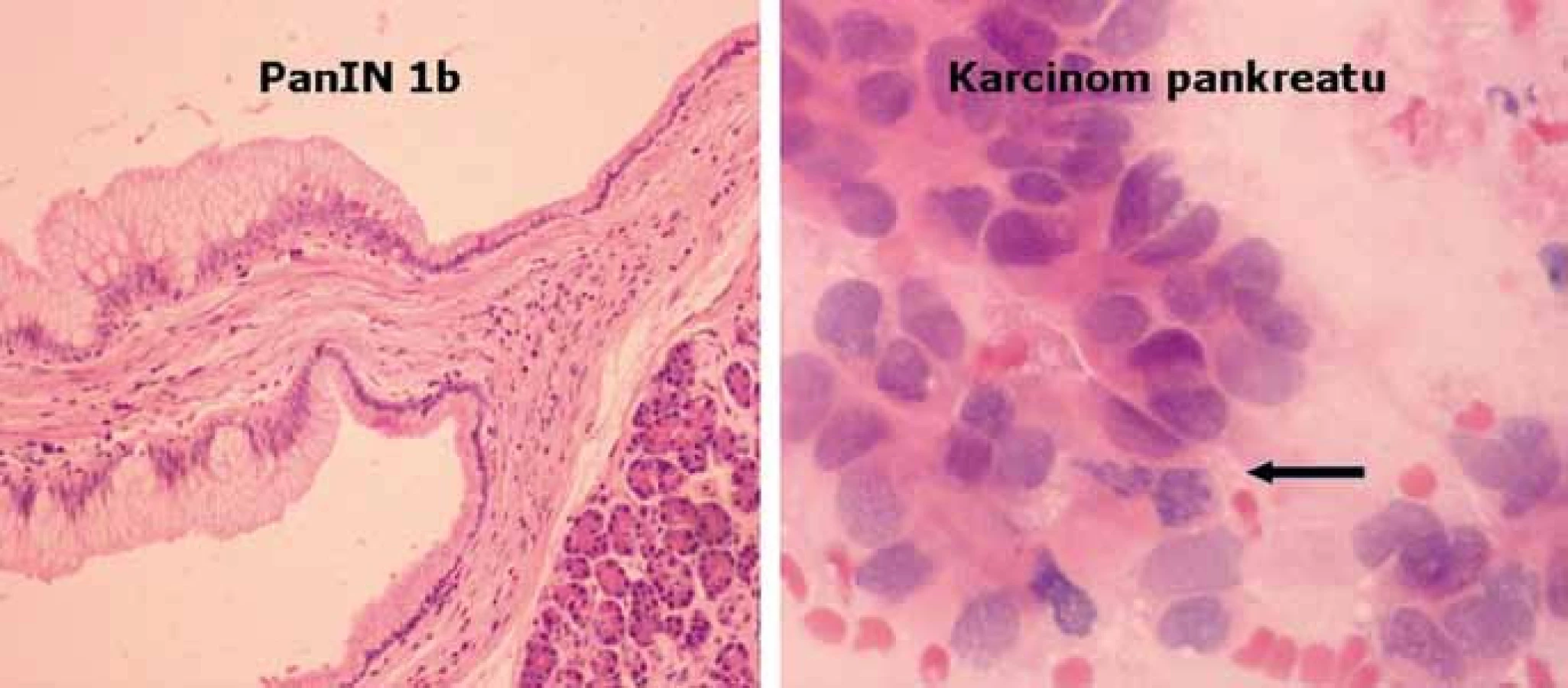
Závěr
Klinický výzkum v oblasti molekulárních mechanizmů nádorového vzniku a proliferace je založen na podrobném sledování jednotlivých signálních drah na úrovni genetických (DNA mutace) nebo epigenetických (DNA metylace) poruch mající vliv na funkce hlavních genů. V některých případech lze tyto aberace účinně využít jako molekulární markery pro časný záchyt nastupujícího onemocnění. Takovými případy jsou vrozené formy kolorektálních karcinomů, kde lze provádět molekulárně‑genetický screening. Na druhé straně se však např. v případě karcinomu pankreatu přes velký postup v poznání principů stále nepodařilo nalézt odpovídající molekulární test pro časný záchyt tohoto onemocnění. Lze očekávat, že v tomto případě se pozornost bude stále více ubírat k biomarkerům na bázi glykoproteinů či endogenních metabolitů specifických pro molekulární pochody v časných fázích a možnosti jejich preventivního vyšetřování v periferní krvi se zaměřením na rizikové skupiny.
Poděkování
Tato práce byla podpořena grantem IGA MZ NS/9809.
Předneseno na XVI. kongresu České internistické společnosti ČLS J. E. Purkyně v Praze ve dnech 13.–16. září 2009
Doručeno do redakce: 30. 9. 2009
MUDr. Petra Mináriková, Ph.D.
www.uvn.cz
e‑mail: petra.minarikova@uvn.cz
Sources
1. Wikipedia – the online encyclopedia, http://en.wikipedia.org/wiki/Precancerous.
2. Epidemiologie zhoubných nádorů v České republice v roce 2007, www.svod.cz.
3. Klein CA. Gene expression sigantures, cancer cell evolution and metastatic progression. Cell Cycle 2004; 3 : 29–31.
4. Toribara NW, Sleisenger MH. Screening for colorectal cancer. N Engl J Med 1995; 332 : 861–867.
5. Barrett N. The lower esophagus lined by columnar epithelium. Surgery 1957; 41 : 881–894.
6. Fléjou J. Barrett’s oesophagus: from metaplasia to dysplasia and cancer. Gut 2005; 54 (Suppl 1): i6–i12.
7. Seewald S, Ang TL, Groth S et al. Detection and endoscopic therapy of early esophageal adenocarcinoma. Curr Opin Gastroenterol 2008; 24 : 521–529.
8. Kaur BS, Khamnehei N, Iravani M et al. Rofecoxib inhibits cyclooxygenase 2 expression and activity and reduces cell proliferation in Barrett’s esophagus. Gastroenterology 2002; 123 : 60–67.
9. Worthley DL, Whitehall VL, Spring KJ et al. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol 2007; 13 : 3784–3791.
10. Lynch HT, Lanspa S, Smyrk T et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I & II). Genetics, pathology, natural history, and cancer control, Part I. Cancer Genet Cytogenet 1991; 53 : 143–160.
11. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61 : 759–767.
12. Capella G, Cronauer‑Mitra S, Pienado MA et al. Frequency and spectrum of mutations at codons 12 and 13 of the c‑K‑ras gene in human tumors. Environ Health Perspect 1991; 93 : 125–131.
13. Jass JR. Colorectal cancer: a multipathway disease. Crit Rev Oncog 2006; 12 : 273–287.
14. Wheeler JM. Epigenetics, mismatch repair genes and colorectal cancer. Ann R Coll Surg Engl 2005; 87 : 15–20.
15. Watson AJ. Apoptosis and colorectal cancer. Gut 2004; 53 : 1701–1709.
16. Osborn NK, Ahlquist DA. Stool screening for colorectal cancer: molecular approaches. Gastroenterology 2005; 128 : 192–206.
17. Tsouma A, Aggeli C, Pissimissis N et al. Circulating tumor cells in colorectal cancer: detection methods and clinical significance. Anticancer Res 2008; 28 : 3945–3960.
18. Anker P, Mulcahy H, Chen XQ et al. Detection of circulating tumour DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev 1999; 18 : 65–73.
19. Hassan MM, Bondy ML, Wolff RA et al. Risk factors for pancreatic cancer: case‑control study. Am J Gastroenterol 2007; 102 : 2696–2707.
20. Otsuki M, Tashiro M. Chronic pancreatitis and pancreatic cancer, lifestyle‑related diseases. Intern Med 2007; 46 : 109–113.
21. Hruban RH, Goggins M, Parsons J et al. Progression model for pancreatic cancer. Clin Cancer Res 2000; 6 : 2969–2972.
22. Salek C, Benesova L, Zavoral M et al. Evaluation of clinical relevance of examining K-ras, p16 and p53 mutations along with allelic losses at 9p and 18q in EUS‑guided fine needle aspiration samples of patients with chronic pancreatitis and pancreatic cancer. World J Gastroenterol 2007; 13 : 3714–3720.
23. Salek C, Minarikova P, Benesova L et al. Mutation status of K‑ras, p53 and allelic losses at 9p and 18q are not prognostic markers in patients with pancreatic cancer. Anticancer Res 2009; 29 : 1803–1810.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 1
Most read in this issue
- Nemocní starší 80 let s de novo akutními myeloidními leukemiemi bez dysplazie v erytroblastické a/nebo megakaryocytární řadě dosahují kompletní remise a delšího přežití po klasické chemoterapii 3+7
- Paraneoplastický tromboembolický syndróm ako prvý príznak zhubného ochorenia
- Dysfunkce pravé komory po implantaci levostranné mechanické srdeční podpory
- Epidemiológia cievnych chorôb