
Více než 10 let trvající kompletní remise monoklonální gamapatie nejistého významu a vymizení nefrotického syndromu vzniklého na podkladně light chain deposition disease po léčbě vinkristinem, adriamycinem a vysokými dávkami dexametazonu (VAD)
More than 10 years of complete remission of monoclonal gammopathy of undetermined significance and cessation of light chain deposition disease‑associated nephrotic syndrome following treatment with vincristine, adriamycin and high‑dose dexamethasone (VAD)
Light chain deposits in the form of amorphous material (light chain deposition disease) damage most frequently kidneys and, less frequently, they affect other organs. The incidence of light chain deposition disease is much lower than that of AL ‑ amyloidosis. Symmetrical swelling of both legs, swelling of the eye lids, erythrocyturia and nephrotic proteinuria were the first signs of light chain deposition disease in our patient. The disease was diagnosed from kidney biopsy performed at the stage of advanced nephrotic syndrome with reduced filtration. The bone marrow aspirate contained 0.8% of plasma cells, serum contained monoclonal immunoglobulin IgG ‑ κ and urine contained free κ chains. Blood count was normal and no osteolytic changes to the skeleton were identified. The patient was, therefore, diagnosed with monoclonal gammopathy of undetermined significance (MGUS) and was treated with 10 cycles of chemotherapy consisting of vincristine, adriamycin and high/ dose dexamethasone (VAD). Following the 10th cycle, the concentration of monoclonal IgG declined below the threshold for quantitative densitometric identification, while the more sensitive immunofixation electrophoresis remained positive. However, 2 months after the completion of chemotherapy, the immunofixation electrophoresis had become negative and thus complete haematological treatment response (remission) was achieved. Restoration of the kidney function was only gradual. Proteinuria declined below 1 g/ l and no erythrocyturia was present 4 years post‑treatment. Proteinuria declined to 0.19 g/ l, i.e. normal values, 9 years post‑treatment completion. Regular follow‑ups in patients with MGUS should seek to identify not only whether MGUS is transforming into malignant disease but also whether monoclonal immunoglobulin is damaging the organism. Treatment of patients with monoclonal immunoglobulin‑associated damage should be initiated early as the restoration of the affected organs function (organ treatment response) after complete haematological remission is only gradual. At present, treatment regimes with high‑dose dexamethasone are recommended for patients with primary systemic AL ‑ amyloidosis. We believe that the same approach is suitable for the treatment of light chain deposition disease in MGUS patients.
Key words:
light chain deposition disease – monoclonal gammopathy of undetermined significance (MGUS) – multiple myeloma – renal insuficiency – nephrotic syndrome – AL ‑ amyloidosis
Authors:
Z. Adam 1; M. Nedbálková 2; M. Krejčí 1; L. Pour 1; K. Hušek 3; K. Veselý 4; Z. Čermáková 5; A. Křivanová 1; J. Mayer 1; R. Hájek 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Vorlíček, CSc. 2II. interní klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta prof. MU Dr. Miroslav Souček, CSc. 3Ústav patologie L
1
Published in:
Vnitř Lék 2010; 56(3): 240-246
Category:
Case Reports
Overview
Depozita lehkých řetězců ve formě amorfních hmot (light chain deposition disease) poškozují nejčastěji ledviny, méně často poškozují jiné orgány. Onemocnění je podstatně vzácnější než AL ‑ amyloidóza. Prvními příznaky light chain deposition disease u našeho nemocného byly symetrické otoky dolních končetin, otoky víček, erytrocyturie a nefrotická proteinurie. Nemoc byla diagnostikována z biopsie ledviny, provedené až ve fázi rozvinutého nefrotického syndromu se snížením filtrace. V aspirátu kostní dřeně bylo 0,8 % plazmocytů, v séru byl přítomen monoklonální imunoglobulin IgG ‑ κ a v moči byly přítomny volné řetězce κ. Krevní obraz byl v normě a nebyly osteolytické změny skeletu. Diagnóza byla tedy uzavřena jako monoklonální gamapatie nejistého významu (MGUS). Pacient byl léčen 10 cykly chemoterapie obsahující vinkristin, adriamycin a vysoké dávky dexametazonu (VAD). Po 10. cyklu poklesla koncentrace monoklonálního IgG pod hranici kvantitativního denzitometrického stanovení, citlivější imunofixační elektroforéza byla však ještě pozitivní. Nicméně za 2 měsíce od ukončení chemoterapie byla i imunofixační elektroforéza negativní, takže bylo dosaženo kompletní hematologické léčebné odpovědi (remise). Reparační změny v ledvinách probíhaly velmi pozvolna. Po 4 letech od ukončení léčby klesla proteinurie pod 1 g/ l a již nebyla přítomna erytrocyturie. Po 9 letech od ukončení léčby klesla proteinurie na 0,19 g/ l, tedy na normální hodnoty. U pacientů s MGUS je při opakovaných kontrolách nutno hledat nejen odpověď na otázku, zda nedochází ke transformaci MGUS do maligního onemocnění, ale také na otázku, zda se nerozvíjí poškození organizmu monoklonálním imunoglobulinem. Pokud monoklonální imunoglobulin poškozuje pacienta, je nutno léčbu zahájit včas, protože obnova funkce postižených orgánů (orgánová léčebná odpověď) probíhá po dosažení kompletní hematologické remise velmi zvolna. Léčebné režimy obsahující vysoké dávky dexametazonu jsou v současnosti doporučovány pro pacienty s primární systémovou AL ‑ amyloidózou. Domníváme se, že pro léčbu light chain deposition disease při MGUS jsou vhodné stejné postupy.
Klíčová slova:
light chain deposition disease – monoklonální gamapatie nejistého významu – mnohočetný myelom – proteinurie – nefrotický syndrom – AL ‑ amyloidóza
Úvod
Monoklonální imunoglobulin může být tvořen jak maligní plazmocytární populací, tak také nemaligním klonem plazmocytů, který je podstatou monoklonální gamapatie nejistého významu, anglickým termínem Monoclonal Gammopathy of Undetermined (Unknown) Significance – MGUS, dříve zvané benigní monoklonální gamapatie.
Maligní či benigní proliferační potenciál plazmocytů však přímo nesouvisí s tím, zda monoklonální imunoglobulin bude poškozovat organizmus, či ne. To je dáno strukturou monoklonálního imunoglobulinu. A jedním z orgánů, který monoklonální imunoglobuliny často poškozují, bývají ledviny. O patofyziologii vzniku poškození ledvin a o léčbě pojednávající četné přehledové články [1 – 5].
Následujícím popisem případu chceme dokumentovat, že:
- chemoterapie obsahující vysoké dávky dexametazonu může u monoklonální gamapatie nejistého významu také dosáhnout kompletní hematologické remise – vymizení monoklonálního imunoglobulinu potvrzené negativním výsledkem imunofixační elektroforézy;
- skutečnost, že po dosažení kompletní hematologické remise mohou reparační mechanizmy v ledvinách velmi pomalu zlepšit jejich funkci a vést ke kompletnímu a dlouhodobému vymizení nefrotického syndromu;
- u pacientů s MGUS je nutné nejen sledovat, jestli se benigní proliferace netransformuje v maligní, ale i jestli se neobjevuje poškození organizmu monoklonálním imunoglobulinem, které při stanovení diagnózy MGUS nemusí být přítomné.
Popis případu
Stanovení diagnózy MGUS v roce 1998
Muž, narozený 1936, měl při preventivní prohlídce v roce 1998, tedy v 62 letech věku, zjištěnu vyšší sedimentaci a následné vyšetření pak odhalilo v séru monoklonální imunoglobulin IgG-κ. Kdošetření monoklonální gamapatie byl poslán na naše pracoviště. Vstupní hodnoty uvádíme přehledně v tab. 1. Počet plazmocytů v aspirátu kostní dřeně (sternální punkce) byl 0,80 %, tedy nezvýšený, a histologické hodnocení aspirátu kostní ze sternální punkce dřeně také neodhalilo vyšší počet plazmatických buněk. Histologické vyšetření válečku kostní dřeně získaného trepanobiopsií nebylo provedeno. RTG vyšetření celého skeletu neodhalilo osteolytická ložiska a kostní denzitometrie prokázala normální kostní hustotu. Kritéria mnohočetného myelomu nebyla naplněna. A tak v prosinci roku 1998 byla diagnóza uzavřena jako monoklonální gamapatie nejistého významu (MGUS), starším termínem benigní gamapatie.
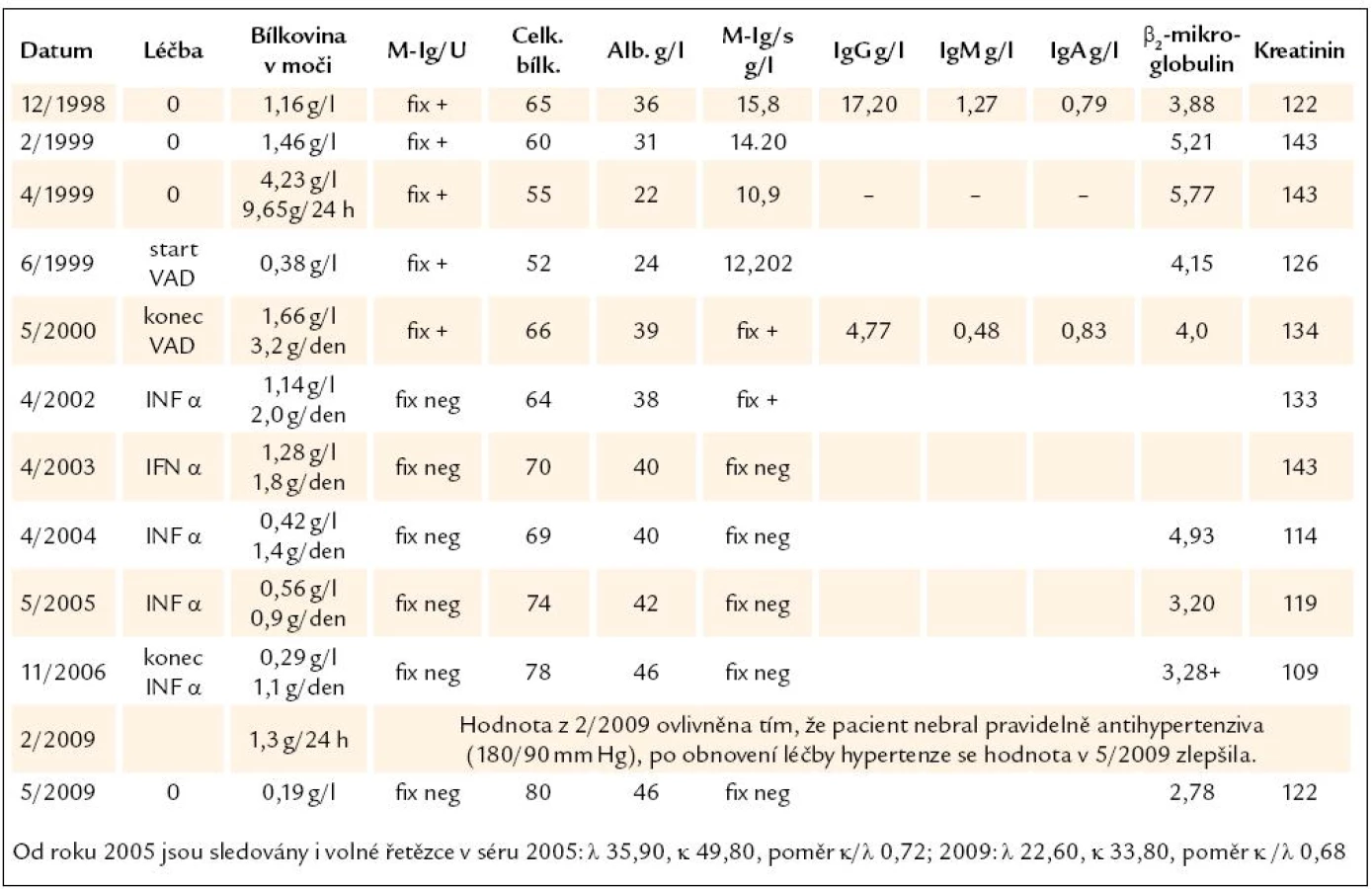
Pacientovi jsme nabídli dispenzari-zaci, protože slovy „gamapatie nejistého (jiným slovem nedeterminovaného) významu“ se míní, že pravděpodobnost transformace do maligního plazmacelulárního či lymfoproliferativního onemocnění je podstatně vyšší než u stejně starých osob bez monoklonálního imunoglobulinu, a navíc je zde riziko poškození orgánů monoklonálním imunoglobulinem, které se může klinicky manifestovat později.
Rozvoj nefrotického syndromu počátkem roku 1999
V únoru roku 1999 si pacient začal stěžovat na otoky kolem očí ráno po probuzení, které jen velmi pomalu mizely, a na symetrické otoky nohou. Měl těstovité otoky po kolena. Laboratorní hodnoty jsou opět uvedeny v tab. 1. Vyšetření v dubnu roku 1999 již potvrdilo výraznou proteinurii 9 g/ den, tato hodnota již odpovídala nefrotickému syndromu. Proto byl odeslán na nefrologické oddělení Fakultní nemocnice u sv. Anny v Brně, kde provedli biopsii ledviny a histologické vyšetření.
Histopatologické nálezy v renální punkci zahrnovaly většinou globoidní hypocelulární mezangiální expanzi se ztluštěním mezangiálních membrán a nesystematickou úsekovitou produkcí dvojitých kontur. Ojediněle byla přítomna segmentální proliferace. Fokálně byla přítomna extrakapilární akumulace PAS pozitivního materiálu a buněčná proliferace nebo fibróza. Doprovodným nálezem byla lehká až střední tubulární atrofie a tomu odpovídající intersticiální fibróza. Byly rovněž přítomny nespecifické arteriosklerotické změny interlobulárních arterií.
Při vyšetření přímou imunofluorescencí byly silně pozitivní glomerulární a tubulární membrány a mezangium v IgG. Amyloid nebyl prokázán. Elektronově mikroskopicky byl vyšetřen jeden glomerulus, s nápadným elektrodenzním zesílením lamina densa bazálních membrán nebo denzním rozšířením lamina densa subendoteliálně. Tubulární bazální membrána byla rovněž s depozity.
Závěr bioptického vyšetření: Histologický i ultrastrukturální obraz včetně imunofluorescenčního vyšetření odpovídá nefropatii s depozity lehkých řetězců (obr. 1 – 3).
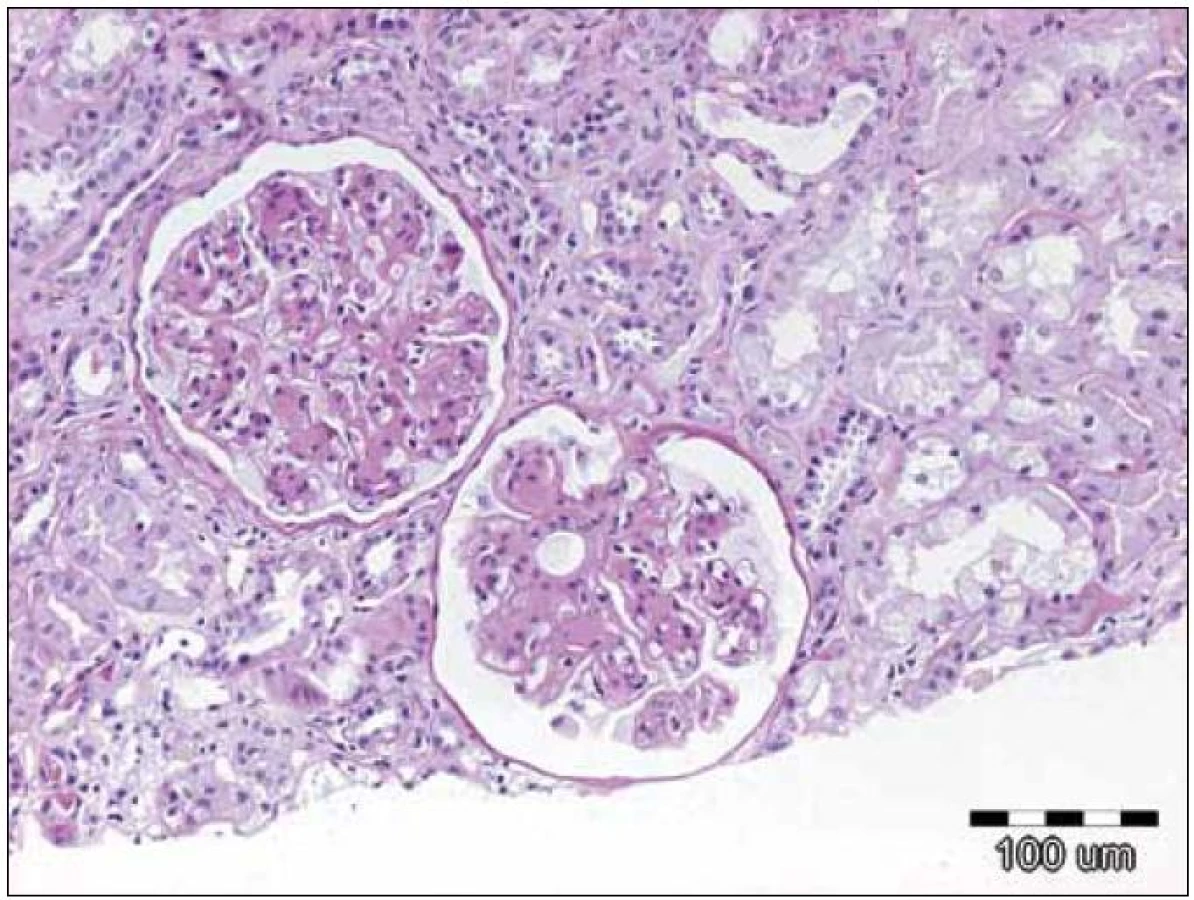
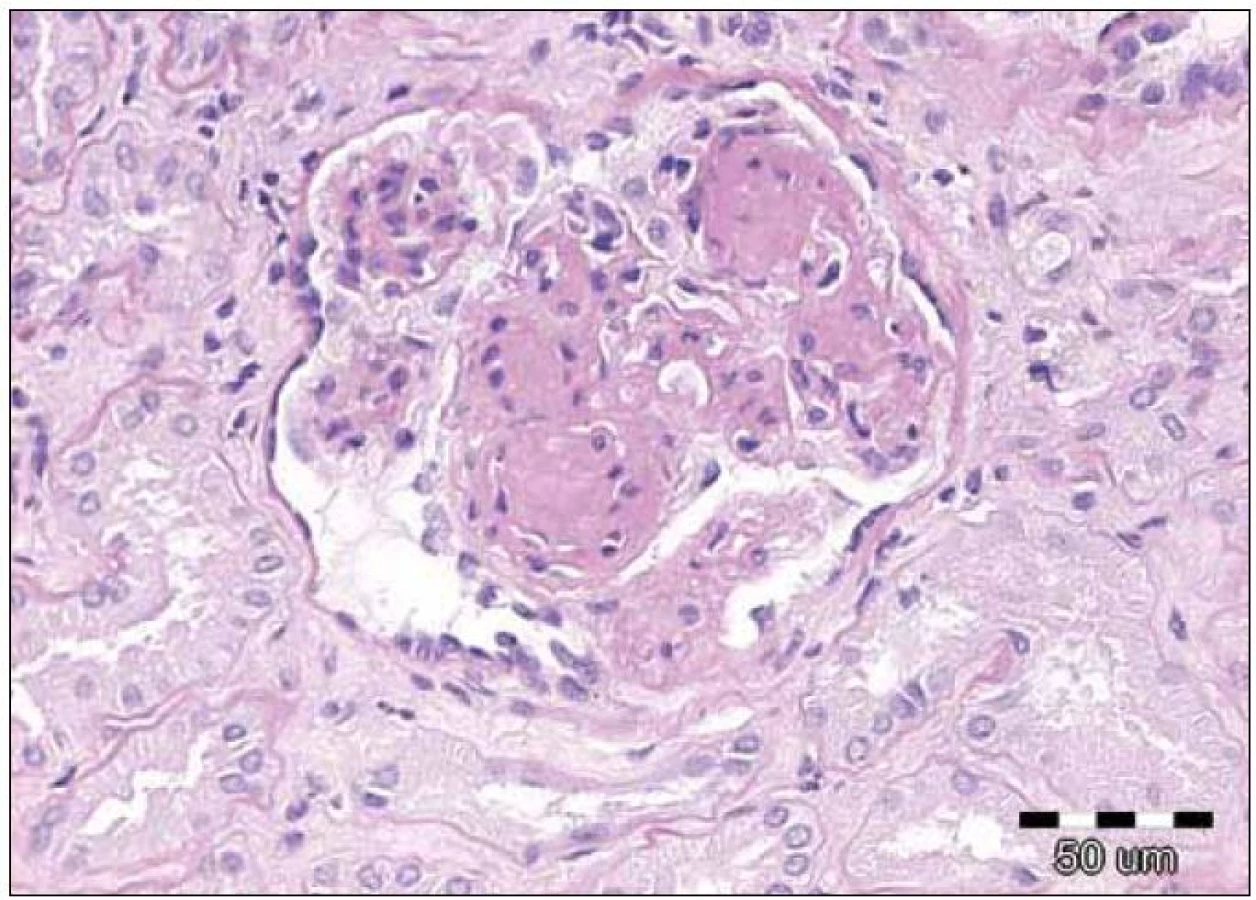
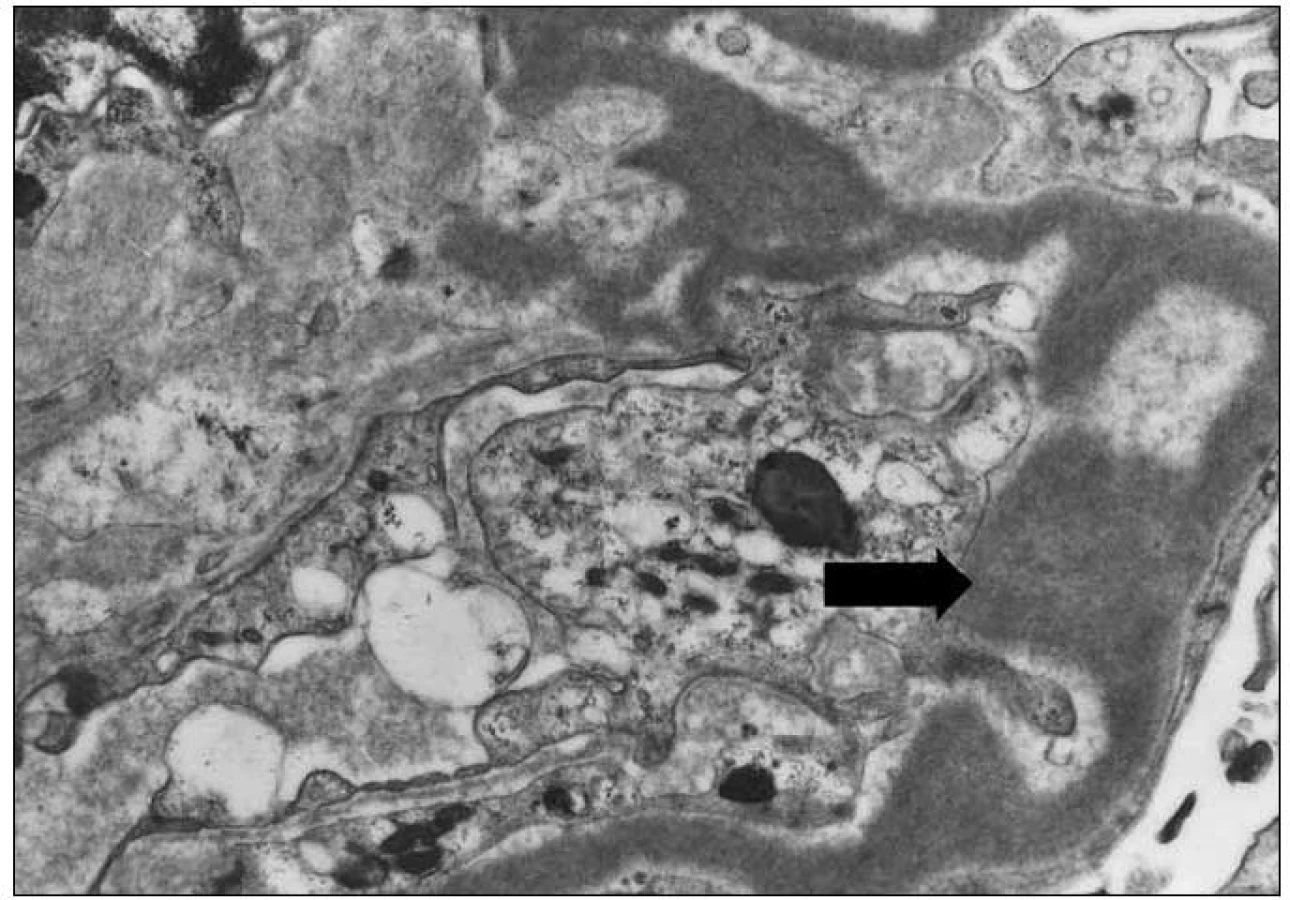
Závěrečná nefrologická diagnóza zněla: Nefrotický syndrom s proteinurií 9 g/ 24 hod, s erytrocyturií, na bazi biopticky verifikované light chain deposition nefropatie, se sníženou hodnotou filtrace (renální insuficiencí), sérový kreatinin se zvýšil na 196 μmol/ l, s hypoalbuminemií, anasarkou, hydrotoraxem, otoky dolních končetin a s hypertenzí.
Kontrolní vyšetření nepotvrdilo přechod monoklonální gamapatie nejistého významu do maligního onemocnění, a tak jsme stáli před otázkou, zda podat pacientovi v té době s benigní gamapatií chemoterapii s cílem potlačit produkci nefrotoxických monoklonálních imunoglobulinů, a zastavit tak další rozvoj nefropatie nefrotickým syndromem.
Zahájení chemoterapie VAD v červnu roku 1999
Po domluvě s pacientem byla v červnu roku 1999 zahájena chemoterapie VAD obsahující vysoké dávky dexametazonu (40 mg p. o. 1. – 4., 10. – 13. a 20. – 23. den),vinkristin (0,5 mg i.v. 1. – 4. den), adriamycin (9 mg/ m2 i.v. 1. – 4. den) ve 28denních intervalech. Laboratorní hodnoty v době zahájení chemoterapie jsou uvedeny v tab. 1. Celkem bylo podáno 10 cyklů této chemoterapie, poslední aplikace byla v dubnu roku 2000. Pak jsme pacientovi začali podávat udržovací léčbu interferonem α (3 mil. jednotek 3krát týdně s.c).
Laboratorní hodnoty po ukončení chemoterapie, z května roku 2000, uvádíme opět v tab. 1. Monoklonální imunoglobulin v séru pokles pod kvantitativně stanovitelné hodnoty (metodou elektroforézy a denzitometrie), ale v moči byla stále metodou imunofixační elektroforézy prokazatelná stopa monoklonálního imunoglobulinu. V době ukončení chemoterapie byla ještě pořád význačná proteinurie, kterou ale máme dokumentovanou pouze koncentrací v ranním vzorku, nikoliv množstvím za 24 hod.
V době ukončení chemoterapie bylo tedy dosaženo velmi dobré parciální remise nemoci s prokazatelným stopovým množstvím monoklonálního imunoglobulinu, ale v tak nízké koncentraci, že byla pod možností kvantitativního stanovení denzitometrií. Po 2 měsících od ukončení chemoterapie však již byla imunofixační elektroforéza v séru i v moči bez průkazu monoklonálního imunoglobulinu, takže bylo dosaženo dlouhodobé kompletní hematologické remise. (Poznámka: V případně monoklonální gamapatie poškozující organizmus rozlišujeme hematologickou léčebnou odpověď, tedy míru snížení tvorby monoklonálního imunoglobulinu, a orgánovou léčebnou odpověď, tedy míru reparace monoklonálním imunoglobulinem poškozeného orgánu.)
Kontroly v průběhu 10 let od zahájení léčby potvrzovaly trvání kompletní hematologické remise a dokumentovaly postupné zmenšování proteinurie
V následujících 10 letech od zahájení léčby se postupně zlepšovala funkce ledvin, zmenšovala se proteinurie (kompletní orgánová léčebná odpověď) a vymizela erytrocyturie. Při poslední kontrole v květnu roku 2009 byl pacient bez průkazu monoklonálního imunoglobulinu, množství bílkoviny v moči je ve fyziologickém rozmezí. Imunofixační elektroforéza byla trvale negativní a v posledních letech byla kompletní remise potvrzena i fyziologickým rozmezím koncentrací a poměru volných lehkých řetězců.
Diskuze
Definice nemoci
Light chain deposition disease je vzácnáforma poškození ledvin (či organizmu obecně), způsobená monoklonálními lehkými řetězci většinou typu κ, které se ukládají v různých tkáních ve formě amorfních hmot, a poškozují tak jejich funkci. Poprvé tento typ poškození organizmu monoklonálního imunoglobulinem popsal Randall v roce 1976 [1]. Nejčastěji jsou tato depozita nalézána v ledvině, vzácněji pak i v dalších tkáních a orgánech, např. v játrech [6 – 8], v plicích [9 – 11] a případně v souvislosti s POEMS syndromem [12]. Klinicky se light chain deposition disease projeví progresivním selháním funkce ledvin (či orgánu, v němž dochází ke vzniku amorfních depozit monoklonálních lehkých řetězců).
Proteinurie nad 1 g/ 24 hod byla popsána u 84 % pacientů s light chain deposition disease a 40 % nemocných mělo proteinurii odpovídající nefrotickému syndromu [2,3]. Do klinického obrazu mimo proteinurii patří také mikrohematurie a hypertenze. Retence dusíkatých látek bývá obvykle závažnější než v případě depozice amyloidových hmot v ledvině [2,3,13,14]. Tomuto popisu odpovídal i stav našeho pacienta v roce 1999, kdy měl plně rozvinutý nefrotický syndrom s erytrocyturií a hypertenzí a zvýšenou hodnotou kreatininu.
Diagnostika
Diagnostikovat lze tuto jednotku pouzepomocí biopsie poškozeného orgánu, nejčastěji to bývají ledviny. V optické mikroskopii je charakteristická přítomnost mezangiálních uzlů, zesílení bazálních membrán glomerulárních kapilár a tubulů. Obraz nodulární sklerózy v základním barvení (H&E, PAS) je histologicky těžko odlišitelný od diabetické glomerulosklerózy. V zásadě u LCDDse noduly vyskytují difuzněji než u diabetické glomerulosklerózy. Histologickýobraz LCDD je však velice variabilní a nepřítomnost mezangiálních uzlů nevylučuje toto onemocnění. Při nepřítomnosti nodulárních formací bývá fokální nebo difuzní mezangiální expanze někdy se zvýšenou buněčnatostí [15 – 21].
Pomocí imunofluorescence lze prokázat depozici monoklonálního imunoglobulinu (monoklonální řetězce κ) v glomerulární bazální membráně, v mezangiu a v tubulární bazální membráně. V elektronmikroskopickém obraze lze znázornit depozita nefibri-lárního materiálu ve zmíněných lokalizacích. V případě mnohočetného myelomu při vyšším kvantu vylučovaných lehkých řetězců může být ledvina poškozována současně amorfními depozity lehkých řetězců a odlitkovými válci. Koexistence těchto dvou patofyziologických podkladů nefropatie byla popsána asi v 1/ 3 případů [3,22].
Výjimečně může být ledvina poškozena jak ukládáním lehkých řetězců v amorfní formě, tak i ve formě amyloidu [23,24].
Diferenciální diagnostika je velice široká. Zahrnuje diabetickou glomerulosklerózu, idiopatickou nodulární glomerulosklerózu, mebranoproliferativní glomerulonefritidu, amyloidózu.
Klinické známky light chain deposition disease se mohou manifestovat za více let od stanovení diagnózy MGUS. V jednom případě se popisuje rozvoj nefrotického syndromu způsobeného light chain deposition disease za 15 let od stanovení diagnózy MGUS [25].
Léčebné postupy
Výsledky léčby light chain deposition disease uváděné literaturou
Light chain deposition disease je vzácná jednotka, a proto lze v literatuře nalézt pouze nečetné klinické studie a popisy případů. V nich byly použity léčebné postupy obdobné jako u mnohočetného myelomu. Léčebnou odpověď však klasická chemoterapie nedosahuje vždy. Heilmann ve skupině 19 pacientů léčených melfalanen a prednisonem pozoroval zmenšení proteinurie jen u 5 z 19. Pokles koncentrace monoklonálního imunoglobulinu, odpovídající parciální remisi, jen u 2 pacientů z 19 nemocných [2].
Pozzi popsal skupinu 19 nemocných, z nichž mělo 18 renální insuficienci a 16 nefrotický syndrom. Extrarenální depozita byla popsána u 12 vyšetřovaných osob (63 %), nejčastěji v srdci, játrech a v periferních nervech. Pacienti byli léčení steroidy a alkylačními cytostatiky. Hematologická léčebná odpověď a zlepšení funkce ledvin bylo dokumentováno pouze u 5 ze 16 hodnocených. Míra proteinurie se však u žád-ného léčeného signifikantně nezmenšila [3,26]. Nevelký efekt klasické chemoterapie, podávané v případě light chain deposition disease při plazmocelulární proliferaci typu mnohočetného myelomu, ale i typu MGUS, popisují i další pracoviště [14], a proto se zde používá také vysokodávkovaná chemoterapie s autologní transplantací kostní dřeně [27 – 33] a případně i ledviny [34]. Dávky melfalanu je však nutno redukovat a aplikaci rozdělit případně i do 2 dnů, protože v případě poškození ledviny depozity lehkých řetězců bylo popsáno i zhoršení funkce po aplikace melfalanu [35].
Primární systémová AL-amyloidóza a light chain deposition disease při MGUS mají nemaligní charakter plazmocelulární proliferace, ale liší se produkovanými lehkými řetězci
Co má primární systémová AL-amyloidóza a light chain deposition disease při MGUS společného a co rozdílného? Společným základem obou jednotek je nemaligní (benigní) proliferace plazmatických buněk. Rozdílné jsou chemicko-fyzikální vlastnosti těmito buňkami tvořených monoklonálních lehkých řetězců a dle toho výsledná forma depozit lehkých řetězců ve tkáních. Z toho odvozujeme, že léčebná doporučení platící pro primární systémovou AL-amyloidózu lze vztáhnout i na pacienty s light chain deposition disease při MGUS. Poškození ledvin typu light chain deposition disease je vzácnější než poškození ledvin při primární systémové AL-amyloidóze, a proto nejsou žádné randomizované studie, které by tuto otázku řešily [36].
Pro kompletnost textu však musíme připomenout, že depozita lehkých řetězců ve formě amyloidu či ve formě amorfních hmot mohou provázet také pacienty s mnohočetným myelomem, tedy s maligní proliferací plazmatických buněk. Tyto případy však označujeme jako AL-amyloidóza při mnohočetném myelomu anebo light chain deposition disease při mnohočetném myelomu.
Pro primární systémovou AL-amyloidózu se doporučují léčebné režimy založené na vysokých dávkách dexametazonu
V roce 1999 bylo poprvé zveřejněno, že léčba primární systémové AL - amyloidózy dosahuje vyššího počtu léčebných odpovědí, pokud se místo běžné dávky prednisonu použije vysoká dávka dexametazonu [37]. Dexametazon samotný navodil u primární systémové AL-amyloidózy 24 % kompletních remisí [38] a kombinace melfalanu a dexametazonu dosáhla u pacientů s primární systémovou AL - amyloidózou 33 % kompletních remisí [39], které byly dlouhodobé [40]. Léčebné režimy založené na vysokých dávkách dexametazonu měly podobný přínos jako vysokodávkovaná chemoterapie [41].
Proto doporučení German Society of Amyloid Disease – http://www.amyloid.de – doporučuje použití vysokých dávek dexametazonu v monoterapii či v kombinaci pro léčbu pacientů s AL-amyloidózou, nevhodných k vysokodávkované chemoterapii s autologní transplantací. Nejvyšší počet orgánových léčebných odpovědí byl zaznamenán u pacientů s dominujícím poškozením ledvin depozity AL-amyloidu (39 %) a nejnižší u pacientů s polyneuropatií způsobenou AL-amyloidózou [42 – 46].
Léčba našeho pacienta s light chain deposition disease při MGUS režimem s vysokými dávkami dexametazonu
Po aplikaci 10 cyklů chemoterapie VAD jsme těsně po posledním cyklu pozorovali velmi dobrou léčebnou odpověď, která za 2 měsíce od ukončení léčby přešla v kompletní remisi s úplným vymizením monoklonálního imunoglobulinu (negativní imunofixace v moči i v krvi), která trvá 10 let. V posledních letech pro sledování aktivity nemoci používáme vyšetřování koncentrací volných lehkých řetězců v séru [47 – 49].
Chemoterapie VAD u našeho pacienta redukovala nejen patologické plazmocyty, ale i fyziologickou lymfocytární a plazmocytární populaci, což je zřetelné z výrazného poklesu polyklonálních imunoglobulinů ostatních tříd, jak dokumentuje tab. 1.
Překvapivá je velmi pomalá orgánová odpověď, velmi pomalé snižování koncentrace bílkovin v moči. Z tab. 1 je však zřetelné, že ačkoliv hematologické kompletní remise bylo dosaženo po 10 měsících léčby, byla maximální orgánová léčebná odpověď dosažena až po mnoha letech od ukončení léčby. Z tab. 1 je však také zřetelné, že v roce 2009 při nedodržení antihypertenzní léčby (Prestarium) a vzestupu krevního tlaku dočasně došlo při kontrole v únoru k nepatrnému vzestupu odpadu bílkovin, po upravení léčby hypertenze se však zase zmenšilo množství bílkovin vyloučených močí. U těchto pacientů je tedy nutné nejen sledovat laboratorní parametry, ale i krevní tlak.
Závěry pro praxi
- Léčba založená na vysokých dávkách dexametazonu je optimálním postupem pro pacienty s MGUS, způsobujícím light chain deposition disease podobně, jako je tomu u primární systémové AL - amyloidózy.
- V případech light chain deposition disease při MGUS, kdy klasickou léčbou nedosáhneme kompletní remise, je na zvážení vysokodávkovaná chemoterapie s autologní transplantací. Pokud je light chain deposition disease způsobena plazmocelulární proliferací typu mnohočetného mye-lomu (nikoliv tedy MGUS), je vysoko-dávkovaná chemoterapie součástí iniciální léčby, jestliže nejsou její kontraindikace.
- Poškození ledviny monoklonálním imunoglobulinem je léčitelné, pokud je léčba zahájena včas před vznikem velmi těžkého orgánového poškození a pokud se dosáhne kompletního vymizení monoklonálního imunoglobulinu. To je předpokladem k tomu, aby reparační změny zlepšily či obnovily funkci poškozeného orgánu. K dosažení maximální orgánové léčebné odpovědi je však třeba více let.
- U pacientů s MGUS je třeba kontrolní vyšetření zaměřit nejen na změny koncentrace monoklonálního imunoglobulinu v séru a moči, ale i sledovat, zda se neobjevuje poškození jednoho či více orgánů monoklonálním imunoglobulinem. Při jakýchkoliv nových zdravotních problémech je třeba si postavit otázku, zda mají, či nemají souvislost s monoklonálním imunoglobulinem.
- Monoklonální imunoglobulin může být odpovědný za velmi různorodé potíže (poškození jednotlivých orgánů, kožní projevy a souvisí např. i s velmi nebezpečným onemocněním, jako je idiopatic capillary leak syndrome) [50]. Proto je vhodné provést vyšetření přítomnosti monoklonálního imunoglobulinu v moči a krvi a vyšetření volných lehkých řetězců u všech pacientů s proteinurií, se známkami poškození ledvin, ale i jiných orgánů, a při jejich přítomnosti pátrat po možné souvislosti [51,52].
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 22. 7. 2009
Přijato po recenzi: 14. 9. 2009
Sources
1. Randall RE, Williamson WC Jr, Mullinax F et al. Manifestation of systemic light chain deposition. Am J Med 1976; 60 : 293 – 299.
2. Heilman RL, Velosa JA, Holley KE et al. Long term follow up and response to chemotherapy in patients with light chain deposition disease. Am J Kidney Dis 1992; 20 : 34 – 41.
3. Pozzi C, Fogazzi GB, Banfi G et al. Renal disease and patient survival in light chain deposition disease. Clin Nephrol 1995; 43 : 281 – 287.
4. Wohl P, Chadimová M, Englis M et al. Light chain deposition disease as a cause of renal failure. Čas Lék Čes 1998; 137 : 721 – 724.
5. Zhu L, Herrera GA, Murphy ‑ Ullrich JE et al. Pathogenesis of glomerulosclerosis in light chain deposition disease. Role for transforming growth factor‑beta. Am J Pathol 1995; 147 : 375 – 385.
6. Bedossa P, Fabre M, Paraf F et al. Light chain deposition disease with liver dysfunction. Hum Pathol 1988; 19 : 1008 – 1014.
7. Girelli CM. Kappa light chain deposition of the liver. Eur J Gastroenterol Hepatol 1998; 10 : 429 – 430.
8. Pelletier G, Fabre M, Attali P et al. Light chain deposition disease presenting with hepatomegaly: an association with amyloid‑like fibrils. Postgrad Med J 1988; 64 : 804 – 808.
9. Kijner CH, Yousem SA. Systemic light chain deposition disease presenting as multiple pulmonary nodules. A case report and review of the literature. Am J Surg Pathol 1988; 12 : 405 – 413.
10. Bhargava P, Rushin JM, Rusnock EJ et al. Pulmonary light chain deposition disease. Report of five cases and review of literature. Am J Surg Pathol 2007; 31 : 267 – 276.
11. Samanez C, Domingo A, Ciberia MT. Development of rapidly progressive liver light chain deposition under VAD chemotherapy in multiple myeloma. Eur J Haematol 2006; 76 : 83 – 85.
12. Lambotte O, Dürrbach A, Ammor M et al. Association of a POEMS syndrome and light chain deposit disease: first case report. Clin Nephrol 2001; 55 : 482 – 486.
13. Merta M, Rysavá R, Zabka J et al. Kidney involvement in light‑chain deposition disease. Sb Lek 2002; 103 : 397 – 403.
14. Lin J, Markowitz GS, Valeri AM et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol 2001; 12 : 1482 – 1492.
15. Preud’homme JL, Aucouturier P, Tou-chard G et al. Monoclonal immunoglobulin deposition disease (Randall type). Relationship with structural abnormalities of immunoglobulin chains. Kidney Int 1994; 46 : 965 – 972.
16. Venkataseshan VS, Faraggiana T, Hughson MD et al. Morphologic variants of light‑chain deposition disease in the kidney. Am J Nephrol 1988; 8 : 272 – 279.
17. Salant DJ, Sanchorawala V, D’Agati VD. A case of atypical light chain deposition disease – diagnosis and treatment. Clin J Am Soc Nephrol 2007; 2 : 858 – 867.
18. Baur A, Stäbler A, Lamerz R et al. Light chain deposition disease in multiple myeloma: MR imaging features correlated with histopathological findings. Skeletal Radiol 1998; 27 : 173 – 176.
19. Ferrario F, Rastaldi MP. Histopathological atlas of renal diseases: light chain deposition disease. J Nephrol 2005; 18 : 499 – 502.
20. Gokden N, Barlogie B, Liapis H. Morphologic heterogeneity of renal light‑chain deposition disease. Ultrastruct Pathol 2008; 32 : 17 – 24.
21. Joh K. Pathology of glomerular deposition diseases. Pathol Int 2007; 57 : 551 – 565.
22. Gu X, Herrera AG. Light chain mediated acute tubular interstitial nephritis: a poorly recognized pattern of renal disease in patients with plasma cell dyscrasia. Arch Pathol Lab Med 2006; 130 : 165 – 169.
23. Hofmann‑Guilaine C, Nochy D, Jacquot C et al. Association light chain deposition disease (LCDD) and amyloidosis. One case. Pathol Res Pract 1985; 180 : 214 – 219.
24. Jacquot C, Saint ‑ Andre JP, Touchard G et al. Association of systemic light‑chain deposition disease and amyloidosis: a report of three patients with renal involvement. Clin Nephrol 1985; 24 : 93 – 98.
25. Okura T, Miyoshi K, Nagao T et al. Light chain deposition disease developing 15 years following the diagnosis of monoclonal gammopathy of undetermined significance. Intern Med 2009; 48 : 101 – 104.
26. Pozzi C, D’Amico M, Fogazzi GB et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factor. Am J Kidney Dis 2003; 42 : 1154 – 1163.
27. Barraclough KA, Dowling JP, Schwarer AP et al. Sequential autologous peripheral blood stem cell transplantation and kidney transplantation of light chain deposition disease. Nephrol Dial Transplant 2007; 22 : 1268 – 1269.
28. Firkin F, Hill PA, Dwyer K et al. Reversal of dialysis dependent renal failure in light chain deposition disease by autologous peripheral blood stem cell transplantation. Am J Kidney Dis 2004; 44 : 551 – 555.
29. Hassoun H, Flombaum C, D’Agati VD et al. High‑dose melphalan and auto ‑ SCT in patients with monoclonal Ig deposition disease. Bone Marrow Transplant 2008; 42 : 405 – 412.
30. Pineda ‑ Roman M, Tricot G. High‑dose therapy in patients with plasma cell dyscrasias and renal dysfunction. Contrib Nephrol 2007; 153 : 182 – 194.
31. Royer B, Arnulf B, Martinez F et al. High dose chemotherapy in light chain or light and heavy chain deposition disease. Kidney Int 2004; 65 : 642 – 648.
32. Sakakima M, Fujigaki Y, Tsuji T et al. High dose chemotherapy and stem cell support in a patient of light ‑ and heavy‑chain deposition disease with abnormal marrow cell surface antigens and no monoclonal protein. Intern Med 2005; 44 : 970 – 974.
33. Wahner ‑ Roedler DL, Kyle R. Heavy chain disease. Best Pract Res Clin Haematol 2005; 18 : 729 – 746.
34. Leung N, Lager DJ, Gertz MA et al. Long‑term outcome of renal transplantation in light‑chain deposition disease. Am J Kidney Dis 2004; 43 : 147 – 153.
35. Leung N, Slezak JM, Bergstralh EJ. Acute renal insufficiency after high dose melphalan in patients with primary systemic amyloidosis during stem cell transplantation. Am J Kidney Dis 2005; 45 : 102 – 111.
36. Ganeval D, Noël LH, Preud’homme JL et al. Light chain deposition disease: Its relation with AL type amyloidosis. Kidney Int 1984; 26 : 1 – 9.
37. Gertz MA, Lacy MQ, Lust JA et al. Phase II trial of high‑dose dexamethasone for untreated patients with primary systemic amyloidosis. Med Oncol 1999; 16 : 104 – 109.
38. Dhodapar MV, Hussein MA, Rasmussen E et al. Clinical efficacy of high dose dexamethasone wit maintenance dexamethasone/ interferon alpha in patients with primary systemic amyloidosis. Blood 2004; 104 : 3520 – 3526.
39. Palladini G, Perfetti V, Obici L et al. Association of melphalan and high dose dexamethasone is effective and well tolerated in patients with AL ‑ amyloidosis, who are ineligible for stem cell transplantation. Blood 2004; 103 : 2936 – 2938.
40. Palladini G, Russso P, Nuvolone M et al. Treatment with oral melphalan plus dexamethasone produces long term remission in AL ‑ amyloidosis. Blood 2007; 110 : 787 – 788.
41. Jaccard A, Moreau P, Leblond V et al. Autologous stem cell transplantation versus oral melphalan and high dose dexametasone in patients with primary AL ‑ amyloidos. Randomized trial. Blood 2005; 106 : 421.
42. Gerz MA, Lacy MQ, Lust JA et al. Phase II trial of high dose dexamethasone for previously treated immunoglobulin light‑chain amyloidosis. Am J Hematol 1999; 61 : 115 – 119.
43. Röcken C, Ernst J, Hund E et al. Interdisciplinary guidelines on the diagnosis of and therapy for systemic amyloidosis. Dtsch Med Wochenschr 2006; 131 (27 Suppl 2): S45 – S66.
44. Comezo RL. Managing systemic light chain amyloidosis. J Natl Compr Canc Netw 2007; 5 : 179 – 187.
45. Lorenz EC, Gertz MA, Fervenza FC et al. Long‑term outcome of autologous stem cell transplantation in light chain deposition disease. Nephrol Dial Transplant 2008; 23 : 2052 – 2057.
46. Palladini G, Perfetti V, Merlini G. Therapy and management of systemic AL ‑ amyloidosis. Swiss Med Wkly 2006; 136 : 715 – 720.
47. Brockhurst I, Harris KP, Chapman CS. Diagnosis and monitoring a case of light‑chain deposition disease in the kidney using a new, sensitive immunoassay. Nephrol Dial Transplant 2005; 20 : 1251 – 1253.
48. Ščudla V, Minařík J, Schneiderka P et al. Význam sérových hladin volných lehkých řetězců imunoglobulinu v diagnostice a hodnocení aktivity mnohočetného myelomu a vybraných monoklonálních gamapatií. Vnitř Lék 2005; 51 : 1249 – 1259.
49. Tichý M, Hrnčíř Z, Urban P et al. Monoklonální imunoglobuliny. Klin Biochem Metabol 2004; 12 : 84 – 87.
50. Doubek M, Brychtova Y, Tomiska M et al. Idiopathic systemic capillary leak syndrome misdiagnosed and treated as polycythemia vera. Acta Haematol 2005; 113 : 150 – 151.
51. Tichý M, Urban P, Matěja P et al. Laboratorní analýza souboru 3049 monoklonálních imunoglobulinů. Klin Biochem Metabol 2002; 10 : 257 – 261.
52. Tichý M. Laboratorní analýza monoklonálních imunoglobulinů (paraproteinů). Český Těšín: Finidr 1997.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 3
Most read in this issue
- Příspěvek k diferenciální diagnostice chronických břišních bolestí
- Pretrvávajúce symptómy, diastolická dysfunkcia a nízka koronárna rezerva u pacientky po úspešnej korekcii rekoarktácie aorty
- Léčba dospělých pacientů s akutní lymfoblastickou leukemií dle protokolu GMALL 07/ 2003 a její výsledky – první zkušenosti v České republice
- Nealkoholová steatóza a steatohepatitida – editorial