
Diabetes mellitus a protrombotická aktivita
Diabetes mellitus and prothrombotic activity
Diabetes mellitus (DM) is defined by significant hyperglycaemia representing a high risk of thrombosis in coronary as well as central and peripheral arteries. The risk of myocardial infarction in patients with type 2 diabetes is 3 – 5 times higher than in non‑diabetics. This is consequent to changes in haemostasis in obese patients with type 2 diabetes, including changes to fibrinolysis, decreased fibrinolytic activity and increased thrombogenic risk, as part of the pluri ‑ metabolic insulin resistance syndrome [1,2]. The REACH study evaluated more than 67 thousands of patients with a risk of arterial thrombosis or with arterial thrombosis. The patients with diabetes were separated and the results subjected to multivariate analysis; differences were confirmed between intensity of treatment in patients with ischemic heart disease and diabetes and with diabetes only. Antithrombotic therapy in diabetic patients with no clinical signs of arterial thrombosis was less intensive [3].
Key words:
diabetes mellitus – arterial thrombosis – insulin resistance thrombosis – prothrombotic activity
Authors:
J. Malý
Authors‘ workplace:
II. interní klinika Lékařské fakulty UK a FN Hradec Králové, přednosta prof. MU Dr. Jaroslav Malý, CSc.
Published in:
Vnitř Lék 2010; 56(4): 284-288
Category:
11th National Diabetes Symposium "Diabetes and Angiology", Hradec Kralove, 5 to 6 June 2009
Overview
Diabetes mellitus (DM) je charakterizován významnou hyperglykemií a vysokým rizikem aterotrombózy v koronárních, centrálních i periferních arteriích. Riziko srdečního infarktu je u diabetiků 2. typu 3 – 5krát vyšší než u nediabetiků. Příčinou jsou změny hemostázy u obézních diabetiků 2. typu, včetně změn fibrinolýzy, snížení fibrinolytické aktivity a zvýšení trombogenního rizika v rámci plurimetabolického syndromu inzulinové rezistence [1,2]. Ve studii REACH bylo vyhodnoceno více než 67 tisíc nemocných s riziky aterotrombózy nebo aterotrombózou. Byla vyčleněna skupina diabetiků a výsledky byly podrobeny multivariační analýze a byly prokázány rozdíly mezi intenzitou léčby nemocných s ischemickou chorobou srdeční a diabetem a pouze diabetem. Intenzita antitrombotické léčby u diabetiků bez klinických projevů aterotrombózy byla nižší [3].
Klíčová slova:
diabetes mellitus – aterotrombóza – syndrom inzulinové rezistence – protrombotická aktivita
Úvod
Diabetes mellitus 2. typu s obezitou ovlivňuje jak složky fibrinolýzy, tak i některé složky koagulačního systému a projevy aktivace destiček a poškození endotelu [4 – 6]. Zvýšení koncentrace PAI-1 je prediktivní faktor infarktu myokardu u nemocných s anginou pectoris. Zvýšení tPa je prognostický faktor pro vznik srdečního infarktu u zdravých osob. Stejně tak je pozitivní korelace mezi hladinou PAI-1 a tPa u nemocných s počínající karotickou stenózou. Všechny tyto epidemiologické studie ukazují, že je úzký vztah mezi sníženými složkami fibrinolytického systému a zvýšeným rizikem arteriálních trombóz a rozvojem vaskulárního postižení [7]. Jiné studie ukazují, že fibrinolytická aktivita, PAI-1 a tPa antigen těsně korelují s četnými komponentami syndromu inzulinové rezistence (IR) (body mass indexem – BMI, poměrem pas-boky, triglyceridy, HDL-cholesterolem, endogenním inzulinem, krevním tlakem atd.) [8,9]. Vzestup PAI-1 koreluje se vzestupem BMI, korekcí hypertenze, hladinou inzulinu, vzestupem triacylglycerolů a poklesem HDL-cholesterolu [10,11]. Diabetes mellitus 2. typu se projevuje protrombogenně i endoteliální dysfunkcí s poklesem syntézy NO a prostacyklinu. DM je provázen poruchami aktivity krevních destiček, poruchy glykace vedou k poruše syntézy fibrinu a poruše funkce fibrinu, který se stává rezistentní na fibrinolýzu. Změny jsou sumarizovány v tab. 1.

Inzulinová rezistence (IR), změny fibrinolýzy a koagulace
Změny fibrinolytických vlastností krve jsou u obézních nemocných s diabetem 2. typu závislé na produkci PAI-1 adipocyty. Metabolický kompartment PAI-1 je tvořen adipocyty a hepatocyty. Hepatocyty produkují jen malé množství PAI-1. Experimentálně se u obézních myší zjistilo, že se až 7krát zvyšuje produkce PAI-1 v adipocytech a jen 2krát v hepatocytech. Hladiny PAI-1 při obezitě a diabetu jsou jednoznačně dány množstvím viscerálního tuku. Lidské kultury omentálních adipocytů tvoří více PAI-1 než podkožní tuková tkáň. Lokální tvorba PAI-1 je důležitá pro regulaci proliferace adipocytů, která je závislá na plazminem aktivované proteolýze. PAI-1 tak nepřímo inhibičně ovlivňuje zvyšování buněčnosti tukové tkáně.
Vztah PAI-1 k tvorbě tukových zásob vysvětluje jeho regulaci různými hormony, růstovými faktory a cytokiny, jako je inzulin, transformační růstový faktor β (TGF β) a TNF α. Inzulin stimuluje tvorbu PAI-1 v adipocytech současně s působením VLDL a volných mastných kyselin. Tuková tkáň je nejdůležitějším zdrojem PAI-1 v závislosti na inzulinu. V experimentu se prokázalo, že aplikace inzulinu zvyšuje expresi mRNK pro PAI-1 více v tuku než v játrech. Ve splanchnickém endotelu se jeho tvorba neindukovala [12].
Adipocyty jsou významným zdrojem lokálních regulátorů množství tuku typu TGF β a TNF aα TGF β je mitogenem pro adipocyty, čímž zvyšuje buněčnost tukové tkáně, zároveň inhibuje jejich diferenciaci na adipocyty. TNF α je naopak fyziologickým adipostatem. Oba faktory indukují tvorbu PAI-1 na úrovni genu, přičemž stimulační účinek TNF α je srovnatelný s inzulinem, zatímco efekt TGF β je několikanásobně vyšší.
Obezita je provázena hyperinzulinemií a zvýšenou koncentrací TNF α v tukové tkáni. TNF α podmiňuje syntézu PAI-1 v kulturách adipocytů. TNF α selektivně zvyšuje syntézu PAI-1 bez současného zvyšování sekrece tPa. Inkubace adipocytů s TNF α nebo inzulinem po 5 dní zvyšuje sekreci PAI-1 [13].
TNF a inhibuje lipoproteinové lipázy a potlačuje diferenciaci adipocytů. Vlivem TNF α dochází k tvorbě kyslíkových radikálů. Hlavním zdrojem tvorby TNF jsou makrofágy v odpovědi na řadu podnětů. TNF α je považován za jednoho z mediátorů inzulinorezistence, a to pro svůj prokázaný vliv na fosforylaci tyrozinu na inzulinovém receptoru, která má za následek zhoršení signalizace po vazbě na inzulin vzhledem k útlumu proteinových kináz. U inzulinorezistence spojené s obezitou byl prokázán vliv solubilního receptoru pro TNF (sTNF-R p55) na substrát inzulinového receptoru IRS-1. Sekundární úlohu u inzulinorezistence při obezitě hraje IL-6. Yudkin [13] shrnuje, že syndrom inzulinové rezistence v sobě neobsahuje pouze klasické rizikové faktory kardiovaskulárních onemocnění, jako je hypertenze a hyperlipoproteinemie, ale je často provázen četnými změnami koagulace a fibrinolýzy. V experimentu je exprese sekrece PAI-1 v hepatocytu a endoteliálních buňkách indukována inzulinem, triglyceridy, oxidovanými LDL a proinzulin like molekulami. U syndromu inzulinové rezistence je zvýšená koncentrace endoteliálního faktoru von Willebrandova, která svědčí o poškození endotelu. Zvýšení fibrinogenu je spojováno se syndromem inzulinové rezistence. Podle posledních pozorování sekrece IL-6 z tukové tkáně, kombinovaná s TNF α u obézních diabetiků indukuje inzulinovou rezistenci a při inzulinové rezistenci dochází k poškození endotelu, koagulopatiím a ischemické chorobě srdeční [14,15].
Galajda et al [6,12] upozornili na to, že hlavní příčinou protrombotického stavu při syndromu IR je hypofibrinolýza při excesu PAI-1. Jde o nejčastější získaný trombofilní stav, který je sledován zvýšeným rizikem arteriální trombózy při AS. Hladiny PAI-1 mají velmi úzký vztah k parametrům IR, ale není zcela jasné, jakým způsobem se tento vztah uplatňuje. Původně navržený model zdůrazňoval klíčovou roli hyperinzulinemie v etipatogenezi hyperfibrinolýzy na základě klinické korelace PAI-1 s inzulinemií a průkazem stimulačního vlivu inzulinu na produkci PAI-1 v hepatocytech in vitro. Galajda et al [12] navrhli jiný model pro vysvětlení hypofibrinolýzy pro syndromu IR. Jde o multikompartmentový a multifaktoriální model, který je v souhlase s názory o komplexnosti etiopatogenezy protrombotického stavu u nemocných s IR a zároveň dovoluje řešit mnohé kontroverzní názory jiných studií. Autoři vyšli z poznatků o polyfunkčnosti plazminového systému, který reguluje nejen fibrinolýzu, ale i růst buněk závislý na pericelulární proteolýze, což má význam při remodelaci cév, ale také v regulaci množství tukové tkáně. Plazmin generovaný vlivem tPa štěpí multimery von Willebrandova faktoru, a moduluje tak i destičkové reakce v podmínkách hemodynamického stresu. Nedávno byla potvrzena významná heterogenita endoteliální produkce tPa, který je tvořen pouze ze 40 % v endotelu, a to ve tkáních citlivých na inzulin. Vzhledem k tomu, že byl prokázán význam viscerálního tuku na řízení koncentrací PAI-1, výrazná produkce tPa ve splanchnickém cévním systému a klíčovou úlohu jater v clearenci komplexů tPa/ PAI-1, je vhodné vyčlenit metabolický kompartment jako zvláštní funkční celek.
PAI-1 je regulátor plazminem aktivované proteolýzy. Metabolický kompartment zahrnuje viscerální adipocyty i hepatocyty a je citlivý na stimulační působení inzulinu, proinzulinu a některých cytokinů. Reguluje postprandiální změny fibrinolýzy důležité pro proliferaci adipocytů a zmnožení tukové tkáně. Tento kompartment je odpovědný za vzestup PAI-1 při obezitě. Vaskulární kompartment produkce PAI-1 reguluje nejen fibrinolýzu, ale i aktivitu destiček v podmínkách hemodynamického stresu. PAI-1 má zásadní význam pro regulaci proliferace endotelu, vyžadující proteolytický fenotyp zprostředkovaný plazminem. Syntéza PAI-1 v endotelu je stimulovaná trombinem, angiotenzinem IV, cytokiny, biologicky aktivními lipidy a oxidačním stresem, zatímco inzulin inhibuje cytokiny indukovanou tvorbu PAI-1. Výsledná hladina PA-1, a tím i výsledná úroveň fibrinolýzy při syndromu IR, je průnikem produkce ve všech kompartmentech.
U nemocných s obezitou a syndromem IR korelují hladiny PAI-1 s inzulinemií a BMI, hlavním stimulačním momentem je zřejmě ovlivnění tvorby PAI-1 v adipocytech pomocí inzulinu. Cévní původ PAI-1 lze prokázat pomocí aktivit endotelových markerů, jako je např. von Willebrandův faktor. Podrobnější rozbor původu zvýšení PAI-1 má klinické důsledky. U nemocných s obezitou, kde pochází PAI-1 z adipocytů, je regulace snížené fibrinolýzy možná redukcí hmotnosti. U nemocných, kde snížená fibrinolýza má endoteliální původ, je třeba komplexnější léčebné ovlivnění funkcí endotelu [16].
Kvasnička a Škrha [4,5] poprvé v České republice popsali změny fibrinolytické aktivity u nemocných s NIDDM a soudili na spoluúčast syndromu IR u těchto nemocných. Kvasnička našel souvislosti mezi zánětlivými ukazateli a fibrinolytickými změnami u IR. Samad a Loskutoff [17] poukázali na to, že u obézních myší je zvýšená prokoagulační aktivita a snížený fibrinolytický potenciál. Zvýšení PAI-1 u geneticky obézních myší bylo způsobeno elevací TNF α, inzulinu a TGF β (transformační růstový faktor β). Vliv těchto působků je směrován především do stimulace tvorby PAI-1 v adipocytu. TGF β, TNF α a inzulin provokují tvorbu PAI-1 v kultuře adipocytů. Metabolizmus tukové tkáně je charakterizován vzájemnými interakcemi mezi zrajícími adipocyty a buňkami stromatu, včetně endoteliálních buněk, hladkých svalových buněk, makrofágů, mezoteliálních buněk a prekurzorů adipocytů. Tukové buňky mají sekreční úlohu. Sekrece může být parakrinní, autokrinní nebo juxtakrinní. Substance uvolňované z tukové tkáně mají vliv na krevní tok i angiogenezu a kontrolují proliferaci a diferenciaci prekurzorů adipocytů.
Fibrinogen u diabetu 2. typu bývá změněn. Předpokládá se, že fibrinogen musí být molekulárně pozměněn tak, aby abnormální struktura fibrinogenu vytvářela abnormální podmínky pro strukturu koagula a zvyšovala riziko trombózy.
Změny struktury fibrinogenu se týkají především změny α a γ řetězce. Změny jsou geneticky podmíněné. Rodiny s mutací struktury fibrinogenu mají vysokou incidenci ischemické choroby srdeční v mladém věku a četný výskyt familiární tromboembolické nemoci. Amiral [6] prokázal, že při mutaci γ řetězce (554 Arg.Cyst) se vytvářejí velmi tenká fibrinová vlákna a že takto změněný fibrin není aktivní k plazminogenu a nemá volné fibrinové konce. Existuje pozitivní korelace mezi hodnotou fibrinogenu v plazmě a kontrolami glykemie, pokles glykemie, resp. udržování glykemie při léčbě diabetu vede k poklesu koncentrace fibrinogenu. Není to zřejmé při léčbě inzulinem, ale významné při léčbě metforminem [18 – 20].
Gentile [21] prokázal, že vyšší koncentrace F VIIa u diabetiků s náhlou cévní příhodou mozkovou má prognostický význam a že hodnoty F VIIa u nediabetiků se stejným cévním postižením jsou nižší. Hodnoty F VIIa korelovaly s kontrolami glykemie, a tedy s kompenzací diabetu.
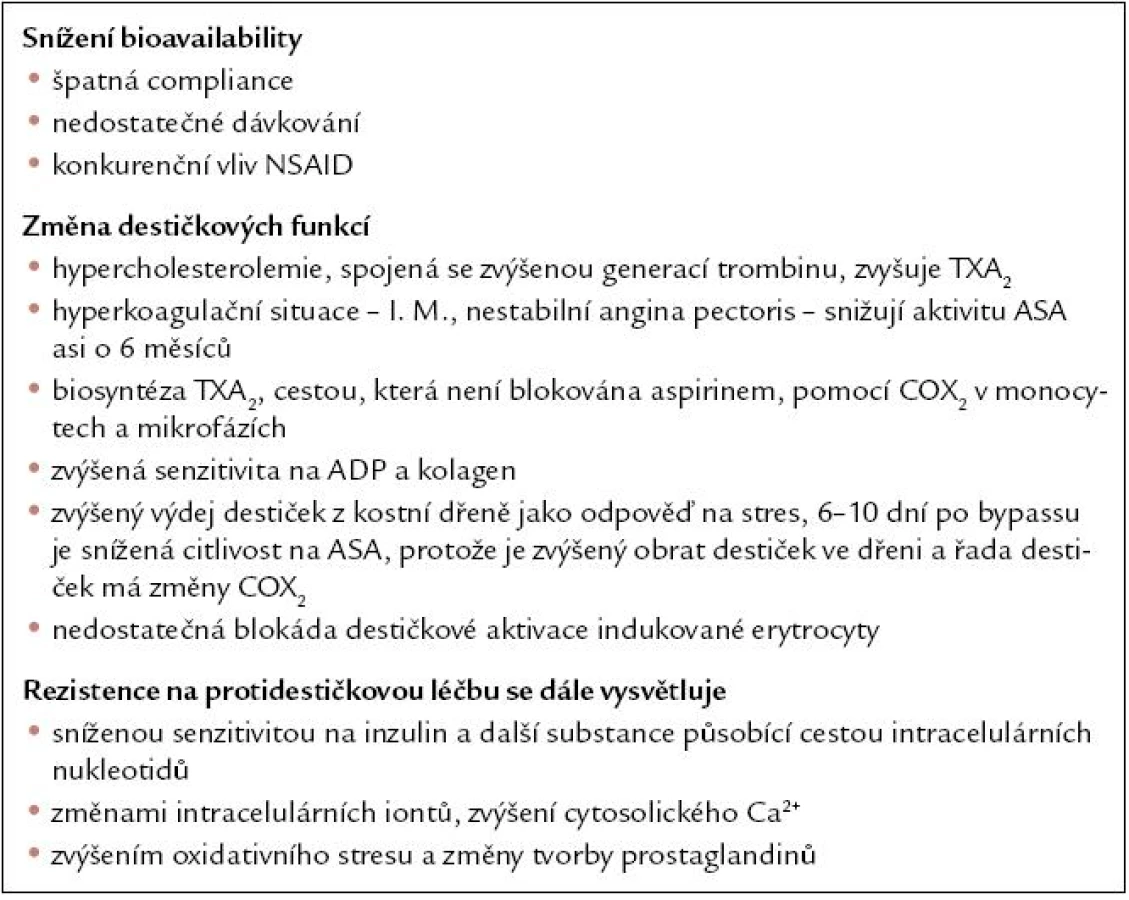
Inzulinová rezistence a změny funkce trombocytů
Ranieri [22] prokázal, že krevní destičky u obézních mužů mají větší střední objem (MPV). Zvětšení MPV je pokládán za projev aktivace destičkových funkcí a provází kardiovaskulární onemocnění, jako je náhlá cévní příhoda mozková, srdeční infarkt, ale i diabetes mellitus [23]. U nemocných s diabetes mellitus byla prokázána snížená propustnost destičkové membrány, změny homeostázy Ca2+ a Mg2+ (zvýšení intracelulárního Ca2+, mobilizace a snížení intracelulárního Mg2+), zvýšení metabolizmu arachidonové kyseliny, zvýšení syntézy TXA2, snížení tvorby prostacyklinu, snížení tvorby NO, snížení tvorby antioxidantů, zvýšení exprese na aktivací závislých adhesních molekul (GpIIb-IIIa, P-selektin).
V řadě studií bylo zkoumáno, zda získané změny krevních destiček, které jsou komplementární součástí zvýšené mortality na kardiovaskulární choroby u diabetiků, jsou důvodem ke změně taktiky protidestičkové léčby [24]. Bylo prokázáno, že GpIIb-IIIa blokátory významně snižují 30denní mortalitu z 6,2 na 4,6 % (p = 0,007) u diabetiků. U 22 tisíc pacientů ve studiích s GP IIb-IIIa blokátory neměly vliv na přežití nemocných, pokud nešlo o diabetiky. Vliv GpIIb-IIIa inhibitorů u diabetiků byl významnější. 1 279 pacientů, kteří měli PTCA GpIIb-IIIa inhibitory, redukovaly 30denní mortalitu z 4,0 na 1,2 % [25].
Hirsh a Bhatt [26] v retrospektivní analýze diabetické podskupiny ve studii CAPRIE prokázali, že při srovnání 1 914 diabetiků s clopidogrelem vs 1 952 diabetiků s kyselinou acetylsalicylovou (ASA) je ve skupině diabetiků s clopidogrelem nižší výskyt vaskulárních komplikací (15,6 vs 17,7 vaskulárních komplikací – p = 0,042). Clopidogrel byl více efektivní v sekundární prevenci u diabetiků, ale ASA je levnější. Angiolillo [27] zkoumal vliv inhibiční efekt receptoru P2Y na destičkovou aktivitu po podání clopidogrelu u diabetiků 2. typu. Prokázal, že nemocní se suboptimální odpovědí na clopidogrel mají změny destičkové prokoagulační aktivity.
V minulosti jsme prokázali také zvýšení cytosolického Ca u nemocných s diabetes mellitus 2. typu. Zvýšení cytosolického Ca vede ke zvýšené aktivitě krevních destiček, zvýšení destičkové agregability [28,29]. Nemocní s inzulinovou rezistencí mají zvýšenou hladinu CAMP, což u obézních vede ke zvýšené rezistenci destiček na protidestičkovou léčbu.
Trombocyty nemocných s diabetem a syndromem inzulinové rezistence vykazují zřetelné známky aktivity. Pacienti s diabetem mají častěji popisovanou rezistenci na protidestičkovou léčbu, a to jak na ASA, tak na clopidogrel [30,31].
Do mechanizmu vzniku rezistence na protidestičkovou léčbu u diabetiků zasahuje řada podnětů. Bylo prokázáno, že hyperlipoproteinemie vede ke zvýšenému počtu destičkových agregátů a ke zvýšení syntézy tromboxanu A2. Dochází ke zvýšení tvorby trombinu, a tím klesá antitrombotický efekt ASA a clopidogrelu [32,33].
Nabízí se otázka, zda nemocní s diabetem by neměli mít protidestičkovou léčbu v primární profylaxi [24]. Primární profylaxe by byla zřejmě výhodná u nemocných:
- s pozitivní rodinnou anamnézou ischemické nemoci srdeční
- kuřáků cigaret
- hypertoniků
- u osob s hmotností > 120 % nad ideální hmotnost
- diabetiků s mikroalbuminurií či makroalbuminurií
- vysokým cholesterolem, vysokým LDL-cholesterolem, nízkým HDL-cholesterolem, vysokými triacylglyceroly
Hyperglykemie, poruchy glykace a rizika trombózy
Jak diabetes 1. typu, tak diabetes 2. typu jsou charakterizovány kolísající glykemií a změnami glykace proteinů. Poruchy glykace proteinů vyvolávají vyšší koncentrace cirkulujícího tkáňového faktoru. Změny glykace u nemocných se špatnou kompenzací diabetu vyvolávají zřejmě i poruchy syntézy fibrinogenu, pokles koncentrace plazminogenu a t-Pa vážících se na fibrin, a tím i nízkou koncentraci plazminu tvořeného na povrchu destiček. Glykace fibrinogenu vede k tvorbě fibrinových vláken rezistentních na plazmin [34].
Podpořeno výzkumným záměrem MZO 00179906.
prof. MUDr. Jaroslav Malý, CSc.
www.fnhk.cz
e-mail: maly@fnhk.cz
Doručeno do redakce: 25. 1. 2010
Sources
1. Juhan ‑ Vague I, Alessi MC. PAI ‑ l, obesity, insulin resistance and risk of cardiovascular events. Thromb Haemost 1997; 78 : 656 – 661.
2. Juhan ‑ Vague I, Morange P, Renucci JF et al. Fibrinogen, obesity and insulin resistance. Blood Coagul Fibrinolysis 1999; 10 (Suppl 1): S25 – S28.
3. Parhofer KG, Zeymer U, Stark RG et al. In germany diabetic patients with coronary artery disease are treated more intensively than diabetic patients with other manifestations of atherothrombosis – results from the REACH registry. Exp Clin Endocrinol Diabetes 2010;118 : 51 – 56.
4. Škrha J, Hodinář A, Kvasnička J et al. Relationship of oxidative stress and fibrinolysis in diabetes mellitus. Diabet Med 1996; 13 : 800 – 805.
5. Kvasnička J, Škrha J, Perušičová J et al. The occurrence of the cardiovascular risk factors – fibrinogen, t ‑ PA, PAI ‑ l, and inflammation in insulin‑dependent diabetes mellitus (IDDM) and in non‑insulin‑dependent diabetes mellitus (NIDDM). Cor Vasa 1997; 39 : 146 – 150.
6. Galajda P, Martinka E, Kubisz P et al. Hemostáza u chorých s diabetes mellitus. Markery dysfunkce endotelu. Vnitř Lék 1996; 42 : 676 – 679.
7. Amiral J. Markers of prethrombotic/ hypercoagulable states of blood and vascular cell origins. In: Hemostasis and Thrombosis: Multicellular processes. Paris: Stago 1996.
8. De Lorenzo F, Mukherjee M, Karziola Z et al. Association of overall adiposity rather than body mass index with lipids and procoagulant factors. Thromb Haemost 1998; 80 : 603 – 606.
9. De Pergola G, De Mitrio V, Giorgino F et al. Increase in both pro‑thrombotic and anti‑thrombotic factors in obese premenopausal women: relationship with body fat distribution. Int J Obes Relat Metab Disord 1997; 21 : 527 – 535.
10. De Pergola G, De Mitrio V, Sciaraffia Met al. Lower androgenicity is associated with higher plasma levels of prothrombotic factors Irrespective of age, obesity, body fat distribution, and related metabolic parameters in men. Metabolism 1997; 46 : 1287 – 1293.
11. Fendri S, Roussel B, Lormeau B et al. Insulin sensitivity, insulin action, and fibrinolysis activity in non diabetic and diabetic obese subjects. Metabolism 1998; 47 : 1372 – 1375.
12. Galajda P, Kubisz P, Mokáň M. Multikompartmentový a multifaktoriálny model produkcie inhibítora plazminogénového aktivátora (PAI‑1). Vnitř Lék 1998; 44 : 718 – 721.
13. Yudkin JS. Abnormalities of coagulation and fibrinolysis in insulin resistance. Evidence for a common antecedent? Diabetes Care 1999; 22 (Suppl 3): C25 – C30.
14. Osende J, Badimon J, Fuster V et al. Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control. J Am Coll Cardiol 2001; 38 : 1307 – 1312.
15. Trovati M, Anfossi G. Influence of insulin and insulin resistance on platelet and vascular smooth muscle cell function. J Diabetes Complications 2002; 16 : 35 – 40.
16. Mokán M, Galajda P, Prídavková D et al. Prevalence of diabetes mellitus and metabolic syndrome in Slovakia. Diabetes Res Clin Pract 2008; 81 : 238 – 242.
17. Samad F, Loskutoff DJ. Hemostatis gene expression and vasculat disease in obesity: insights from studies of genetically obese mice. Thromb Haemostas 1999; 82 : 742 – 747.
18. Kvasnička J, Škrha J, Perušičová J et al. Koncentrace tkáňového aktivátoru plazminogenu (T ‑ PA), jeho inhibitoru (PAI ‑ l) a fibrinogenu v krevní plazmě pacientů s diabetes mellitus 1. a 2. typu. Čas Lék Čes 1996; 135 : 174 – 177.
19. Dunn E, Ariëns R, Grant P. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 2005; 48 : 1198 – 1209.
20. Grant PJ. Beneficial effects of metformin on haemostasis and vascular function in man. Diabetes Metab 2003; 29 : 6S44 – 6S52.
21. Gentile NT, Vaidyula VR, Kanamalla U et al. Factor VIIa and tissue factor procoagulant activity in diabetes mellitus after acute ischemic stroke: impact of hyperglycemia. Thromb Haemost 2007; 98 : 1007 – 1013.
22. Ranieri R et al. Mean platelet volume in obesity its relation to visceral fat and to cardiovascular risk factor. Int J Obesity 1996; 20 (Suppl 4): 115S.
23. Chu S, Becker RC, Berger PB et al. Mean platelet volume as a predictor of cardiovascular risk: a systematic review and meta‑analysis. J Thromb Haemost 2010; 8 : 148 – 156.
24. Vinik A, Erbas T, Park T et al. Platelet dysfunction in type II diabetes. Diabetes Care 2001; 24 : 1476 – 1485.
25. Grant PJ. Diabetes mellitus as a prothrombotic condition. J Intern Med 2007; 262 : 157 – 172.
26. Hirsh J, Bhatt DL. Comparative benefits of clopidogrel and aspirin in high‑risk patient populations: lessons from the CAPRIE and CURE studies. Arch Intern Med 2004; 164 : 2106 – 2110.
27. Angiolillo DJ, Suryadevara S. Aspirin and clopidogrel: efficacy and resistance in diabetes mellitus. Best Pract Res Clin Endocrinol Metab 2009; 23 : 375 – 388.
28. Malý J, Pecka M, Pleskot M et al. Změny aktivity destiček při invazivních kardiologických výkonech. Vnitř Lék 1996; 42 : 314 – 319.
29. Baldi S, Natali A, Buzzigoli G et al. In vitro effect of insulin on intracellular calcium concentrations: relation to insulin resistance. Metabolism 1996; 45 : 1402 – 1407.
30. Anfossi G, Russo I, Trovati M. Resistance to aspirin and thienopyridines in diabetes mellitus and metabolic syndrome. Curr Vasc Pharmacol 2008; 6 : 313 – 328.
31. Anfossi G, Mularoni E, Burzacca S et al. Platelet resistance to nitrates in obese NIDDM, and normal platelet sensitivity to both insulin and nitrates in lean NIDDM. Diabetes Care 1998; 21 : 121 – 126.
32. Betteridge D, El Tahir K, Reckless J et al. Platelets from diabetic subjects show diminished sensitivity to prostacycline. Eur J Clin Invest 1982; 12 : 395 – 398.
33. Westerbacka J, Yki ‑ Järvinen H, Turpeinen A et al. Inhibition of platelet ‑ collagen interaction. An in vivo action of insulin is abolished by insulin resistance in obesity. Arterioscler Thromb Vasc Biol 2002; 22 : 167 – 172.
34. Enomoto M, Adachi H, Yamagishi SI et al. Positive association of serum advanced glycation end products with thrombogenic markers in humans. Metabolism 2006; 55 : 912 – 917.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 4
Most read in this issue
- Angiopatie a oko
- Ischemická choroba dolních končetin a diabetes
- Chirurgická léčba ischemické choroby srdeční a diabetes mellitus
- Diabetes mellitus a ischemická choroba srdeční