
Skrytá cystická fibróza u nemocného se sarkoidózou
Hidden cystic fibrosis in patient suffering from sarcoidosis
We describe a case of an adult patient, whom sarcoidosis was diagnosed at first and cystic fibrosis was also discovered 2 years later on. Cystic fibrosis and sarcoidosis are uncommon diseases that only rarely occur together. On the other hand, the coincidence of sarcoidosis and cystic fibrosis is possible, and, moreover, one of the diseases can remain undiagnosed long time. Relationship between sarcoidosis and cystic fibrosis seems not to be probable, in accordance with recent medical reports, and the coincidence of both conditions appears as sporadic.
Key words:
sarcoidosis – cystic fibrosis – CFTR gene – diagnosis
Authors:
M. Doubková; I. Binková; J. Skřičková
Authors‘ workplace:
Klinika nemocí plicních a tuberkulózy Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednostka prof. MUDr. Jana Skřičková, CSc.
Published in:
Vnitř Lék 2012; 58(4): 329-334
Category:
Case Reports
Overview
Popisujeme případ dospělého pacienta, u něhož byla diagnostikována sarkoidóza, a až po dalších 2 letech byla u téhož pacienta zjištěna i atypická varianta cystické fibrózy. Cystická fibróza a sarkoidóza jsou méně běžné nemoci, které se jen vzácně vyskytují společně, což dokazuje i omezený počet publikovaných případů koincidence těchto nemocí v zahraniční literatuře. Na druhou stranu koincidence je možná, a navíc jedna z těchto nemocí může zůstat dlouho nerozpoznána. Možná souvislost mezi oběma nemocemi se podle dostupných informací zdá méně pravděpodobná a společný výskyt obou nemocí se jeví jako náhodný.
Klíčová slova:
sarkoidóza – cystická fibróza – CFTR gen – diagnóza
Úvod
Sarkoidóza je méně běžné granulomatózní onemocnění, které patří do skupiny intersticiálních plicních procesů (IPP) neboli difuzních parenchymatózních onemocnění plic čítajících přibližně 200 nosologických jednotek [1,2]. Sarkoidóza je systémové onemocnění neznámé etiologie s plicními a mimoplicními projevy. Historie poznávání nemoci spadá do 19. století [3]. Název sarkoidóza pochází ze složeniny 2 slov – „sark“ a „oid“, což znamená „podobný masu“, a název pravděpodobně vychází z prvních histopatologických popisů nemoci [4]. Toto onemocnění patří mezi záhady medicíny nejen nedostatečně poznanou etiologií, ale i pestrostí klinických příznaků.
Sarkoidóza je granulomatózní systémový zánětlivý proces s celosvětovým výskytem a postihuje jedince všech etnik a věkových skupin. Sarkoidóza patří k IPP neznámé etiologie. Zvažován je infekční původ nemoci, zejména mykobakterie, propionibakterie a jiné, jakož i genetická predispozice (variabilita v oblasti genu pro molekuly hlavního histokompatibilního systému HLA I. a II. třídy). Je velmi pravděpodobné, že nemoc se vyvíjí po expozici specifickým antigenům zevního prostředí u geneticky predisponovaných jedinců. Epidemiologická data jsou nepřesná, protože sarkoidóza může probíhat až u 1/3 pacientů zcela asymptomaticky nebo může dojít v závislosti na stadiu nemoci ke spontánnímu vyléčení [5]. V ČR dosahuje incidence kolem 3,1/100 000, prevalence všech forem je 63,1/100 000 [6]. Onemocnění převážně postihuje ženy a poměr muži : ženy je 1 : 2,35 [5]. Mortalita je nízká a udává se mezi 3 a 4 % [5].
Deskriptivní definice sarkoidózy zní: sarkoidóza je systémové onemocnění neznámé příčiny. Obvykle postihuje dospělé pacienty mladého a středního věku a často se projevuje jako oboustranná hilová lymfadenopatie, plicní infiltrace, oční a kožní léze. Postiženy mohou být také játra, slezina, lymfatické uzliny, slinné žlázy, srdce, nervový systém, svaly, kosti a jiné orgány. Diagnózu sarkoidózy je možné stanovit, když klinické a radiologické nálezy podpoří průkaz nekazeifikujícího granulomu. Granulomy jiných známých příčin a lokální sarkoidní reakce mají být vyloučeny (prohlášení expertů American Thoracic Society – ATS, European Respiratory Society – ERS, World Association of Sarcoidosis and Other Granulomatous – WASOG o sarkoidóze z roku 1999) [1,2]. Diagnóza sarkoidózy může být pravděpodobná při nepřítomnosti histologického ověření, pokud klinika, nálezy radiologické a nálezy při vyšetření bronchoalveolární tekutiny korelují s diagnózou sarkoidózy [7].
Morfologická definice sarkoidózy zní: nemoc je charakterizována granulomy z epiteloidních buněk v různých orgánech a tkáních, které vznikají akumulací T-lymfocytů a transformovaných makrofágů. V granulomech nedochází ke kazeifikaci, ačkoli fibrinoidní nekróza může být v jejich centru přítomna. Granulomy vedou k úplné rezoluci nebo k hyalinní fibróze [1,2].
Cystická fibróza (CF), ve starším pojmenování mukoviscidóza, je vrozené autozomálně recesivní dědičné, multisystémové a letální onemocnění. CF je v současné době sice stále nevyléčitelné, ale léčitelné onemocnění. Se zlepšením terapeutických možností se prodlužuje délka života nemocných s CF. Medián přežití je 33–35 let [8,9]. CF postihuje orgány s epiteliálním povrchem. Je charakterizována postupně se zhoršujícím postižením plic z důvodu opakovaných infekcí dýchacích cest nebo chronické infekce dýchacích cest, vysokou koncentrací chloridů v potu, celkovým neprospíváním při insuficienci zevní sekrece pankreatu a až u 98 % mužů neplodností při obstruktivní azoospermii. Etiopatogeneze nemoci, na rozdíl od sarkoidózy, je známa. Jde o mutaci CFTR genu (cystic fibrosis transmembrane conductance regulator) [10–12] objevenou v roce 1989. Lokalizace genu je na dlouhém raménku chromozomu 7 (7q). Bílkovinný produkt genu je chloridový kanál v apikální membráně epiteliálních buněk. U cystické fibrózy je neprůchodný pro chloridové ionty. Celkem je dosud známo více než 1 902 mutací, které se dělí do tříd podle závažnosti, s jakou se manifestují. Mutace I., II. a III. třídy jsou takzvané „těžké, závažné“ mutace, charakterizované minimální aktivitou a nepřítomností proteinu CFTR. Jsou spojeny s klasickou formou CF, zvýšením koncentrace chloridů v potu, nedostatečnou zevní sekrecí pankreatu, postupně se zhoršujícím sinopulmonálním onemocněním a mužskou neplodností. Mutace IV. a V. třídy jsou takzvané „mírné“ mutace, jsou spojeny s vyšší variabilitou klinického průběhu oproti třídám I–III a signifikantně častěji se nacházejí u atypických, monosymptomatických forem CF [8,9]. Pokud je u nemocného přítomna alespoň jedna z mírných mutací, pak nemusí být přítomna nedostatečnost zevní sekrece pankreatu a koncentrace chloridů v potu může být jen hraniční. U těchto pacientů bývá zjišťován pozdní začátek nemoci a méně závažné projevy postižení dýchacího ústrojí [8]. Molekulárně genetické vyšetření potvrdí diagnózu CF nalezením 2 mutací [8,13,14]. Nejčastější z nich, F508del, je přítomna na 68 % chromozomů českých nemocných s CF. CF postihuje stejně obě pohlaví a poměr nemocných na živě narozené je 1 : 2 500–4 500. V České republice se narodí s CF každý rok 28–40 dětí. Diagnostikována je necelá polovina [8,15]. Každý 25.–30. z nás je nosičem. Každé 676. manželství je manželstvím 2 nosičů genu CFTR. V současné době je diagnostikováno v České republice do 500 nemocných [15]. Předpokládaný medián přežití dětí narozených v 21. století je více než 50 let [13].
Diagnostická kritéria pro cystickou fibrózu jsou tato: 1 a více projevů onemocnění, které zahrnují respirační příznaky, gastrointestinální příznaky, poruchy výživy, syndrom ztráty solí + + pozitivní rodinná anamnéza a pozitivní novorozenecký screening, současně 2 a více pozitivních potních testů, 2 „klasické“ mutace CFTR genu [8,13–19]. CF nemůžeme vyloučit ani tehdy, pokud u nemocného s charakteristickými klinickými příznaky a 2 mutacemi typickými pro CF prokážeme jen hraniční potní test. Rovněž nemůžeme CF vyloučit u nemocného, který má klinické příznaky spojené s CF, má pozitivní potní test, ale nenajdeme u něj žádnou z mutací typických pro CF. V současné době se vyšetřuje standardně 30–40 CFTR mutací [8,13–19].
Cystická fibróza a sarkoidóza jsou onemocnění, která se jen vzácně vyskytují společně. Pestrost projevů může v některých případech vést k tomu, že v terénu sarkoidózy jsou přehlédnuta jiná onemocnění. Naší kazuistikou chceme demonstrovat možnost současného výskytu dlouhodobě nediagnostikované cystické fibrózy u nemocného se sarkoidózou.
Popis případu
Muž, 28 let, bývalý silný kuřák, byl odeslán k plicnímu vyšetření pro 1/2 roku trvající dušnost při námaze, kašel a patologický RTG nález hrudníku. Pracovní anamnéza prokázala expozici prachu oxidu křemičitého (5 let byl řezač skla) a expozici při výrobě plastů (3,5 roku). Jeden z bratrů nemocného zemřel v 50 letech z neznámých příčin, starší bratr a matka pacienta mají problémy se slinivkou břišní. Fyzikálním vyšetřením bylo zjištěno dýchání alveolární, bez vedlejších fenoménů. Při celkové inspekci nebyla nalezena patologie. Nebyly přítomny paličkovité prsty. Plicním funkčním vyšetřením byla potvrzena kombinovaná ventilační porucha těžkého stupně [FVC – forsírovaná plicní kapacita byla 2,56 (53 %), FEV1 – jednosekundová plicní kapacita je 1,75 (42 %), IVC – inspirační vitální kapacita 2,73 (54 %), TLC – celková plicní kapacita 4,56 (68 %), FEV1/FVC 68 %], těžká hyperinflace plicního parenchymu [RV – reziduální objem 3,40 (211 %) a poměr RV/TLC – reziduální objem/celková plicní kapacita je 55] a lehká porucha plicní difuze (DLCO – difuzní plicní kapacita je 77 %, DL/VA – difuzní koeficient byl 114 %) s lehkou parciální respirační nedostatečností (pO2 – parciální tlak kyslíku 8,93 kPa, pCO2 – parciální tlak oxidu uhličitého 5,20). Na vstupním RTG snímku hrudníku byly patrné nehomogenní splývavé infiltráty oboustranně navazující na plicní hilus (obr. 1).

Laboratorní vyšetření prokázala zvýšené CIK (cirkulující imunokomplexy), slabě pozitivní ANA (antinukleární protilátky) a silně pozitivní anti-dsDNA. Byla zjištěna výrazně zvýšená hodnota ACE v séru (angiotenzin konvertující enzym) 188 U/l (normální hodnoty 12–68 U/l). Vyšetřením kalciového metabolizmu byla zjištěna mírná hyperkalcemie 2,60 mmol/l (norma 2,15–2,55 mmol/l) a hyperkalciurie 10,6 mmol/24 hod (norma 2,4–7,5 mmol/24 hod). Byly zjištěny lehce zvýšené jaterní testy, a to ALT 1,60 µkat/l (alkalická transamináza, norma 0,17–0,60 µkat/l) a GGT 1,21 µkat/l (gama glutamyltransferáza). Krevní obraz a diferenciální rozpočet včetně eozinofilů byl v normě. Průkaz Aspegillového antigenu v séru a candidového antigenu v séru metodou ELISA (Enzyme-Linked ImmunoSorbent Essay) byl negativní. Vzhledem k RTG plicním nálezům bylo vysloveno podezření na intersticiální plicní proces a v diferenciální diagnostice byla zvažována silikóza, sarkoidóza, systémové postižení pojiva. Infekční a alergická příčina nebyla zjištěna. Bylo indikováno HRCT hrudníku (výpočetní tomografie s vysokou rozlišovací schopností, hight resolution computed tomography). Na HRCT hrudníku byla popisována oboustranně v plicním parenchymu výrazně porušená architektonika plicní tkáně s maximem nálezu v horních lalocích, nodulární zesílení intersticia v peribronchovaskulární, ale i intralobární distribuci, retikulární zesílení v oblasti pravého hrotu. Změny byly lokalizovány subpleurálně, v S1/2 vlevo pod ventrální stěnou hrudní. V horních lalocích byly popisovány okrsky výrazné kondenzace až infiltrace plicního parenchymu s negativním bronchogramem, dorsobazálně oboustranně s dutinovými projasněními (kavitace) do velikosti 2 cm. V dolních lalocích byly zaznamenány rozsáhlé infiltráty s negativním bronchogramem, pleura byla zde zesílená a povytažená. Pohrudniční dutiny byly bez výpotků. V mediastinu byly četné lymfatické uzliny velikosti 2 cm (obr. 2).
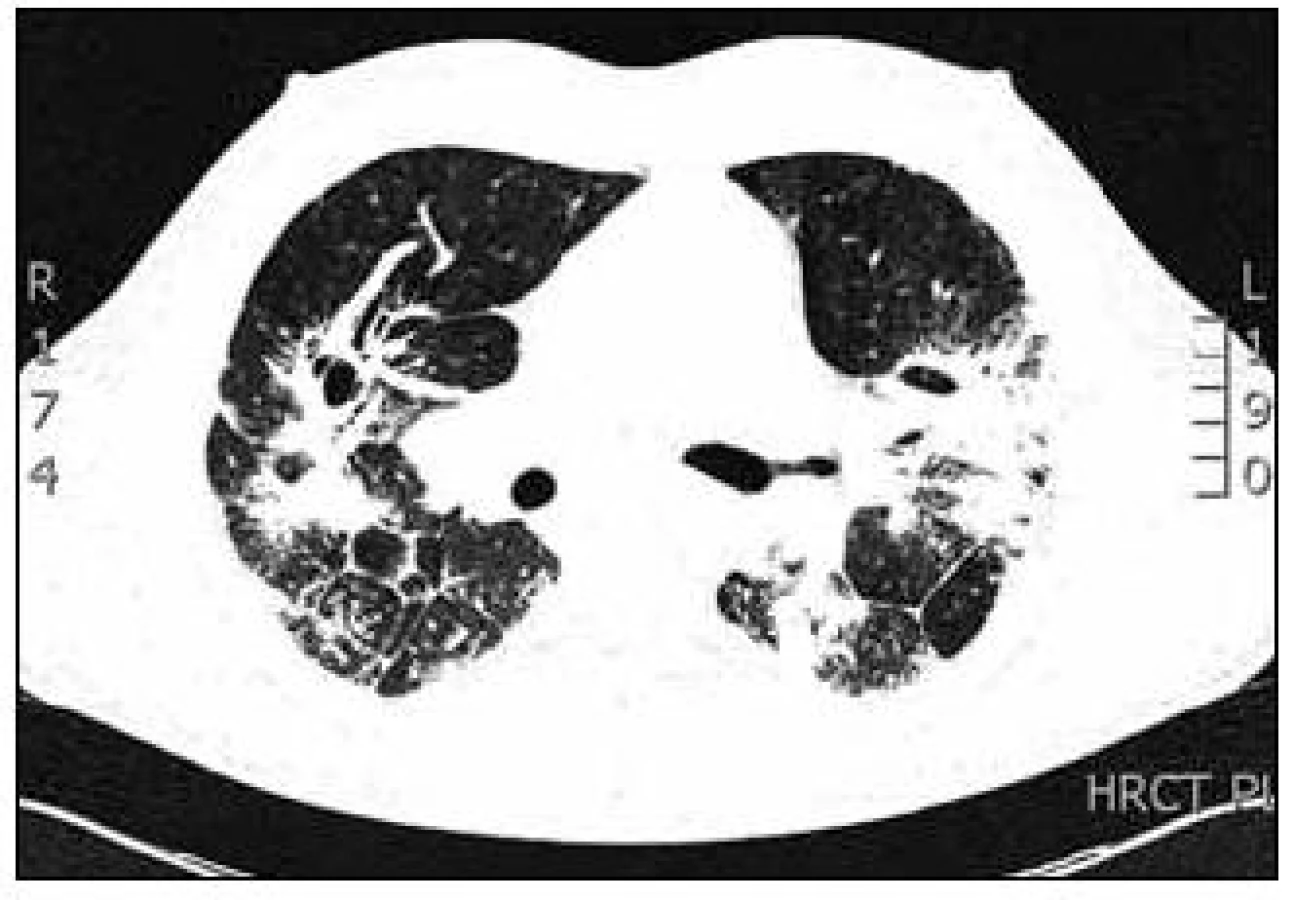
Na základě závěru HRCT hrudníku byl na prvním místě zvažován granulomatózní proces včetně sarkoidózy, silikózy či kolagenózy s nasedajícími zánětlivými změnami. Bronchoskopicky byly zjištěny difuzně známky zánětu v bronchech. Vyšetřením bronchoalveolární tekutiny (BALT) byla zjištěna neutrofilní alveolitida, makrofágy 75 %, neutrofily 14,2 %, lymfocyty 9,7 % a eozinofily 0,8 %. Imunoregulační index (IRI, poměr subpopulací T-lymfocytů CD4+ a CD8+) byl 1,788. Mikrobiologické vyšetření bronchoalveolární tekutiny bylo negativní. Transbronchiální plicní biopsií ze středního a horního pravého bronchu byla potvrzena granulomatózní reakce a v diferenciální diagnostice zvažována sarkoidóza, ale i silikóza nebo jiná onemocnění s granulomatózní komponentou. Na základě těchto výsledků bylo doporučeno provedení videoasistované torakoskopie (VATS) s odběrem vzorku plicní tkáně v celkové anestezii, kterou pacient podstoupil v únoru roku 2008. Histologicky excizí z horního laloku levé plíce a dolního laloku levé plíce byl potvrzen granulomatózní zánět mající morfologické znaky sarkoidózy. Vyšetřeními nebylo zjištěno jiné mimoplicní postižení sarkoidózou kromě hyperkalcemie, hyperkalciurie a lehce elevovaných jaterních testů bez prokázané patologie na ultrazvuku břicha. Na základě korelace výše uvedených výsledků byla diagnóza uzavřena jako sarkoidóza II. stadia a v dubnu roku 2008 byla zahájena systémová léčba glukokortikoidy, prednisonem v dávce 40 mg denně s postupným snižováním na dlouhodobou udržovací dávku. Pro obstrukční plicní funkční poruchu byla pacientovi nasazena inhalační bronchodilatancia (anticholinergika a β2-mimetika). Po 1/2 roce léčby byla vidět na zadopředním snímku hrudníku (obr. 3) a kontrolním CT a HRCT hrudníku částečná regrese intersticiálních změn a mediastinální lymfadenopatie (obr. 4).
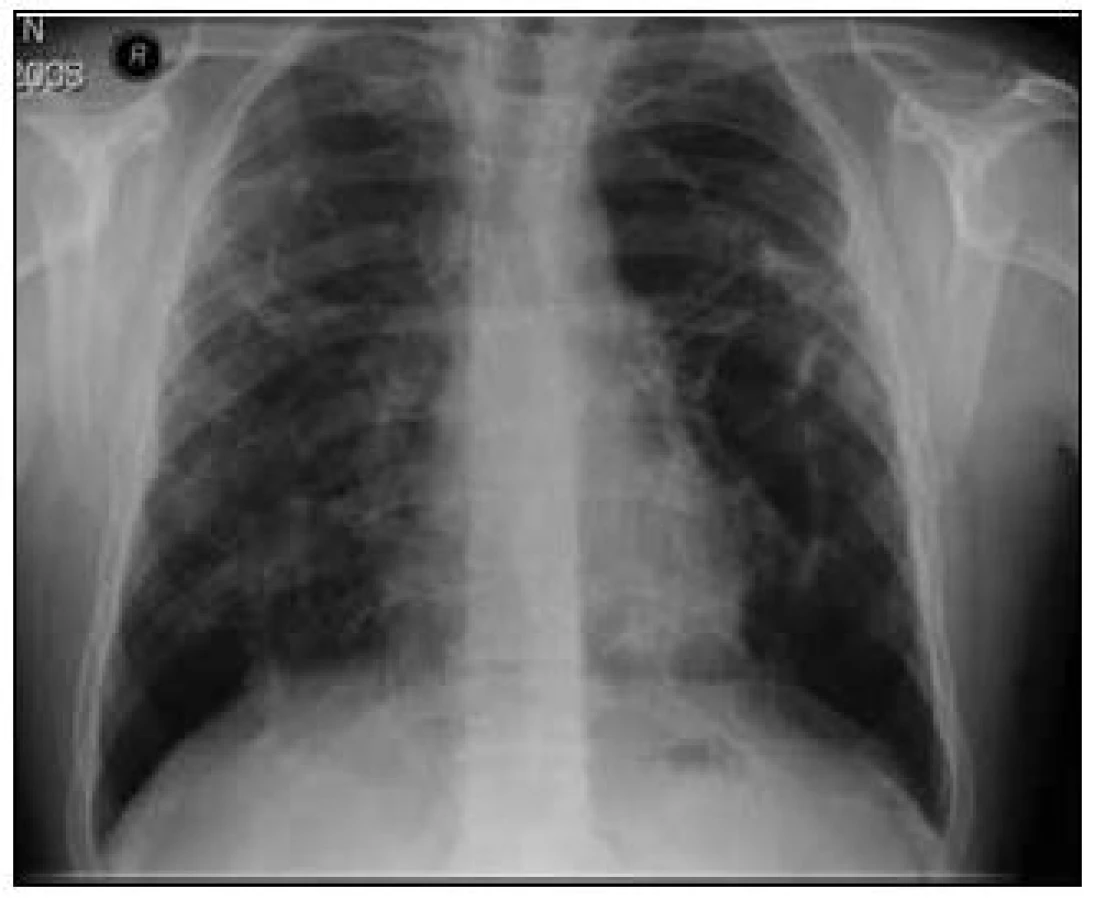
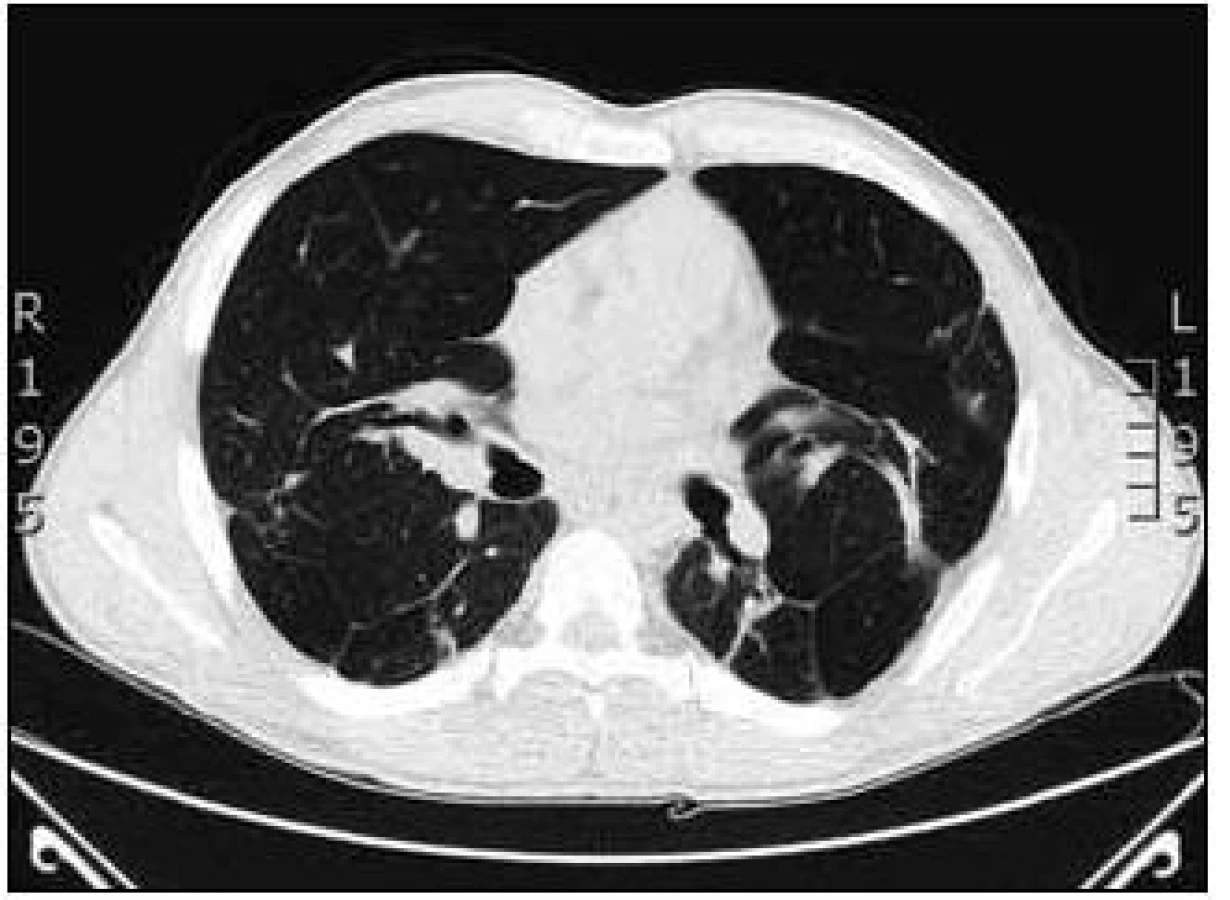
Maximum nálezu bylo popisováno v horních lalocích a bazálně oboustranně s patrným nodulárním zesílením intersticia jak v peribronchovaskulární, tak i intralobulární distribuci i retikulární zesílení. Změny byly lokalizovány subpleurálně. Nebyly patrné kondenzace či infiltrace plicního parenchymu. Ojedinělá byla dutinová projasnění (kavity) do velikosti 2 cm. Pohrudniční dutiny byly bez výpotků. Mediastinum nebylo rozšířeno. Plicní funkční vyšetření prokázalo zlepšení z těžké kombinované ventilační poruchy na středně těžkou obstrukční ventilační poruchu (IVC 2,99 (59 %), FVC 3,32 (68 %), index FEV1//FVC 67 %, FEV1 2,22 (54 %), TLC 5,73 (86 %), RV 169 %, RV/TLC 48 %). Došlo k normalizaci hodnot ACE (39,7 U/l), jaterních enzymů (ALT 0,63 µkat/l, GGT 0,93 µkat/l), kalcia v séru (2,34 mmol/l) a v moči (7,2 mmol/l). Subjektivní stav pacienta byl zlepšen, ale přetrvávala dušnost při větší námaze. Pacient byl 4krát po dobu sledování přeléčen pro infekci dolních cest dýchacích běžnými antibiotiky. V srpnu roku 2008 byl nemocný hospitalizován pro akutní pankreatitidu a byl nasazen přechodně preparát pancreatinum (Kreon) substituující pankreatickou nedostatečnost. Současně byl zjištěn diabetes mellitus s nutností zahájení inzulinové léčby. Při udržovací léčbě glukokortikoidy bylo popisováno na kontrolním CT a HRCT hrudníku v září roku 2010 postižení a zesílení intersticia, zejména v peribronchovaskulární distribuci s trakčními bronchiektaziemi bazálně výraznějšími, zesílení interlobulárního intersticia až nodulární rozsev, zejména paraseptálně. Periferněji i subpleurálně bylo popisováno intersticiální zesílení, difuzně zvýšená transparence až obraz emfyzému; bez infiltrátů charakteru acinózního postižení. V mediastinu byly popisovány jen ojedinělé drobné lymfatické uzlinky. Oproti minulému vyšetření z roku 2008 byly popisovány výraznější bronchiektazie až obraz emfyzému. Plicní funkční vyšetření prokázalo lehkou obstrukční ventilační poruchu (IVC 4,23 (81 %), FVC 3,74 (75 %), index FEV1/FVC 71 %, TLC 7,00 101 %, RV 3,80 (223 %), RV//TLC 54 %), lehkou poruchu difuze (TLco 73 %, Kco 103 %) a lehkou parciální respirační nedostatečnost. Pacient i nadále užíval 10 mg Prednisonu denně. I přes histologicky potvrzenou diagnózu sarkoidózy jsme uvažovali o koincidenci jiné plicní nemoci. Na základě nálezů na HRCT hrudníku, popisující lehce výraznější bronchiektazie až obraz emfyzému, diabetu, proběhlé pankreatitidy a rodinné anamnézy bylo vysloveno podezření na cystickou fibrózu. Podrobnější anamnézou bylo zjištěno, že pacient má problémy s neplodností. Vyšetřením reprodukčního systému transrektální ultrasonografií prostaty byly zjištěny gracilní semenné váčky s hypoplazií. Byl prokázán abnormální spermiogram. Další vyšetření již směřovala k vyloučení či potvrzení diagnózy cystické fibrózy. První potní test byl hraniční, chloridy v potu byly 56,82 mmol/l (referenční mez 0,00–40,00 mmol/l). Druhý potní test byl rovněž hraniční, 54 mmol/l. Pacient byl pankreaticky suficientní (elastáza ve stolici byla 280 µg/1 g, norma 200–1 000 µg/1 g). Molekulárně genetické vyšetření potvrdilo mutace G542X (těžká mutace, I. třída) na jedné alele genu CFTR a mutace R117C (mírná mutace, IV. třída) na druhé alele tohoto genu (smíšený heterozygot). Na základě těchto vyšetření jsme diagnostikovali atypickou variantu cystické fibrózy.
Diskuze
Cystická fibróza a sarkoidóza jsou onemocnění, která se jen vzácně vyskytují společně. Sporadické jsou popisy výskytu obou nemocí v zahraniční literatuře, v českém písemnictví jsme žádnou práci na toto téma nenašly. Problematice koincidence CF a sarkoidózy se věnuje především pediatrická literatura. U naprosté většiny případů diagnóza CF předchází diagnóze sarkoidózy [20–23]. Naše práce ukazuje, že tomu může být naopak a že některé CF mohou být skryty pod jiným plicním onemocněním, např. sarkoidózou.
V práci Dobbina et al byla popsána sarkoidóza plic a Crohnova nemoc u dospělého pacienta s CF [24]. Soden et al ve své práci popisují případ pacienta dětského věku s CF a polyartritidou při sarkoidóze [23]. Práce z roku 1987 popisuje případ chlapce s CF, u kterého byla sarkoidóza diagnostikována v 11 letech [22]. Burton et al ve své práci popisují první případ 13leté dívky s CF, publikovaný v Německu, u které se následně vyvinula plicní sarkoidóza [20].
Mutace CFTR genu byly popsány jak u pacientů s typickou nebo atypickou variantou CF, CF related diseases (CFTR patie – většinou monosymptomatická onemocnění, hraniční potní testy, mírné mutace CFTR genu, sinusitidy, bronchiektazie, alergická bronchopulmonální aspergilóza, idiopatická chronická pankreatitida, obstruktivní azoospermie při kongenitální bilaterální absenci vas deferens CBAVD), tak i sporadicky u nemocných se sarkoidózou [25–33].
Dle dostupných informací se nejeví, že by obě nemoci měly cokoli společného, až na systémové postižení. Sarkoidóza je systémové onemocnění postihující prakticky všechny orgány a orgánové systémy v lidském těle. Rovněž CF je systémové onemocnění. CF ale na rozdíl od sarkoidózy ve své systémovosti „vynechává“ a nepostihuje primárně centrální ani periferní nervový systém. Není postiženo ani hladké svalstvo a vzácně je postiženo příčně pruhované svalstvo. Typická forma CF zahrnuje současné postižení respiračního (těžké sinopulmonální onemocnění) a gastrointestinálního traktu (pankreatická nedostatečnost, diabetes mellitus, prolapsy rekta, mekoniový ileus), mužskou neplodnost (obstrukční azoospermie) a pozitivní potní test (60–120 mmol/l). Diagnóza typické formy bývá obvykle v časném dětství. Atypická forma CF zahrnuje variabilní tíži sinopulmonálního onemocnění, buď respirační, nebo zažívací potíže nebo pouze recidivující sinusitidy, obvykle pankreatickou dostatečnost, diabetes mellitus, obstrukční azoospermii (congenital bilateral absence of the vas deference – CBAVD), chloridy v potu 30–59 mmol/l, nepřítomnost mekoniového ileu a hepatopatie. Zjištění diagnózy atypické formy CF bývá spíše později, a to i v adolescenci a dospělosti. CF related diseases (CFTR patie) nesplňují diagnostická kritéria CF a projevují se jako monosymptomatická onemocnění s mírnými mutacemi CFTR genu (jedna nebo žádná mutace CFTR genu), bronchiektaziemi, alergickou bronchopulmonální aspergilózou, sinusitidami, idiopatickou chronickou pankreatitidou, sklerotizující cholangitidou a obstrukční azoospermií. Diagnóza v tomto případě často uniká [29].
Vyšetření vedoucí k diagnostice sarkoidózy zahrnují klinické vyšetření s důrazem na systémové postižení sarkoidózou. Laboratorní vyšetření často prokazuje zvýšené hodnoty kalcia v séru a v moči. Rovněž bývá zvýšen sérový angiotenzin konvertující enzym ACE. Tyto zvýšené hodnoty jsou z důvodu aktivace makrofágů v sarkoidních granulomech [5].
Plicní funkční vyšetření u sarkoidózy nemusí prokázat ventilační poruchu, popřípadě se setkáváme s restriktivní ventilační poruchou nebo kombinací restrikce s obstrukcí. U pacientů s CF se nejčastěji setkáváme s obstrukční ventilační plicní poruchou se známkami hyperinflace plicního parenchymu [8,15].
Zadopřední snímek hrudníku u sarkoidózy je důležitý pro diagnózu, určení stadia nemoci a prognózu. Máme celkem 5 RTG stadií. Stadium 0 znamená, že na RTG snímku hrudníku je normální nález. Stadium I znamená oboustranné zvětšení plicních hilů. Stadium II značí oboustranné zvětšení plicních hilů + oboustranně symetricky přítomné retikulonodulární zastínění zejména ve středních plicních polích. U stadia III vidíme pouze změny oboustranně v plicních třetinách (plicních polích), ale bez zvětšení plicních hilů. A u stadia IV jsou přítomny změny v plicním parenchymu mající charakter plicní fibrózy, apikalizace plicních hilů, distenze mediastina, buly, bronchiektazie, splývající zastínění, voština. Nejčastějším nálezem na HRCT hrudníku u sarkoidózy jsou nodulární zastínění viditelná podél lymfatik peribronchiálně, subpleurálně, obraz mléčného skla, bronchiektazie, distorze cév a bronchů, pruhovitá zastínění [5]. U cystické fibrózy bývají přítomny na zadopředním snímku hrudníku ložisková projasnění (okrsková hyperinflace), pruhovité stíny (atelektázky), okrouhlá projasnění, polycyklické rozšíření mediastina a hilů, perihilární lineární stíny a známky plicního emfyzému (oploštění bránice, inspirační postavení hrudníku, kapkovité vertikálně uložené srdce, zvětšený retrokardiální a retrosternální prostor). Na HRCT hrudníku u CF vidíme obláčkovitá zastínění, retikulární zastínění při fibrotizaci plicního intersticia, bronchiektazie, bronchiektatické cysty, střídání okrsků hyper - a hypoinflace, drobné atelektázy, obraz „kvetoucího stromu“ při vyplnění terminálních bronchiolů hlenovými zátkami, tenkostěnné kavitace [8]. RTG nálezy u obou nemocí se mohou překrývat. Don et al v práci věnující se radiologickým nálezům zjistili, že u 52 % pacientů (z celkového souboru 48 nemocných) s CF je přítomná hilová a mediastinální lymfadenopatie. Přítomnost adenopatie byla pozorována u nemocných se závažným plicním postižením CF. Adenopatie byla chronická a pomalu progredující [34].
Bronchoskopický nález u sarkoidózy prokazuje fyziologický nález nebo makroskopicky na sliznicích viditelné překrvení či nažloutlé uzlíky. Bronchoalveolární tekutina získaná bronchoalveolární laváží prokazuje v případě sarkoidózy obvykle lymfocytární alveolitidu s poměrem CD4+ T-lymfocytů (pomocné T-lymfocyty) a CD8+ T-lymfocytů (cytotoxické T-lymfocyty) větší než 3,5. Normální nebo nízký imunoregulační index (IRI) však diagnózu sarkoidózy nevylučuje. U vyšších stadií III a IV, s přechodem do fibrózy je navíc mírné až střední zastoupení neutrofilů [5]. Bronchoskopické vyšetření u CF se využívá ve zhodnocení aktivity zánětu, změn průchodnosti dýchacích cest a cíleném odběru materiálu na mikrobiologické vyšetření.
Histologické vyšetření plicní tkáně u sarkoidózy prokáže granulomatózní zánět charakteru sarkoidózy. Je třeba vyloučit jiné infekční i neinfekční granulomatózní procesy [5].
Podobně jako u sarkoidózy, tak i u CF neexistuje dosud kauzální léčba. Sarkoidóza má tendenci ke spontánnímu vyléčení v závislosti na charakteru a stupni postižení. Pokud se rozhodneme pro léčbu, pak lékem první volby jsou systémové kortikosteroidy, jejichž účinek je protizánětlivý a protigranulomatózní. Terapie CF je symptomatická a můžeme ji rozdělit na léčbu respiračního onemocnění (péče o dobrou průchodnost dýchacích cest inhalacemi a následnou fyzioterapií, potlačení infekce a zánětu agresivní protizánětlivou antibiotickou léčbou), léčbu gastrointestinálního onemocnění (péče o dobrý stav výživy, tj. vysokokalorická strava a užívání trávicích enzymů) a léčbu komplikací [19].
Prognóza obou nemocí je rozdílná. Sarkoidóza má poměrně dobrou prognózu s tendencí ke spontánnímu vyléčení a jen u malého procenta pacientů přechází do chronicity. Cystická fibróza je letální onemocnění [19]. Nejnepříznivější prognózu mají nemocní s nedostatečnou zevní sekrecí pankreatu. Pacienti s „mírnými“ mutacemi CFTR genu přežívají podstatně déle.
Závěr
Jako první byla u našeho nemocného diagnostikována sarkoidóza, granulomatózní zánět charakteru sarkoidózy byl potvrzen histologicky. Histologické nálezy byly korelovány s ostatními nálezy a klinikou. Indicie k možnému společnému výskytu cystické fibrózy alespoň zpočátku nebyly přítomny. Pacient nebyl pankreaticky insuficientní. Po nasazení léčby kortikoidy docházelo k částečnému zlepšení jak plicních funkcí, tak i nálezů na zadopředním snímku hrudníku a HRCT hrudníku. Vzhledem ke klinickému nálezu, hraničním potním testům a přítomnosti mutací, z nichž jedna spadá mezi těžké mutace I. třídy a druhá mutace na 2. alele téhož genu patří mezi mírné mutace IV. třídy, se u našeho pacienta jedná o atypickou variantu CF v koincidenci se sarkoidózou.
MUDr. Martina Doubková
www.fnbrno.cz
e-mail: doubkovamartina@seznam.cz
Doručeno do redakce: 23. 9. 2011
Přijato po recenzi: 9. 1. 2012
Sources
1. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999; 160 : 736–755.
2. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999; 16 : 149–173.
3. Hutchinson J. Anomolous disease of skin the fingers: case of livid papillary psoriasis. In: Illustration of clinical surgery.London: J and A Churchill 1878.
4. Kolek V et al. Sarkoidóza – známé a neznámé. Praha: Grada Publishing 1998.
5. Baughman H et al. Sarcoidosis. New York: Inform a Healthcare USA 2010.
6. Kolek V, Hutyrová B, Lošťáková V. MORSA – multicentrická moravská studie epidemiologických trendů sarkoidózy. Studia pneumologica et phtiseologica cechoslovaca 2005; 65 : 17–20.
7. Bradley B, Branley HM, Egan JJ et al. British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008; 63 (Suppl 5): v1–v58.
8. Vávrová V et al. Cystická fibróza. 1. vyd. Praha: Grada Publishing 2006.
9. Hodson MF, Geddes DM (eds). Cystic Fibrosis. 2nd ed. London: ARNOLD 2000.
10. Kerem B, Rommens JM, Buchanan JA et al. Identification of the cystic fibrosis gene. Genetic analysis. Science 1989; 245 : 1073–1080.
11. Riordan JR, Rommens JM, Kerem B et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245 : 1066–1073.
12. Rommens JM, Ianuzzi MC, Kerem BS et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245 : 1059–1065.
13. Farell PM, Rosenstein BJ, White TB et al. Cystic Fibrosis Foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatr 2008; 153: S4–S14.
14. Davies JC, Alton EW, Bush A. Cystic fibrosis. BMJ 2007; 335 : 1255–1259.
15. Fila L, Sedlák V, Binková I et al. Dvacet let péče o dospělé nemocné s cystickou fibrózou v České republice. Vnitř Lék 2009; 55 : 542–548.
16. Castellani C, Cuppens H, Macek M Jr et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 2008; 7 : 179–196.
17. Castellani C, Southern KW, Brownlee K et al. European best practice guidelines for cystic fibrosis neonatal screening. J Cyst Fibros 2009; 8 : 153–173.
18. Ratjen F, Döring G. Cystic fibrosis. Lancet 2003; 361 : 681–689.
19. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis. A concensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr 1998; 132 : 589–595.
20. Burton CM, Pressler T, Milman N. Pulmonary sarcoidosis in a child with cystic fibrosis. Pediatr Pulmonol 2005; 39 : 473–477.
21. Rettinger S, Trulock E, Mackay B et al. Case report: sarcoidosis in an adult with cystic fibrosis. Thorax 1989; 44 : 829–830.
22. Cooper TJ, Day AJ, Keller PH et al. Sarcoidosis in two patient with cystic fibrosis: a fortuitous association? Thorax 1987; 42 : 818–820.
23. Soden M, Tempany E, Bresnihan B. Case report: sarcoid arhropaty in cystic fibrosis. Br J Rheumatol 1989; 28 : 341–343.
24. Dobbin CJ, Moriarty C, Bye PT. Granulomatous diseases in a patient with cystic fibrosis. J Cyst Fibros 2003; 2 : 35–37.
25. Bombieri C, Luisetti M, Belpinati F et al. Increased frequency of CFTR gene mutations in sarcoidosis: a case/control association study. Eur J Hum Genet 2000; 8 : 717–720.
26. Bombieri C, Belpatini F, Pignati P et al. Comment on CFTR gene station in sarcodosis. Eur J Hum Genet 2003; 11 : 553–554.
27. Hughes D, Dörk T, Stuhrmann M et al. Mutation and haplotype analysis of the CFTR gene in atypically mild cystic fibrosis patient from Nothern Ireland. J Med Genet 2001; 38 : 136–139.
28. Iannuzzi MC, Rybicki BA. Genetics of sarkoidosis: candidate genes and genome scans. Proc Am Thorac Soc 2007; 4 : 108–116.
29. Luisetti M, Pignatti PF. Genetics of idiopathic diseminated bronchiektasis. Semin Respir Crit Care Med 2003; 24 : 179–184.
30. Makrythanasis P, Tzetis M, Rapti A et al. Cystic fibrosis conductance regulator, tumor necrosis factor, interferon alfa-10, interferon alfa-17, and interferon gamma genotyping as potential risk markers in pulmonary sarcoidosis pathogenesis in greek patients. Genet Test Mol Biomarkers 2010; 14 : 577–584.
31. Pier GB, Grout M, Zaidi TS. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aerugionosa from the lung. Proc Natl Acad Sci USA 1997; 94 : 12088–12093.
32. Schürmann M, Albrecht M, Schwinger E. CFTR gene mutations in sarcoidosis. Eur J Hum Genet 2002; 10 : 729–732.
33. Spector WG, Heesom N. The production of granulomata by antigen-antibody complexes. J Pathol 1969; 98 : 31–39.
34. Don CJ, Dales RE, Desmarais RL et al. The radiographic prevalence of hilar and mediastinal adenopathy in adult cystic fibrosis. Can Accos Radiol J 1997; 48 : 265–269.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2012 Issue 4
Most read in this issue
- Akutní infarkt myokardu navozený požitím drogy pervitin
- Význam biomarkerů NGAL a cystatinu C u kardiovaskulárních onemocnění
- Akutní intoxikace mědí při suicidiálním pokusu
- Nové poznatky v patogenezi Crohnovy choroby