
Naše zkušenosti s léčbou multicentrické plazmocelulární Castlemanovy choroby s projevy vaskulitidy – popis případu a přehled literatury
Our experience in treatment of multicentric plasma-cell Castleman disease associated with vasculitis manifestations – case report and literature review
Castleman disease is a rare idiopathic non-neoplastic lymphoproliferative disorder with 2 clinical (unicentric and multicentric) and 3 histomorphological (hyaline-vascular, plasma-cell and mixed) forms identified. The case report given here describes the 3-year experience with therapy in a patient, male born 1961, diagnosed with multicentric plasma-cell Castleman disease (HIV and HHV-8 negative) with the finding of generalized lymphadenopathy and splenomegaly. During first line treatment (R-CHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone, 3 cycles in total, 12/2008–2/2009) the development of bilateral upper and lower limb edemas with clinical manifestation of vasculitis occurred and a restaging computed tomography (CT) examination revealed a stable finding of the lymphadenomegaly. Greater success was achieved with thalidomide regimen (CTD: cyclophosphamide, thalidomide, dexamethasone, 10 cycles, 3/2009–1/2010) leading to reduction in the size of the hypervascularized lymph nodes (almost by 50%) as well as their radiopharmaceutical (fluorodeoxyglucose) uptake as seen on a combined positron emission tomography and computed tomography (PET/CT) scan imaging. Thalidomide was given daily at doses between 100 and 200 mg. We returned to the CTD regimen again in April 2010 after a short period of monoclonal antibody tocilizumab treatment (400 mg intravenous in 2-week intervals with 50% dose reduction due to a limited supply of the drug, 5 doses in total) during which edemas reoccurred with a CT scan finding of stable lymphadenomegaly. However, the renewed regimen with thalidomide was stopped after 2.5 cycles due to adverse effects of thalidomide (neuropathy) and corticoids (Cushing syndrome). In September 2010, after enrollement in the Celgene’s Compassionate Use Program we were able to start treating the patient with the derivative of thalidomide, lenalidomide, at a dosage of 25 mg on days 1–21 in a 28-day cycle, 15 cycles in total (10/2010–12/2011). The monotherapy with lenalidomide was very well tolerated by the patient without any effects of myelotoxicity, thromboembolism or relapses of edemas and vasculitis, additionally now with apparent improvement of fatic disorder and the patient’s motor abilities. Thus, lenalidomide represents an attractive alternative agent for patients with Castleman disease after rituximab and cytostatics failures. It has a favourable safety profile and could be therefore considered for administering in first line treatment.
Key words:
Castleman disease – glucocorticoids – chemotherapy – rituximab – thalidomide – monoclonal antibody – tocilizumab – lenalidomide – positron emission tomography – computed tomography
Authors:
P. Szturz 1; Z. Adam 1; M. Moulis 2; L. Šmardová 1; M. Klincová 3; R. Šlaisová 4; R. Koukalová 5; Z. Řehák 5; P. Volfová 1; J. Chovancová 3; O. Stehlíková 1; J. Mayer 1
Authors‘ workplace:
Interní hematologická a onkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Josef Feit, CSc.
2; Lékařská fakulta Masarykovy univerzity, Brno, děkan prof. MUDr. Jiří Mayer, CSc.
3; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil Válek, CSc.
4; Oddělení nukleární medicíny PET Centra Masarykova onkologického ústavu Brno, přednosta prim. MUDr. Karol Bolčák
5
Published in:
Vnitř Lék 2012; 58(9): 679-690
Category:
Case Reports
Overview
Castlemanova choroba je vzácné idiopatické ne-neoplastické lymfoproliferativní onemocnění, u něhož byly vymezeny 2 klinické (unicentrická a multicentrická) a 3 histomorfologické (hyalinně-vaskulární, plazmocelulární a smíšená) formy. Tato kazuistika popisuje 3leté zkušenosti s léčbou pacienta, muže narozeného v roce 1961, u něhož byla diagnostikována multicentrická plazmocelulární Castlemanova choroba (HIV a HHV-8 negativní) s nálezem generalizované lymfadenopatie a splenomegalie. Během podávání léčby první linie (R-CHOP: rituximab, cyklofosfamid, doxorubicin, vinkristin, prednison, celkem 3 cykly, prosinec roku 2008–únor roku 2009) došlo k rozvoji oboustranného otoku rukou a nohou s klinickými projevy vaskulitidy a restagingové vyšetření počítačovou tomografií (CT) ukázalo neměnný rozsah lymfadenomegalie. Větších úspěchů bylo dosaženo režimem s thalidomidem (CTD: cyklofosfamid, thalidomid, dexametazon, 10 cyklů, březen roku 2009–leden roku 2010), který vedl k regresi velikosti (celkově téměř o 50 %) i míry akumulace radiofarmaka (fluorodeoxyglukózy) hypervaskularizovaných zvětšených lymfatických uzlin dle vyšetření pozitronovou emisní tomografií v kombinaci s výpočetní tomografií (PET/CT). Dávky thalidomidu se pohybovaly mezi 100 a 200 mg denně. K režimu CTD jsme se opět vrátili v dubnu roku 2010 po krátkém období léčby monoklonální protilátkou tocilizumabem (400 mg i.v. každé 2 týdny, tedy redukce dávky na 50 % vzhledem k omezenému množství léku, celkem 5 dávek), při níž došlo k recidivě edémů při stacionárním nálezu lymfadenomegalie na CT vyšetření. Obnovený režim s thalidomidem byl však po 2,5 cyklech ukončen pro výskyt nežádoucích účinků thalidomidu (neuropatie) i kortikoidů (Cushingův syndrom). V září roku 2010 jsme po registraci do programu CUP (Compassionate Use Program) firmy Celgene získali možnost zahájit u pacienta léčbu s lenalidomidem, derivátem thalidomidu, v dávkování 25 mg ve dnech 1 až 21 v rámci 28denního cyklu, celkem 15 cyklů (říjen roku 2010–prosinec roku 2011). Monoterapie lenalidomidem byla velmi dobře tolerována bez jakýchkoliv projevů myelotoxicity, tromboembolizmu či recidivy edémů a vaskulitidy, navíc s patrným zlepšením fatické poruchy a motorických schopností pacienta. Lenalidomid tedy představuje atraktivní léčebnou alternativu u pacientů s Castlemanovou chorobou po selhání rituximabu a cytostatik. Má velmi příznivý bezpečnostní profil, a lze proto zvážit jeho podání i v rámci první linie léčby.
Klíčová slova:
Castlemanova choroba – glukokortikoidy – chemoterapie – rituximab – thalidomid – monoklonální protilátka – tocilizumab – lenalidomid – pozitronová emisní tomografie – výpočetní tomografie
Úvod
Castlemanova choroba (Castleman disease – CD) je neklonální lymfoproliferativní onemocnění pojmenované podle amerického lékaře, patologa z Massachusetts General Hospital, Dr. Benjamina Castlemana, který jako první popsal charakteristický histologický nález v oblasti mediastinální uzlinové hyperplazie [1,2]. Bylo tomu v roce 1954 formou kazuistiky a o 2 roky později formou popisu souboru 13 pacientů [3,4]. Za necelá 2 desetiletí, v roce 1972, pak Keller klinicky a histopatologicky definoval hyalinně--vaskulární a méně častý plazmocelulární typ [5], který však jako variantu bohatou na plazmatické buňky popsali již v roce 1969 Flendrig a Schillings [6].
U CD můžeme rozlišovat 3 histomorfologické typy (hyalinně-vaskulární, plazmocelulární a smíšený) a 2 typy klinické (unicentrický a multicentrický). Projevy CD jsou tedy heterogenní a sahají na jedné straně od benigní ložiskové lymfadenomegalie až k rekurentním epizodám generalizované lymfadenopatie sdružené s celkovými příznaky a abnormálními laboratorními nálezy. Zatímco unicentrická, lokalizovaná, forma CD je vyléčitelná excizí postižených lymfatických uzlin, multicentrický typ je často refrakterní k léčbě, a to i přes použití kortikoidů nebo chemoterapie [7]. Prevalence tohoto onemocnění není známa, odhadovaný počet případů v USA sahá od 30 000 k 100 000 [8]. Toto onemocnění je však často poddiagnostikované nebo špatně diagnostikované [9].
Tato kazuistika popisuje naše zkušenosti s 3letou léčbou pacienta s multicentrickou plazmocelulární CD a pomocí radiologických a nukleárně medicínských nálezů dokumentuje průběh onemocnění. Rutinní laboratorní testování bylo rozšířeno o sledování cytokinové odpovědi organizmu, kterým dokládáme účinnost zcela ojedinělé terapie lenalidomidem.
Popis případu
Stanovení diagnózy a léčba 1. linie
Pacient, 46letý muž s pravostrannou frustní hemiparézou a expresivní fatickou poruchou po opakovaných cévních mozkových příhodách, byl poprvé vyšetřen na našem pracovišti v červnu roku 2008 pro generalizovanou (krční, axilární, mediastinální, retroperitoneální a tříselnou) lymfadenopatii, mírnou splenomegalii a histologický nález Castlemanovy choroby z exstirpované (červen roku 2008) zvětšené lymfatické uzliny v pravém třísle. Dle vstupních laboratorních výsledků byla přítomna mírná leukocytóza (13,9 × 109/l), normochromní normocytární anémie (127 g/l) a trombocytóza (577 × 109/l), zvýšená hladina D-dimerů (1,29 µg/ml), fibrinogenu (4,7 g/l), C-reaktivního proteinu (35,4 mg/l) a sedimentace erytrocytů (16 mm za 1. hod, 30 mm za 2. hod). Ostatní parametry včetně jaterních testů, renálních funkcí a hodnoty celkové bílkoviny v séru byly v normě. Trepanobiopsie z lopaty kosti kyčelní prokázala normální trilineární hematopoézu. Kromě neurologického postižení byl muž s anamnézou nikotinizmu a prodělané hepatitidy B sledován s arteriální hypertenzí. Etiologie cévních mozkových příhod u tohoto relativně mladého muže velmi pravděpodobně souvisela s vaskulitidou, která tuto nemoc provázela (viz dále), a s laboratorními projevy hyperkoagulace (trombocytóza, zvýšená koncentrace D-dimerů a fibrinogenu).
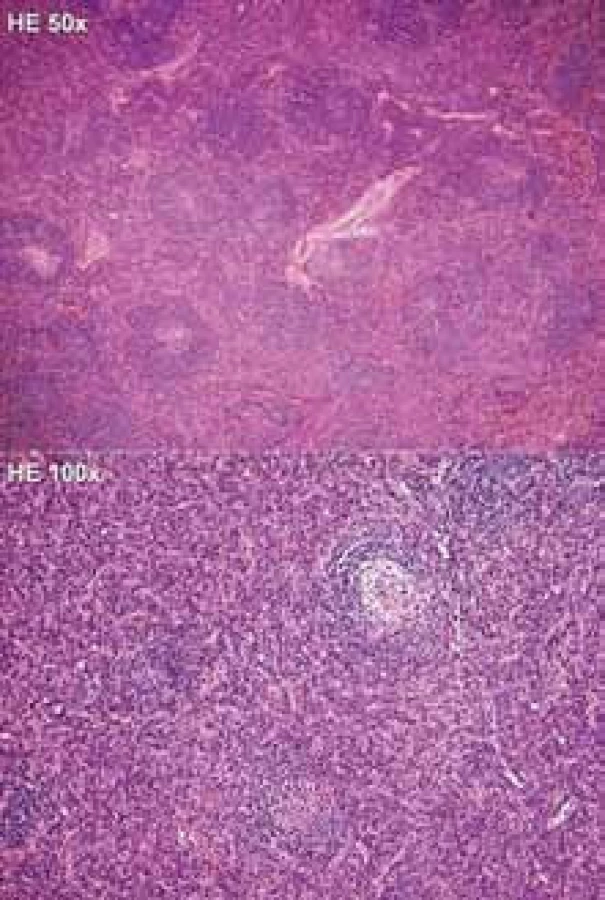
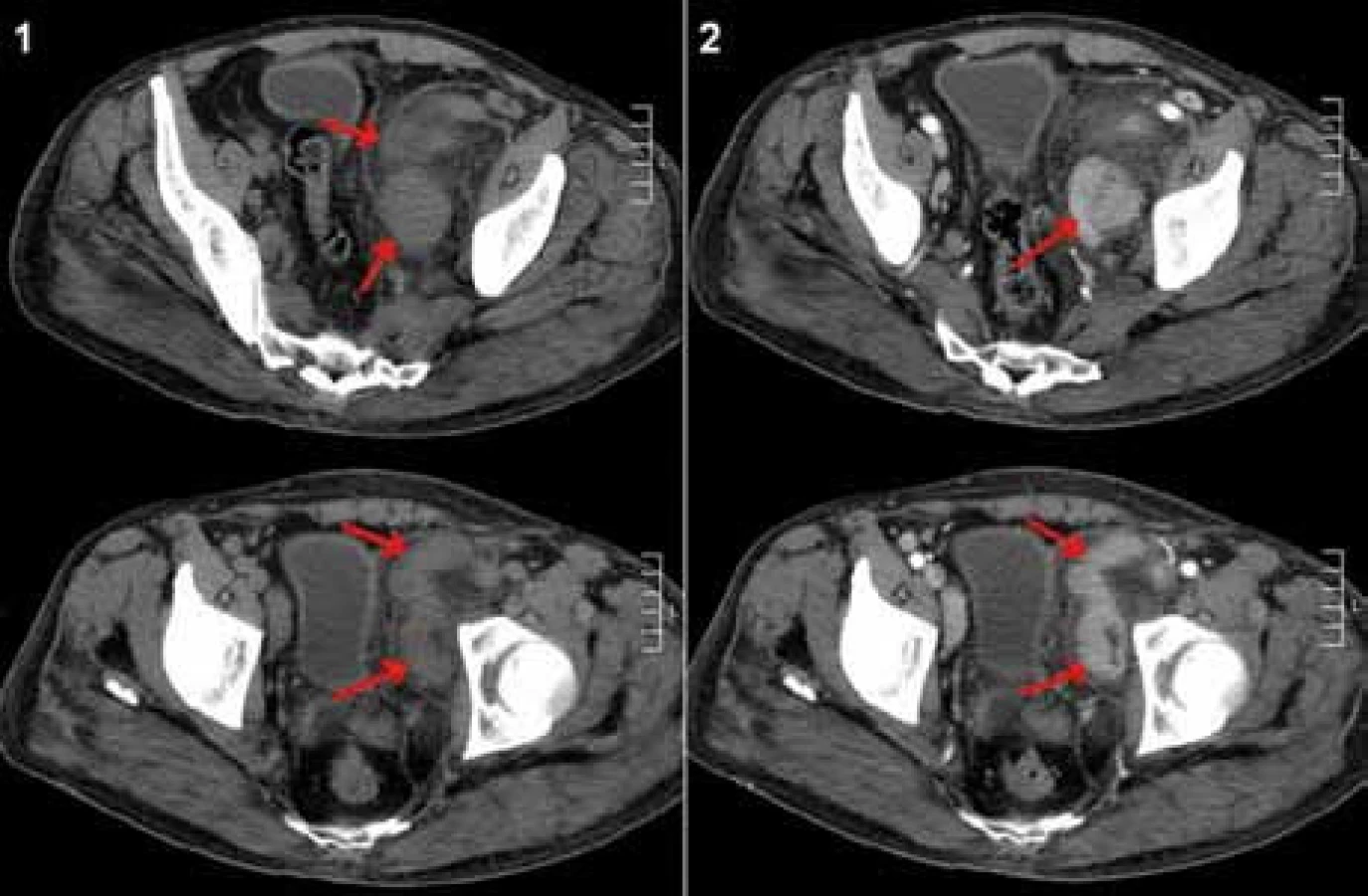
K ověření vzácné diagnózy podstoupil pacient opakované bioptické vyšetření lymfatické uzliny z levého třísla (červenec roku 2008) a retroperitonea (říjen roku 2008), které v obou případech toto vzácné onemocnění potvrdilo (obr. 1). Přešetření na případnou infekční nebo revmatologickou etiologii v rámci diferenciální diagnostiky bylo negativní. Pro přítomnost velké uzlinové masy v malé pánvi utlačující okolní struktury (obr. 2) byl pacient i přes absenci celkových příznaků indikován k podání systémové léčby.
Po prefázi kortikoidy (dexametazon 40 mg p.o. den 1–4, 11–14, 21–24) byl v prosinci roku 2008 zahájen chemoterapeutický režim R-CHOP (rituximab 375 mg/m2 i.v. den 1, cyklofosfamid 750 mg/m2 i.v. den 1, adriamycin 50 mg/m2 i.v. den 1, vinkristin 2 mg i.v. den 1, prednison 100 mg p.o. den 1–5, opakování po 21 dnech, celkem podány 3 cykly). Léčba rituximabem, monoklonální protilátkou proti antigenu CD20, byla opodstatněna nálezem CD20 pozitivních buněk v histologickém preparátu. Dávky cyklofosfamidu, adriamycinu a prednisonu v tomto léčebném schématu byly vzhledem k celkovému stavu pacienta a ke komorbiditám v 1. cyklu redukovány.
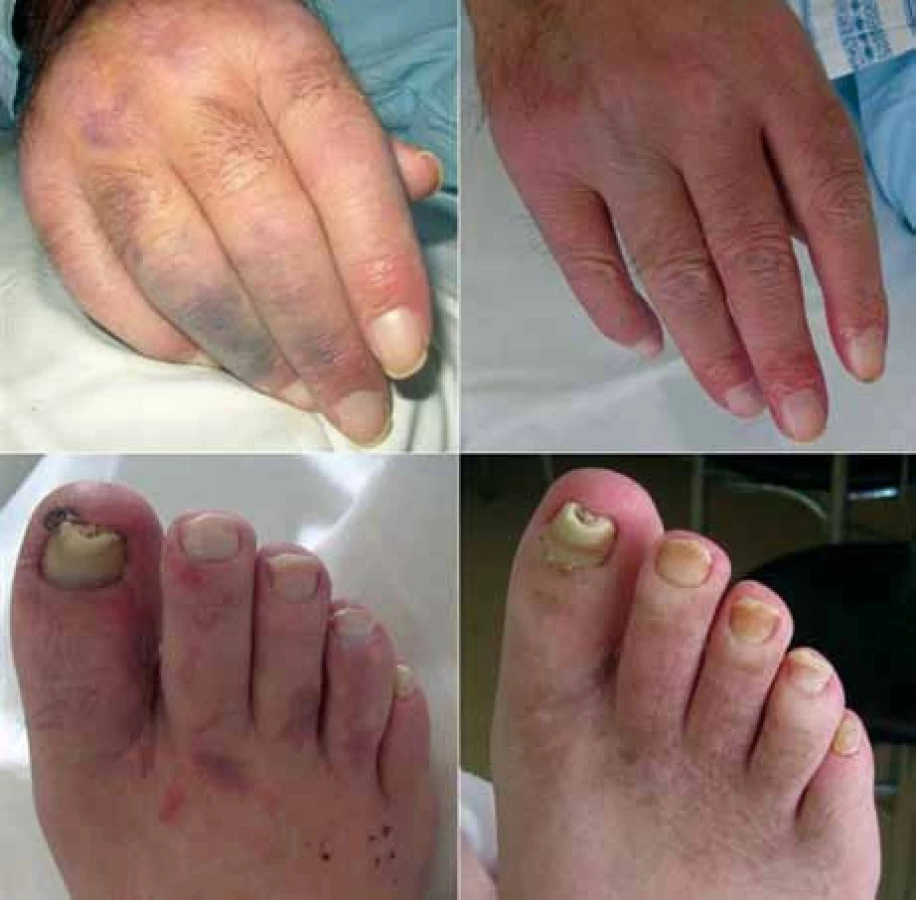
Na zavedené léčbě se však celkový stav pacienta postupně zhoršoval s rozvojem dyspepsií a progredujícího oboustranného otoku rukou a nohou s lividním zbarvením prstů. Tato kožní manifestace byla na základě klinického nálezu a výborného účinku antiagregační a vazodilatační léčby uzavřena jako vaskulitida (obr. 3), i když histologicky byly popsány změny odpovídající erythema multiforme. Kontrolní CT vyšetření po podání 3 cyklů ukázalo neměnný rozsah lymfadenomegalie. Tento režim s použitím rituximabu se tedy i přes pozitivitu CD20 antigenu u našeho pacienta neosvědčil, a byl proto v březnu roku 2009 ukončen.
Thalidomid v léčbě 2. linie
Vzhledem k přetrvávajícím otokům dolních končetin a celkovému chřadnutí pacienta nebyla další léčba odkládána a již v březnu roku 2009 byl zahájen 1. cyklus režimu s thalidomidem (režim CTD senior, cyklofosfamid 50 mg p.o. den 1–28, thalidomid 100 mg p.o. den 1–28, dexametazon 20 mg p.o. den 1–4 a 15–18, opakování po 28 dnech, podáno 10 cyklů). Během 1. týdne léčby byla patrná postupná regrese otoků končetin, v dalším průběhu se celkový stav pacienta postupně zlepšoval a pro dobrou toleranci byly od V. do VIII. cyklu navýšeny dávky thalidomidu na 150–200 mg denně. Dexametazon byl naopak od 4. cyklu pro opakované dekompenzace glykemií při nově diagnostikovaném diabetes mellitus redukován na podání pouze v intervalu 1.–4. dne každého cyklu. Restagingová PET/CT vyšetření ukázala postupnou regresi velikosti (celkově téměř o 50 %) i míry akumulace radiofarmaka patologicky hypervaskularizovaných zvětšených lymfatických uzlin. Již při PET/CT vyšetření v červnu roku 2009, po 3 cyklech léčby CTD, přetrvávala jen mírná zvýšená metabolická aktivita v postižených uzlinách, orientačně v dolním pásmu malignity (obr. 4).

Použití monoklonální protilátky a návrat k thalidomidu
Na začátku ledna roku 2010 obdržela naše klinika formou daru 5 ampulí (v každé po 400 mg) monoklonální protilátky proti receptoru pro interleukin-6 (IL-6, tocilizumab). Jedná se dosud o jednu z nejúčinnějších dostupných terapeutických možností pro pacienty s multicentrickou formou CD. Při klasickém dávkování se každé 2 týdny podává 800 mg formou krátkodobé i.v. infuze. S ohledem na omezené množství tohoto léku, které jsme měli k dispozici, jsme upravili dávkovací schéma (400 mg i.v. každé 2 týdny, redukce dávky na 50 %) s plánem 10týdenní léčby. Aplikace tocilizumabu byla zahájena v lednu roku 2010 a probíhala vždy po premedikaci kortikoidy, antihistaminiky a antipyretiky jako prevence případných nežádoucích reakcí (obdobně jako u rituximabu).
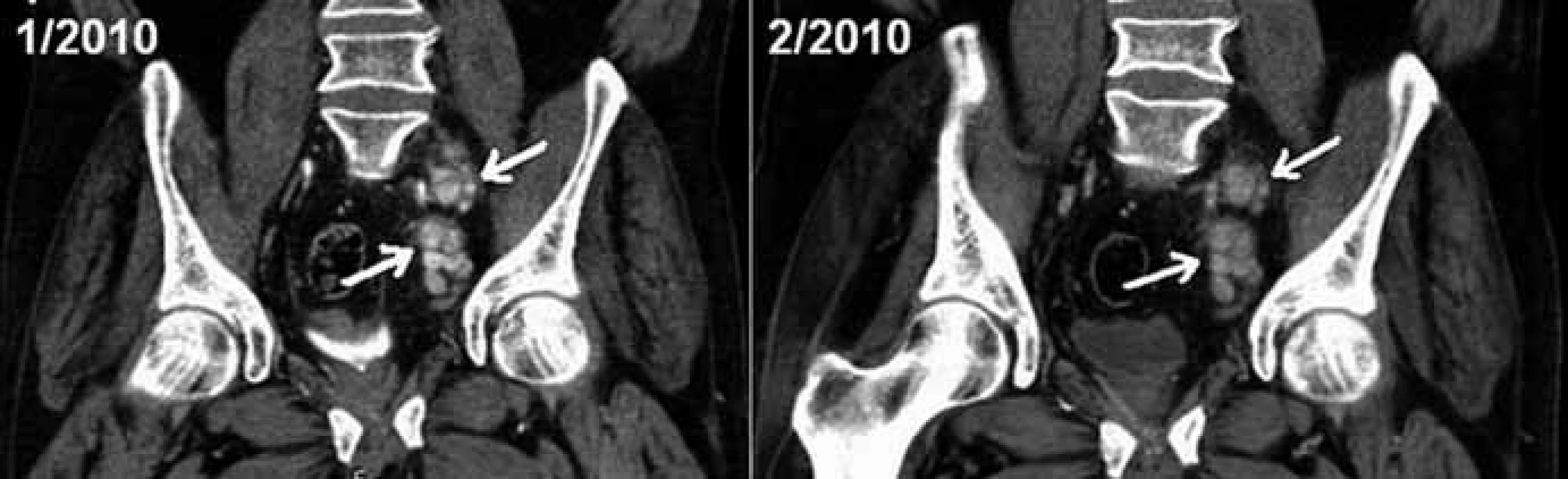
Ačkoliv restagingové CT vyšetření po 2 dávkách léku ukázalo obraz stacionární retroperitoneální lymfadenopatie (obr. 5), u pacienta došlo ke zhoršení klinického stavu s recidivou otoků dolních končetin. Podávání tocilizumabu bylo proto po 5 dávkách zastaveno a v dubnu roku 2010 jsme se vrátili k režimu CTD (cyklofosfamid 50 mg p.o. den 1–28, thalidomid 100 mg p.o. den 1–28, dexametazon 20 mg p.o. den 1–2 a 15–16, opakování po 28 dnech, podáno dalších 2,5 cyklu). Třebaže stran základního onemocnění byl účinek obnoveného režimu s thalidomidem uspokojivý, léčbu jsme byli nuceni opět ukončit, a to pro projevy nežádoucích účinků thalidomidu (neuropatie) i kortikoidů (Cushingův syndrom).
Monoterapie lenalidomidem a sledování cytokinové odpovědi organizmu
V září roku 2010 se naše pracoviště zaregistrovalo do programu CUP (Compassionate Use Program) firmy Celgene, a získalo tak možnost podat u sledovaného muže léčbu s lenalidomidem, derivátem thalidomidu. Na základě našich zkušeností s terapií mnohočetného myelomu, u něhož je lenalidomid standardně využíván, jsme se rozhodli pro dávkovací schéma 25 mg ve dnech 1–21 v rámci 28denního cyklu. S ohledem na riziko tromboembolizmu při podávání léku jsme doplnili pravidelnou medikaci o profylaktické subkutánní podávání nízkomolekulárních heparinů [10]. Do prosince roku 2011 bylo podáno 15 cyklů (říjen roku 2010 až prosinec roku 2011).
Během léčby jsme nepozorovali žádné známky myelosuprese. Hladiny IL-6, které jsme v rámci sledování cytokinové odpovědi organizmu v pravidelných intervalech monitorovali, kolísaly během prvního cyklu léčby mezi 3,1 a 10,7 pg/ml a klesly počínaje 2. cyklem pod hranici detekce. Analogický byl vývoj hodnot C-reaktivního proteinu, které v průběhu terapie poklesly (tab. 1). Restagingové PET/CT vyšetření ukázalo zmenšení postižených lymfatických uzlin doprovázené poklesem patologického hypermetabolizmu glukózy (obr. 4). Při pravidelných kontrolách bylo dále patrné zlepšení fatické poruchy a motorických schopností pacienta. Zatímco vstupně byl muž na ambulanci přivážen na invalidním vozíčku, na zahájení 12. cyklu již přišel v doprovodu a sám nyní ujde vzdálenost 50 metrů. Jedinou interkurencí monoterapie lenalidomidem byla nekomplikovaná bronchitida, která byla dobře zvládnuta empirickou antibiotickou léčbou za hospitalizace.

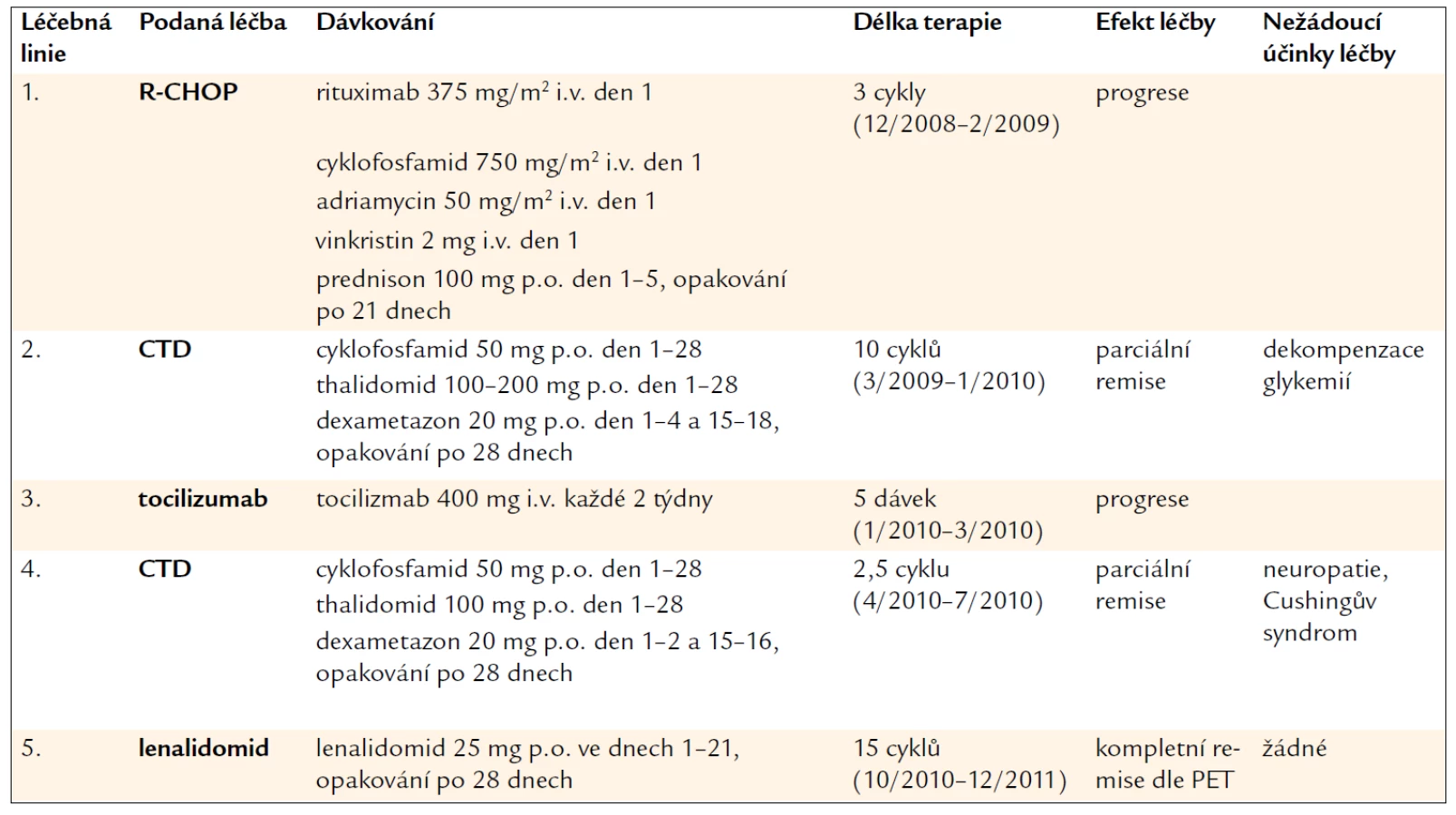
Souhrnný pohled na 3letý průběh léčby pacienta ukazuje tab. 2.
Diskuze
CD může být limitována na jednu uzlinovou oblast, nebo ji může onemocnění přesahovat. Na základě toho pak rozlišujeme unicentrickou (lokalizovanou) a multicentrickou (generalizovanou) formu CD. Uvážíme-li navíc existenci 3 histopatologických forem onemocnění (hyalinně-vaskulární, plazmocelulární a smíšený typ), můžeme se setkat minimálně s 6 typy onemocnění s více či méně podobnými projevy. Na pomyslné škále agresivity by se na straně benigních lézí nacházela hyalinně-vaskulární unicentrická CD a na protilehlé straně torpidní plazmocelulární multicentrická varianta s typickými systémovými příznaky.
Hyalinně-vaskulární typ, někdy také označovaný jako angiofolikulární typ, je morfologicky charakterizován lymfatickými folikuly s malými hyalinizovanými germinálními centry s interfolikulární proliferací kapilár [11]. Histologický obraz bývá někdy přirovnáván ke slupce cibule (koncentricky uspořádaná rozšířená plášťová zóna folikulů) či lízátku (plášťová zóna společně s prominující sklerotickou cévou). V roce 1993 popsali Danon et al podtyp hyalinně-vaskulární formy bohatý na stromu (stroma-rich variant), u něhož dominuje zastoupení interfolikulární oblasti zaujímající více než 50 % lymfatické uzliny [12].
Na rozdíl od hyalinně-vaskulární varianty má plazmocelulární CD méně charakteristické rozlišovací histologické znaky, typická je interfolikulární akumulace polyklonálních plazmatických buněk [1,9,13]. Jelikož znaky plazmocelulární CD nejsou specifické, je nezbytné vyloučení dalších nozologických jednotek, jako jsou B lymfomy, vzácné plazmocytomy, reaktivní lymfadenopatie asociované s infekcí, autoimunitní choroby, jako je revmatoidní artritida, nebo reaktivní lymfadenopatie asociované s jinými imunodeficity [1,14,15].
Patogeneze
Ačkoliv bylo navrženo několik imunologických mechanizmů zahrnujících dysregulaci cytokinové sítě a herpesvirotické infekce, není etiopatogeneze onemocnění dosud zcela objasněna. Většina autorů se však přiklání k názoru, že CD je chronická nespecifická zánětlivá reakce na neznámý stimul [4,5,16]. Rozdílný fenotyp hyalinně-vaskulární a plazmocelulární varianty CD pak lze vysvětlit buď jako různou hostitelsky specifickou imunitní odpověď na tentýž stimul, kdy oba subtypy CD tvoří součást nedělitelného kontinua, anebo se jedná o reakce na 2 různé ale vzájemně příbuzné stimuly [16].
Interleukin-6
IL-6 je cytokin s širokým rozsahem biologických účinků. V současné době je věnováno hodně pozornosti jeho nadprodukci v plášťové zóně postižených lymfatických uzlin. Jeho role v systémové manifestaci CD se opakovaně potvrdila [17–20]. Tento pleiotropní interleukin stimuluje B buněčnou proliferaci a je nezbytný pro vyzrávání aktivovaných B buněk do plazmatických buněk, což je charakteristické pro plazmocelulární variantu CD. IL-6 rovněž podněcuje vaskulární proliferaci, což je zase charakteristické pro hyalinně-vaskulární variantu, a to tak, že stimuluje sekreci vaskulárního endoteliálního růstového faktoru (vascular endothelial growth factor – VEGF) [14,21]. IL-6 je dále dáván do souvislosti s maligními lymfoproliferacemi [11,22]. Sérové hladiny IL-6 u pacientů s CD jsou však signifikantně vyšší než u pacientů s Hodgkinovým lymfomem, difuzním velkobuněčným B lymfomem nebo mnohočetným myelomem [16]. Bylo zjištěno, že hladina IL-6 nejvíce koreluje s aktivitou multicentrické CD [11].
Mikrocytární anémie, typická pro multicentrickou plazmocelulární variantu, zřejmě souvisí s IL-6 a hepcidinem, tedy stejně jako u ostatních anémií typu chronických chorob, a má vztah k probíhajícímu chronickému zánětu v organizmu [23]. IL-6 přímo zvyšuje jaterní produkci hepcidinu, klíčového regulátoru v metabolizmu železa [24,25]. Hepcidin blokuje uvolňování železa z jaterních makrofágů a snižuje absorpci železa ve střevě, čímž omezuje dodávku železa do vyvíjejících se erytrocytů v kostní dřeni [23,26]. Nadměrná produkce hepcidinu je zřejmě odpovědná za mikrocytární anémii u pacientů s CD a tato produkce může být tlumena blokováním signalizační cesty IL-6 pomocí tocilizumabu (viz dále) [27].
Lidský herpesvirus 8
Lidský herpesvirus 8 (human herpes virus type 8 – HHV-8, někdy označovaný jako herpesvirus Kaposiho sarkomu) má rovněž úzký vztah k CD, zejména k multicentrické CD u HIV (virus lidského imunodeficitu, human immunodeficiency virus) pozitivních pacientů [7]. HHV-8 pozitivita byla nalezena téměř u 100 % publikovaných případů HIV pozitivních pacientů s CD a v 50 % HIV negativních pacientů s multicentrickou CD [16]. Dosud byla prokázána etiologická souvislost HHV-8 se 2 malignitami, Kaposiho sarkomem a primárním efuzním lymfomem. Zajímavé bylo zjištění, že v případě CD dochází k expresi jak latentních, tak i lytických proteinů HHV-8, zatímco u Kaposiho sarkomu a primárního efuzního lymfomu jsou exprimovány především proteiny lytické fáze [28].
Jedním z těchto lytických proteinů je i virusový analog IL-6 (50% podobnost genu), který má přímý efekt, ale také zvyšuje hladinu lidského IL-6 u infikovaného hostitele [29,30]. Virusový IL-6 sdílí funkční vlastnosti s lidským korelátem, a aniž by tvořil komplex s receptorem, může se vázat s glykoproteinem gp130, což je vlastní přenašeč receptorového signálu IL-6. Tocilizumab (monoklonální protilátka proti receptoru pro IL-6, viz dále) není schopen blokovat tuto formu nereceptorové aktivace gp130. Přesto léčba s tocilizumabem byla s to zlepšit příznaky a laboratorní parametry HHV-8 pozitivních pacientů s CD. Lze tedy usuzovat, že i když virusový IL-6 může být exprimován u těchto pacientů, je to stále lidský IL-6, který je dominantně odpovědný za systémové příznaky u HHV-8 pozitivních pacientů s multicentrickou CD [31].
Klasifikace
Ačkoliv byl na začátku tohoto textu popsán odlišný klinický a morfologický pohled na CD, tato 2 dělení se navzájem překrývají, neboť ve většině případů unicentrické formy je nalézán obraz hyalinně-vaskulární CD a multicentrická varianta je typická manifestace plazmocelulární, případně smíšené formy CD. Charakteristiky onemocnění jsou přehledně uvedeny v tab. 3.
![Základní charakteristiky 2 klinických forem CD. Upraveno podle [16].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c0846767cba75d7ca0af7a21dd9d4534.png)
Unicentrická forma
Tento subtyp onemocnění podle starších literárních údajů odpovídá asi za 80 % CD, novější zdroje uvádějí asi 60% podíl [16]. Tento zdánlivý nesoulad je dán zejména dostupností a významnými pokroky v radiodiagnostice a nukleární medicíně, na které spoléháme při posuzování postižených oblastí.
Lokalizovaná forma se klinicky manifestuje v podobě benigního, nebolestivého, pomalého zvětšování lymfatické uzliny. Příznaky však může působit při kompresi přilehlých struktur (bolest, dušnost, kašel, chrapot, porucha polykání, průjmy a další) nebo je její nález náhodný při rutinní lékařské prohlídce [9]. Systémová manifestace včetně laboratorních abnormalit (anémie, zvýšená sedimentace erytrocytů, hypergamaglobulinemie) není častá, a pokud je přítomna, je s velkou pravděpodobností způsobena plazmocelulární variantou [7]. Podle různých studií mělo 31–50 % pacientů asymptomatickou masu, u 33–69 % se vyskytly lokální nebo systémové příznaky [9,32,33].
Unicentrická forma CD postihuje v 46–70 % případů mediastinum, v 3–39 % břicho a v 10–15 % periferní uzlinové oblasti [5,9,34–36]. Úplné chirurgické odstranění má kurativní potenciál [9]. Medián nejčastějšího výskytu nemoci leží mezi 20 a 35 lety věku [34,37], muži i ženy jsou postiženy rovnoměrně. Postižené uzliny dosahují velikosti asi 6–7 cm (rozptyl 1–25 cm) [7]. Hyalinně-vaskulární varianta CD bývá nalézána u 74–91 % pacientů s lokalizovanou formou CD a manifestuje se solitárně zvětšenou lymfatickou uzlinou, zatímco minoritně zastoupená plazmocelulární forma obvykle postihuje skupinu mízních uzlin tvořících agregát [5,9,15,38,39]. U této formy onemocnění nebyla popsána zvýšená smrtnost [9].
Multicentrická forma
Generalizovaná forma CD byla jako multicentrická označena v roce 1985 [40]. Poprvé však byla popsána již v roce 1978 s charakteristickými znaky v podobě lymfadenopatie, systémových příznaků, organomegalie, progresivního klinického průběhu s maligním potenciálem [33]. Jedná se tedy o atypické lymfoproliferativní onemocnění charakterizované systémovou lymfadenopatií a celkovými zánětlivými příznaky [31].
Multicentrická forma je méně častý subtyp CD, nemocní jsou většinou starší než v případě unicentrické formy, pohlaví jsou zastoupena stejně [7]. Medián věku pacientů leží podle různých autorů mezi 48 a 57 lety [9,34,37]. Histologicky jsou ve většině případů nalézány změny svědčící pro plazmocelulární CD. Nejčastějším příznakem je horečka a asi 1/2 nemocných trpí hubnutím a nočními poty [41,42]. Často bývá přítomna celková slabost a nevůle, bolesti kloubů a kožní svědění. Asymptomatický průběh byl popsán v méně než 10 % případů [32,43].
Splenomegalie je vyjádřena asi u 33–79 %, hepatomegalie se vyskytuje téměř výlučně se splenomegalií a pozorujeme ji asi u 63 % pacientů [7]. Asi u 1/2 pacientů se setkáme s otoky nebo výpotky (fluidotorax, ascites). Laboratorní odchylky jsou konstantním nálezem. U všech nemocných s multicentrickou CD byla zjištěna zvýšená sedimentace erytrocytů, v 90 % anémie (chronických chorob nebo autoimunitní), trombocytopenie nebo elevace transamináz u 2/3 vyšetřených pacientů a polyklonální hypergamaglobulinemie v 85 % případů [32,41,43,44]. Dále byl popsán výskyt hypalbuminemie, snížené hladiny feritinu, zvýšených jaterních testů a mnohých autoprotilátek (zejména antinukleárních) [16,32,43,44].
V literatuře se uvádí, že onemocnění je často doprovázeno imunodeficiencí, sekundární amyloidózou, neuropatií, kožními lézemi, Kaposiho sarkomem (13 %), nehodgkinovými lymfomy (18 %), hemangiomy, plazmocytomy, karcinomy kolon, ledvin, štítné žlázy, dále i smíšenou chorobou pojiva, ale i POEMS syndromem (polyneuropatie, organomegalie, endokrinopatie, monoklonální gamapatie, kožní léze – skin) (15 %) [20,45–48]. Podle studie zveřejněné v roce 1985 došlo v průběhu nemoci k rozvoji příznaků z postižení centrálního nervového systému (včetně záchvatů a afázie) u 40 % pacientů [40]. Toto číslo však mohlo být zkresleno nerozpoznanou přítomností HIV pozitivity a následným rozvojem syndromu získané poruchy imunity (AIDS).
Diagnostika
Po vyloučení reaktivních lymfadenopatií u infekcí a systémových chorob pojiva je pro určení diagnózy onemocnění rozhodující histologický nález [49]. Na CD by se mělo myslet vždy u pacientů s Kaposiho sarkomem a POEMS syndromem, tedy ještě před biopsií uzlin, které jinak bývají tím prvním, co nás navede ke správné diagnóze [7].
Pro odlišení unicentrické od multicentrické CD mají zásadní význam radiodiagnostika a metody nukleární medicíny pro zjištění případné krční, hrudní, břišní a pánevní lymfadenopatie. Vstupní laboratorní testování zahrnuje vyšetření krevního obrazu, sedimentace erytrocytů, C reaktivního proteinu, jaterních testů, sérového kreatininu, elektroforézy séra, případné pozitivity virusu HIV a dále i pozitivity virusu HHV-8, která však není rutinně stanovována ve všech laboratořích. Měření cytokinového profilu organizmu (zejména hladin IL-6) je zatím rezervováno pro vědecké účely.
PET/CT přes svoji vysokou senzitivitu při diagnostice neoplazií není vhodné pro odlišení CD od jiných benigních nebo maligních lymfoproliferací [50]. CD jakožto benigní choroba vykazuje mírnou až střední akumulaci radiofarmaka nad lymfatickými uzlinami [50,51]. Lymfomy s nižším stupněm malignity však mohou také někdy vykazovat nízké vychytávání značené glukózy na PET skenech [52]. PET/CT je cenné vyšetření při odlišování unicentrické od multicentrické CD [50].
Terapie
Doporučení k léčbě CD lze jen obtížně formulovat, neboť neexistuje žádná standardní terapie. Literatura je tvořena popisy jednotlivých případů a malých souborů a až na výjimky nejsou k dispozici výsledky randomizovaných studií. Pro volbu optimální léčebné strategie je v první řadě rozhodující rozlišení unicentrické a multicentrické formy a dále pak stanovení HIV statusu pacienta v případě multicentrické CD [16].
Unicentrická forma
Kompletní chirurgická excize patologické masy je v případě unicentrické choroby téměř vždy kurativní, a to jak u hyalinně-vaskulární, tak u plazmocelulární varianty [53–60]. 5leté přežití pak dosahuje 100 % [5,61]. K recidivě však může dojít po subtotální nebo parciální resekci. V jedné studii podstoupilo všech 48 pacientů s lokalizovanou CD kompletní chirurgickou resekci a přežilo s excelentní prognózou, po dobu sledování (22–115 měsíců) se neobjevily žádné příznaky nemoci [9]. Po chirurgické resekci plazmocelulární CD dochází k remisi i případných asociovaných paraneoplastických a autoimunitních poruch. U operativy hyalinně-vaskulárního CD je třeba velké opatrnosti pro přítomnost rozvinuté vaskularizace tumoru, kdy bývá resekce často spojena s velkými perioperačními ztrátami krve [53].
Radioterapie byla použita s různou mírou úspěchu u neresekabilních nádorů [42,62]. I v monoterapii je schopna navodit kompletní remisi, ovšem ne u všech pacientů. Může být alternativou pro nemocné, kteří nemohou podstoupit chirurgický výkon [7].
Rituximab je monoklonální protilátka proti povrchovému antigenu B buněk, proteinu CD20, která cílové buňky vybírá pro destrukci buď cestou komplementu, nebo přes cytotoxické buňky [63]. Tato látka se osvědčila u hyalinně-vaskulární formy jak v monoterapii [64], tak i při neoadjuvantním použití u primárně neresekabilních případů [65].
Multicentrická forma, HIV negativní status
Výsledky chirurgického debulkingu u multicentrické CD nedosáhly dle dostupné literatury významnějších léčebných odpovědí [37]. Jeden pacient s hyalinně-vaskulární formou multicentrické CD však přežil dlouhodobě (sledování 82 měsíců) po cervikální a axilární disekci lymfatických uzlin [9].
V několika případech bylo dosaženo léčebné odpovědi radioterapií [16]. U dvou pacientů vedlo ozáření největší skupiny lymfatických uzlin dokonce k regresi i ve vzdálenějších lokalizacích [66,67].
Alkylační cytostatika jako součást intenzivní chemoterapie bývají často používána v rámci léčby první linie. Vzorem pro cytoredukční léčbu se staly režimy určené pro terapii nehodgkinových lymfomů [7]. Mezi nejčastěji používané režimy se řadí CHOP (cyklofosfamid, vinkristin, doxorubicin, prednison) a CVAD (cyklofosfamid, vinkristin, doxorubicin, dexametazon), oba využívané se smíšenými úspěchy. Publikované malé studie udávají počet léčebných odpovědí 50 % u CHOP a 67 % u CVAD [37,42].
Dle publikovaných popisů případů mají kortikosteroidy potenciál navodit u některých nemocných remisi onemocnění [63]. Frizzera et al popisují 6 pacientů, kteří byli léčeni glukokortikoidy, u 2 se podařilo dostat nemoc pod kontrolu při dlouhodobém podávání kortikoidů, u 4 nebylo dosaženo žádné trvalé odpovědi [40]. Dlouhodobé užívání kortikosteroidů však může být spojeno s vysokým rizikem bakteriální infekce u pacientů s multicentrickou formou, kdy bylo popsáno mnoho případů úmrtí na sepsi při terapii [32,40].
Cílená biologická léčba CD monoklonálními protilátkami proti receptoru pro IL-6 nebo proti samotnému cytokinu dosahuje u HIV negativních pacientů excelentních terapeutických výsledků [2]. Tyto látky výrazně zmírňují příznaky a upravují biochemické abnormality multicentrické CD, ačkoliv se projevy většinou po vysazení léčby vrátí [31,68–70]. Tocilizumab je humanizovaná monoklonální protilátka proti receptoru pro IL-6. První prospektivní studie popisuje zkušenosti s 60týdenní léčbou u 28 pacientů. Aplikováno bylo vždy 8 infuzí po 8 mg/kg tocilizumabu každé 2 týdny. Po 16 týdnech došlo ke zmenšení lymfadenomegalie, poklesu zánětlivých parametrů, vzestupu hemoglobinu, albuminu, celkového cholesterolu, HDL a BMI (body mass indexu), únava se snížila [31]. Intravenózně aplikovaný tocilizumab v dávce 8 mg/kg každé 2 týdny je schválený pro terapii multicentrické CD v Japonsku [68].
Rovněž bylo popsáno úspěšné použití rituximabu po selhání předchozí léčby pomocí kortikosteroidů [71], chemoterapie [72], ale např. i u HIV a HHV-8 negativního muže s CD asociovanou s autoimunitní hemolytickou anémií a Raynaudovým fenoménem [73]. S úspěchem jsou využívány kombinované režimy s klasickou chemoterapií [74].
Léčebné úspěchy byly zaznamenány i při použití imunomodulačních látek u malých skupin pacientů. V literatuře lze nalézt popisy případů, u nichž měla léčba interferonem α nebo all-trans retinovou kyselinou výborný terapeutický efekt [63]. Thalidomid navíc snižuje produkci IL-6 a má rovněž antiangiogenní vlastnosti. Výhoda podávání thalidomidu spočívá v dobrém bezpečnostním profilu léku, absenci významné myelotoxicity, a to i při několikaletém podávání, limitující však může být rozvoj periferní neuropatie [63,75,76]. Vynikající účinky thalidomidu jsou doloženy několika kazuistikami. U jedné pacientky došlo ke kompletní remisi cytopenie, ascitu a perikardiálního výpotku po dobu 40 měsíců na monoterapii thalidomidem, přetrvávala pouze lymfadenopatie. Po tuto dobu byla pacientka asymptomatická, začala opět pracovat, aniž ji neuropatie omezovala [77,78]. Ke zlepšení klinického i laboratorního statusu a regresi lymfadenomegalie a hepatosplenomegalie došlo dále u pacientky s POEMS syndromem asociovaným s hyalinně-vaskulární CD, kde byla zpočátku léčba doplněna o dexametazon [79]. Thalidomidem bylo dosaženo remise ne-frotického syndromu v jednom případě CD [80] a úspěch byl zaznamenán i u pacientky s CD asociovaným s paraneoplastickým pemfigem [81].
Lenalidomid je funkční a strukturální analog thalidomidu s protizánětlivými, antiangiogenními a imunomodulačními účinky. Stejně jako thalidomid snižuje produkci IL-6. Je schválen pro léčbu mnohočetného myelomu a myelodysplastického syndromu, navíc byly popsány jeho pozitivní účinky u pacientů s rezistentní chronickou lymfatickou leukemií, folikulárním lymfomem, lymfomem z plášťových buněk, difuzním velkobuněčným B lymfomem, ale i u solidních tumorů. Na rozdíl od thalidomidu je hlavním nežádoucím účinkem lenalidomidu jeho myelotoxicita [82,83]. Použití lenalidomidu u CD je dosud zcela ojedinělé. Tímto popisem našeho případu nejen dokazujeme jeho léčebné účinky u této diagnózy, ale demonstrujeme i vynikající bezpečnostní profil léku, a to i při dlouhodobém podávání, které u našeho pacienta dosáhlo 15 měsíců, tedy déle než v případě hůře tolerovaného thalidomidu. Absenci typické myelotoxicity lenalidomidu vysvětlujeme přítomností funkční, nepostižené kostní dřeně pacienta. Podobné zkušenosti máme i při použití lenalidomidu u pacienta s refrakterní multisystémovou histiocytózou z Langerhansových buněk, kde rovněž nebyla dřeň nemocí postižena a během 11 cyklů se v krevním obrazu neobjevily známky myelotoxicity [84].
Multicentrická forma, HIV pozitivní status
S ohledem na preexistující těžkou imunosupresi u těchto pacientů je další medikamentózní imunosuprese riziková. Na rozdíl od vynikajících výsledků u Kaposiho sarkomu po podání vysoce účinné antiretrovirové terapie (HAART) k regresi CD většinou nedochází. Ačkoliv bylo dále prokázáno, že některá antivirotika (foscarnet, ganciklovir, cidofovir) mají in vitro potenciál přerušit replikaci virusu HHV-8, úspěchy v klinické praxi při použití těchto léků byly smíšené [16]. U 3 HIV a HHV-8 pozitivních pacientů s multicentrickou CD bylo dosaženo ganciklovirem (nebo derivátem pro orální aplikaci, valganciklovirem) klinické remise a poklesu HHV-8 viremie [85].
Formou kazuistik a popisů malých souborů pacientů byly zveřejněny účinky chemoterapie [86,87] a interferonu α [88]. Největší studie s rituximabem zahrnovala 21 HIV a HHV-8 pozitivních pacientů, z nichž u 20 bylo dosaženo remise onemocnění a u 14 měla tato remise i radiologický korelát. Celkové 2leté přežití bylo 95 %. Po léčbě rituximabem poklesla i HHV-8 virusová nálož v plazmě. Hlavním nežádoucím efektem byla reaktivace Kaposiho sarkomu [44]. Stary et al dosáhli kombinovaným režimem s thalidomidem (200 mg, později přechod na 100 mg) a rituximabem kompletní klinické a radiologické remise plazmocelulární CD u HIV a HHV-8 pozitivního muže [89]. U jednoho HIV a HHV-8 pozitivního muže s Kaposiho sarkomem došlo ke zlepšení celkového stavu, zvýšení počtu destiček a negativnímu restagingovému vyšetření z kostní dřeně po 38 týdnech léčby s 200 mg thalidomidu denně, která byla na začátku doplněna o etoposid [90].
Průběh a prognóza onemocnění
Lokalizovaná forma CD většinou negativně ovlivňuje organizmus pouze tlakem na okolní tkáně i orgány a kompletní chirurgická resekce přináší konečné řešení včetně odeznění případných systémových příznaků. Prognóza je příznivá i v případě parciální resekce, kdy postižené oblasti mohou být dlouhodobě v klinické remisi, bez známek progrese nemoci [5,32,42]. Na druhé straně i přes kompletní resekci je unicentrická varianta sdružena se zvýšeným rizikem sekundární amyloidózy [91] a lymfomů [32,37,42]. Je proto nezbytné tyto pacienty dlouhodobě dispenzarizovat. Casper doporučuje u unicentrické CD radiologické zhodnocení lymfadenopatie za 6–12 měsíců po skončení terapie [7].
Prognóza multicentrické CD je bez léčby špatná [68]. Většina pacientů s multicentrickou CD zemře na fulminantní infekce, renální selhání, progresi nemoci nebo příbuzné malignity (Kaposiho sarkom, folikulární dendritický buněčný nádor, nehodgkinův nebo Hodgkinův lymfom) [1,11,41]. Smrtnost při sepsi dosahuje až 50 % [49]. Ve 4 největších souborech pacientů s multicentrickou CD byl medián přežití 14–30 měsíců. Někteří pacienti však žili pouze několik týdnů po stanovení diagnózy, jiní přežívali až 20 let [37,40,92,93]. Weisenburger et al rozdělili průběh multicentrické CD do 4 skupin:
- stabilní nemoc,
- chronicky relabující nemoc,
- agresivní choroba,
- rozvoj maligní lymfoproliferace [92].
V případě HIV pozitivity je významně vyšší riziko progrese do lymfomu a průběh CD je prudší [16,94].
Závěry pro praxi
- CD je důležitou součástí diferenciální diagnostiky lymfadenopatie, mikrocytární anémie, horečky nejasného původu a dalších B symptomů (hubnutí, noční poty).
- PET/CT vyšetření má význam při stagingovém hodnocení rozsahu CD u pacientů, jak dokládáme v přiložené obrazové dokumentaci.
- U všech pacientů s CD, včetně pacientů s unicentrickou hyalinně--vaskulární variantou po kurativní resekci, je nezbytná dlouhodobá dispenzarizace pro zvýšené riziko rozvoje maligní lymfoproliferace.
- Testování na případnou pozitivitu HIV by mělo být standardním vstupním vyšetřením, zejména u pacientů s multicentrickou formou.
- Lenalidomid v běžném dávkování představuje atraktivní léčebnou alternativu s velmi příznivým bezpečnostním profilem, a lze proto zvážit jeho podání i v rámci první linie léčby.
Poděkování
Léčba pacienta se uskutečnila za laskavé podpory firmy Celgene, s.r.o., z projektu Compassionate Use Program určeného pro pacienty se vzácnými hematoonkologickými onemocněními.
Tato publikace byla připravena v rámci aktivity následujících grantů: IGA MZd NT13492, NT12130, MUNI/A/0784/2011 a za institucionální podpory výzkumné organizace poskytnuté Ministerstvem zdravotnictví ČR v roce 2012.
MUDr. Petr Szturz, Ph.D.
www.fnbrno.cz
e-mail: petr.szturz@fnbrno.cz
Doručeno do redakce: 21. 12. 2011
Přijato po recenzi: 6. 5. 2012
Sources
1. Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol 2009; 16: 236–246.
2. Van Rhee F, Stone K, Szmania S et al. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol 2010; 8: 486–498.
3. Castleman B, Towne VW. Case records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. Cabot. N Engl J Med 1954; 251: 396–400.
4. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956; 9: 822–830.
5. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972; 29: 670–683.
6. Flendrig JA, Schillings PM. Benign giant lymphoma: the clinical signs and symptoms. Folia Medica Neerlandica 1969; 12: 119–120.
7. Casper C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol 2005; 129: 3–17.
8. Moore DF, Preti A, Tran SM. Prognostic implications following an indeterminate diagnostic work-up of lymphoma. Blood 1996; 88 (Suppl 1): 229a.
9. Ye B, Gao SG, Li W et al. A retrospective study of unicentric and multicentric Castleman’s disease: a report of 52 patients. Med Oncol 2010; 27: 1171–1178.
10. Kessler P. Profylaxe a léčba tromboembolické nemoci v onkologii. Vnitř Lék 2009; 55: 219–222.
11. Choi JH, Jo YJ, Gong SJ et al. Unicentric Castleman disease is not clearly distinguished from multicentric type: a case report. Clin Lymphoma Myeloma 2008; 8: 256–259.
12. Danon AD, Krishnan J, Frizzera G. Morpho--immunophenotypic diversity of Castleman’s disease, hyaline-vascular type: with emphasis on a stroma-rich variant and a new pathogenetic hypothesis. Virchows Arch A Pathol Anat Histopathol 1993; 423: 369–382.
13. Ioachim HL, Medeiros LJ. Ioachim’s lymph node pathology. Philadelphia: Lippincott Williams & Wilkins 2008.
14. McClain KL, Natkunam Y, Swerdlow SH. Atypical cellular disorders. Hematology Am Soc Hematol Educ Program 2004: 283–296.
15. McCarty MJ, Vukelja SJ, Banks PM et al. Angiofollicular lymph node hyperplasia (Castleman’s disease). Cancer Treat Rev 1995; 21: 291–310.
16. Dispenzieri A. Castleman disease. In: Ansell SM (ed.). Rare Hematological Malignancies. Boston: Springer Science + Business Media 2008: 293–330.
17. Leger-Ravet MB, Peuchmaur M, Devergne O et al. Interleukin-6 gene expression in Castleman’s disease. Blood 1991; 78: 2923–2930.
18. Yoshizaki K, Matsuda T, Nishimoto N et al. Pathogenic significance of interleukin-6 (IL-6//BSF-2) in Castleman’s disease. Blood 1989; 74: 1360–1367.
19. Hsu SM, Waldron JA, Xie SS et al. Expression of interleukin-6 in Castleman’s disease. Hum Pathol 1993; 24: 833–839.
20. Oksenhendler E, Carcelain G, Aoki Y et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood 2000; 96: 2069–2073.
21. Gloddek J, Pagotto U, Paez Pereda M et al. Pituitary adenylate cyclase-activating polypeptide, interleukin-6 and glucocorticoids regulate the release of vascular endothelial growth factor in pituitary folliculostellate cells. J Endocrinol 1999; 160: 483–490.
22. van Kooten C, Rensink I, Aarden L et al. Effect of IL-4 and IL-6 on the proliferation and differentiation of B-chronic lymphocytic leukemia cells. Leukemia 1993; 7: 618–624.
23. Vinzio S, Ciarloni L, Schlienger JL et al. Isolated microcytic anemia disclosing a unicentric Castleman disease: The interleukin-6/hepcidin pathway? Eur J Intern Med 2008; 19: 367–369.
24. Nemeth E, Valore EV, Territo M et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101: 2461–2463.
25. Nemeth E, Rivera S, Gabayan V et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271–1276.
26. Ganz T. Hepcidin – a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol 2005; 18: 171–182.
27. Kawabata H, Tomosugi N, Kanda J et al. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman’s disease. Haematologica 2007; 92: 857–858.
28. Katano H, Sato Y, Kurata T et al. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 2000; 269: 335–344.
29. Moore PS, Boshoff C, Weiss RA et al. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996; 274: 1739–1744.
30. Veldhuis GJ, van der Leest AH, de Wolf JT et al. A case of localized Castleman’s disease with systemic involvement: treatment and pathogenetic aspects. Ann Hematol 1996; 73: 47–50.
31. Nishimoto N, Kanakura Y, Aozasa K et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005; 106: 2627–2632.
32. Bowne WB, Lewis JJ, Filippa DA et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer 1999; 85: 706–717.
33. Gaba AR, Stein RS, Sweet DL et al. Multicentric giant lymph node hyperplasia. Am J Clin Pathol 1978; 69: 86–90.
34. Ko SF, Wan YL, Ng SH et al. Imaging features of atypical thoracic Castleman disease. Clin Imaging 2004; 28: 280–285.
35. Gangopadhyay K, Mahasin ZZ, Kfoury H. Pathologic quiz case 2. Castleman disease (giant lymph node hyperplasia). Arch Otolaryngol Head Neck Surg 1997; 123: 1137–1139.
36. Johkoh T, Müller NL, Ichikado K et al. Intrathoracic multicentric Castleman disease: CT findings in 12 patients. Radiology 1998; 209: 477–481.
37. Herrada J, Cabanillas F, Rice L et al. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med 1998; 128: 657–662.
38. Frizzera G. Castleman’s disease and related disorders. Semin Diagn Pathol 1988; 5: 346–364.
39. Menke DM, Camoriano JK, Banks PM. Angiofollicular lymph node hyperplasia: a comparison of unicentric, multicentric, hyaline vascular, and plasma cell types of disease by morphometric and clinical analysis. Mod Pathol 1992; 5: 525–530.
40. Frizzera G, Peterson BA, Bayrd ED et al. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol 1985; 3: 1202–1216.
41. Peterson BA, Frizzera G. Multicentric Castleman’s disease. Semin Oncol 1993; 20: 636–647.
42. Chronowski GM, Ha CS, Wilder RB et al. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer 2001; 92: 670–676.
43. Sagaert X, De Wolf-Peeters C. De ziekte van Castleman: twee afzonderlijke ziekte-entiteinten. Tijdschr Geneesk 2004; 60: 949–956.
44. Bower M, Powles T, Williams S et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med 2007; 147: 836–839.
45. Feigert JM, Sweet DL, Coleman M et al. Multicentric angiofollicular lymph node hyperplasia with peripheral neuropathy, pseudotumor cerebri, IgA dysproteinemia, and thrombocytosis in women. A distinct syndrome. Ann Intern Med 1990; 113: 362–367.
46. Bitter MA, Komaiko W, Franklin WA. Giant lymph node hyperplasia with osteoblastic bone lesions and the POEMS (Takatsuki’s) syndrome. Cancer 1985; 56: 188–194.
47. Mandler RN, Kerrigan DP, Smart J et al. Castleman’s disease in POEMS syndrome with elevated interleukin-6. Cancer 1992; 69: 2697–2703.
48. Bélec L, Mohamed AS, Authier FJ et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood 1999; 93: 3643–3653.
49. Wang SH, Ruan Z, Huang HL et al. A rare case of Castleman disease presenting as pulmonary mass mimicking central pulmonary malignancy. Chin Med J (Engl) 2009; 122: 990–991.
50. Halac M, Ergul N, Sager S et al. PET/CT findings in a multicentric form of Castleman’s disease. Hell J Nucl Med 2007; 10: 172–174.
51. Murphy SP, Nathan MA, Karwal MW. FDG-PET appearance of pelvic Castleman’s disease. J Nucl Med 1997; 38: 1211–1212.
52. Leskinen-Kallio S, Ruotsalainen U, Någren K et al. Uptake of carbon-11-methionine and fluorodeoxyglucose in nehodgkin’s lymphoma: a PET study. J Nucl Med 1991; 32: 1211–1218.
53. Mohanna S, Sanchez J, Ferrufino JC et al. Characteristics of Castleman’s disease in Peru. Eur J Intern Med 2006; 17: 170–174.
54. Seco JL, Velasco F, Manuel JS et al. Retroperitoneal Castleman’s disease. Surgery 1992; 112: 850–855.
55. Larroche C, Cacoub P, Godeau P. Castleman’s disease. Rev Med Interne 1996; 17: 1003–1013.
56. Ebisuno S, Yamauchi T, Fukatani T et al. Retroperitoneal Castleman’s disease: a case report and brief review of tumors of the pararenal area. Urol Int 1989; 44: 169–172.
57. Takihara H, Yamakawa G, Baba Y et al. Castleman disease. Unusual retroperitoneal location indistinguishable from malignant tumor in preoperative angiographic appearance. Urology 1993; 41: 162–164.
58. Kiguchi H, Ishii T, Ishikawa Y et al. Castleman’s disease of the abdomen and pelvis: report of three cases and a review of the literature. J Gastroenterol 1995; 30: 661–666.
59. Bartkowski DP, Ferrigni RG. Castleman’s disease: an unusual retroperitoneal mass. J Urol 1988; 139: 118–120.
60. Skolnik G, Wiklund LM, Risberg B. Castleman’s tumor with retroperitoneal location: a malignant-appearing benign tumor. J Surg Oncol 1985; 28: 153–155.
61. d’Agay MF, Miclea JM, Clauvel JP et al. Castleman’s disease: a well defined histological pattern for a widely divergent clinical spectrum. Nouv Rev Fr Hematol 1989; 31: 145–148.
62. Nordstrom DG, Tewfik HH, Latourette HB. Giant lymph node hyperplasia: a review of literature and report of two cases of plasma cell variant responding to radiation therapy. Int J Radiat Oncol Biol Phys 1978; 4: 1045–1048.
63. Adam Z, Krejčí M, Vorlíček J. Hematologie: přehled maligních hematologických nemocí. Praha: Grada Publishing 2008.
64. Estephan FF, Elghetany MT, Berry M et al. Complete remission with anti-CD20 therapy for unicentric, non-HIV-associated, hyaline-vascular type, Castleman’s disease. Cancer Invest 2005; 23: 191.
65. Bandera B, Ainsworth C, Shikle J et al. Treatment of unicentric Castleman disease with neoadjuvant rituximab. Chest 2010; 138: 1239–1241.
66. Marti S, Pahissa A, Guardia J et al. Multicentric giant follicular lymph node hyperplasia. Favorable response to radiotherapy. Cancer 1983; 51: 808–810.
67. Sethi T, Joshi K, Sharma SC et al. Radiation therapy in the management of giant lymph node hyperplasia. Br J Radiol 1990; 63: 648–650.
68. Matsuyama M, Suzuki T, Tsuboi H et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman’s disease. Intern Med 2007; 46: 771–774.
69. Nishimoto N, Sasai M, Shima Y et al. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000; 95: 56–61.
70. Beck JT, Hsu SM, Wijdenes J et al. Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med 1994; 330: 602–605.
71. Ide M, Ogawa E, Kasagi K et al. Successful treatment of multicentric Castleman’s disease with bilateral orbital tumour using rituximab. Br J Haematol 2003; 121: 818–819.
72. Gholam D, Vantelon JM, Al-Jijakli A et al. A case of multicentric Castleman’s disease associated with advanced systemic amyloidosis treated with chemotherapy and anti-CD20 monoclonal antibody. Ann Hematol 2003; 82: 766–768.
73. Ocio EM, Sanchez-Guijo FM, Diez-Campelo M et al. Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am J Hematol 2005; 78: 302–305.
74. Fragasso A, Mannarella C, Ciancio A et al. Complete remission and virologic response to combined chemoimmunotherapy (R-CVP) followed by rituximab maintenance in HIV-negative, HHV-8 positive patient with multicentric Castleman disease. Leuk Lymphoma 2008; 49: 2224–2226.
75. Adam Z, Krejčí M, Tichý M et al. Léčba selhání ledvin u mnohočetného myelomu. Vnitř Lék 2009; 55: 570–582.
76. Fishman SJ, Feins NR, D’Amato RJ et al. Long-term remission of Crohn’s disease treated with thalidomide: a seminal case report. Angiogenesis 1999; 3: 201–204.
77. Lee FC, Merchant SH. Alleviation of systemic manifestations of multicentric Castleman’s disease by thalidomide. Am J Hematol 2003; 73: 48–53.
78. Starkey CR, Joste NE, Lee FC. Near-total resolution of multicentric Castleman disease by prolonged treatment with thalidomide. Am J Hematol 2006; 81: 303–304.
79. Kim SY, Lee SA, Ryoo HM et al. Thalidomide for POEMS syndrome. Ann Hematol 2006; 85: 545–546.
80. Menegato MA, Canelles MF, Tonutti E et al. Remission of nephrotic syndrome after thalidomide therapy in a patient with Castleman’s disease. Clin Nephrol 2004; 61: 352–356.
81. Miltenyi Z, Toth J, Gonda A et al. Successful immunomodulatory therapy in castleman disease with paraneoplastic pemphigus vulgaris. Pathol Oncol Res 2009; 15: 375–381.
82. Palumbo A, Rajkumar SV. Multiple myeloma: chemotherapy or transplantation in the era of new drugs. Eur J Haematol 2010; 84: 379–390.
83. Vallet S, Palumbo A, Raje N et al. Thalidomide and lenalidomide: Mechanism-based potential drug combinations. Leuk Lymphoma 2008; 49: 1238–1245.
84. Szturz P, Adam Z, Rehák Z et al. Lenalidomide proved effective in multisystem Langerhans cell histiocytosis. Acta Oncol 2012; 51: 412–415.
85. Casper C, Nichols WG, Huang ML et al. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood 2004; 103: 1632–1634.
86. Oksenhendler E, Duarte M, Soulier J et al. Multicentric Castleman’s disease in HIV infection: a clinical and pathological study of 20 patients. AIDS 1996; 10: 61–67.
87. Loi S, Goldstein D, Clezy K et al. Castleman’s disease and HIV infection in Australia. HIV Med 2004; 5: 157–162.
88. Kumari P, Schechter GP, Saini N et al. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis 2000; 31: 602–604.
89. Stary G, Kohrgruber N, Herneth AM et al. Complete regression of HIV-associated multicentric Castleman disease treated with rituximab and thalidomide. AIDS 2008; 22: 1232–1234.
90. Jung CP, Emmerich B, Goebel FD et al. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol 2004; 75: 176–177.
91. Lachmann HJ, Gilbertson JA, Gillmore JD et al. Unicentric Castleman’s disease complicated by systemic AA amyloidosis: a curable disease. QJM 2002; 95: 211–218.
92. Weisenburger DD, Nathwani BN, Winberg CD et al. Multicentric angiofollicular lymph node hyperplasia: a clinicopathologic study of 16 cases. Hum Pathol 1985; 16: 162–172.
93. Oksenhendler E, Duarte M, Soulier J et al. Multicentric Castleman’s disease in HIV infection: a clinical and pathological study of 20 patients. AIDS 1996; 10: 61–67.
94. Dupin N, Diss TL, Kellam P et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000; 95: 1406–1412.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2012 Issue 9
Most read in this issue
- Povrchová tromboflebitida, neprávem podceňovaná choroba – je čas změnit názor?
- Upozornění na nebezpečí invazivních infekcí u splenektomovaných pacientů. Zkušenosti z FN Brno 2011
- Morbus Weil – kazuistika a princípy
- Hyperventilační echokardiografie v diagnostice vazospastické anginy pectoris