
Léčba AL-amyloidózy v roce 2012, přínos nových léků (bortezomibu, thalidomidu a lenalidomidu). Přehled publikovaných klinických studií
Treatment of AL amyloidosis in 2012; the benefit of new drugs (bortezomib, thalidomide, and lenalidomide). Summary of published clinical trials
Until 2011, the gold standard of treatment for patients with AL amyloidosis was the combination of alkylating cytostatics (melphalan or cyclophosphamide) and dexamethasone. For a selected group of patients under 65 years of age with only moderate damage to their body caused by amyloid and with good cardiac function (EF> 40%), high-dose chemotherapy with autologous hematopoietic cell transplantation seems to be optimal. Patients with AL amyloidosis and low bone marrow plasma cell count generally undergo the harvest of hematopoietic cells from peripheral blood, followed by high-dose chemotherapy immediately after they are diagnosed. In contrast to multiple myeloma, high-dose chemotherapy is not preceded by several months of conventional treatment. The year 2012 witnessed a release of reports about extensive experience with new drugs that were used in Phase I and Phase II clinical trials, and in isolated cases also in Phase III, for the treatment of patients with AL amyloidosis. Based on these studies it can be concluded that among the new available drugs (bortezomib, thalidomide and lenalidomide) bortezomib is the drug with the greatest curative effect in patients with AL amyloidosis; it achieved 24–37% of complete remissions in monotherapy. The greatest number of treatment responses was reported during the treatment that combined bortezomib, alkylating cytostatics and dexamethasone. This treatment showed significantly more treatment responses during the first-line drug therapy than during therapies that followed. Clinical trials with lenalidomide combined with other drugs saw a lower number of treatment responses than the number described in treatment with bortezomib combined with other drugs. That is the reason why lenalidomide combinations are not considered the optimal first-line therapy, with the exception of AL amyloidosis with bortezomib contraindication (severe neuropathy caused by AL amyloidosis). It was confirmed that lenalidomide combined with other drugs could cause remission in patients whose disease was resistant to the initial bortezomib therapy. Lenalidomide (or alternatively also thalidomide) can therefore be used as second-line therapy if bortezomib therapy proves unsuccessful, with the possibility of achieving a complete remission. The increase in the number of complete remissions brought about by bortezomib therapies in patients with AL amyloidosis poses a question about which treatment should be used for younger patients with only moderate damage to their body, i.e. high-dose chemotherapy with autologous hematopoietic cell transplantation or combined treatment with bortezomib. Additional comparative studies are required to be able to answer that question and determine which of the aforesaid therapy modalities is optimal. A question still remains whether the increase in the number of complete remissions due to bortezomib will also bring about longer survival comparable to the results of high-dose chemotherapy treatment with autologous hematopoietic cell transplantation.
Key words:
AL amyloidosis – bortezomib – lenalidomide – thalidomide – high-dose chemotherapy with autologous transplantation
Authors:
Z. Adam 1; V. Ščudla 2; M. Krejčí 1; Z. Čermáková 3; L. Pour 1; Z. Král 1
Authors‘ workplace:
Interní hematologická a onkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; III. interní klinika - nefrologie, revmatologie a endokrinologie Lékařské fakulty UP a FN Olomouc, přednosta prof. MUDr. Josef Zadražil, CSc.
2; Oddělení klinické biochemie FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Milan Dastych, CSc., MBA
3
Published in:
Vnitř Lék 2013; 59(1): 37-58
Category:
Review
Overview
Zlatým standardem léčby pro pacienty s AL-amyloidózou byla až do roku 2011 kombinace alkylačního cytostatika (melfalanu či cyklofosfamidu) s dexametazonem. Pro vybrané nemocné mladší 65 let, s nevelkým poškozením organizmu amyloidem a s dobrou srdeční funkcí (EF > 40 %), je považována za optimální vysokodávkovaná chemoterapie s autologní transplantací krvetvorných buněk. U pacientů s AL-amyloidózou s nízkým počtem plazmocytů v kostní dřeni se provádí sběr krvetvorných buněk z periferní krve a následná vysokodávkovaná chemoterapie ihned po stanovení diagnózy. Na rozdíl od mnohočetného myelomu nepředchází vysokodávkované chemoterapii několikaměsíční konvenční léčba. V roce 2012 byly zveřejněny rozsáhlé zkušenosti s novými léky, které byly použity pro léčbu pacientů s AL-amyloidózou v rámci klinických studií fáze I a II a ojediněle i fáze III. Vyplývá z nich, že z dostupných nových léků (bortezomib, thalidomid a lenalidomid) má u pacientů s AL-amyloidózou nejvyšší léčebnou účinnost bortezomib, který dosáhl v monoterapii 24–37 % kompletních remisí. Nejvyšší počet léčebných odpovědí přináší léčba kombinací bortezomibu s alkylačním cytostatikem a s dexametazonem. V první léčebné linii přináší tato léčba podstatně více léčebných odpovědí než při jejím použití v rámci dalších léčebných linií. V klinických studiích s lenalidomidem v kombinaci s dalšími léky byl zaznamenán menší počet léčebných odpovědí, než je popisováno při léčbě bortezomibem v kombinaci s dalšími léky. Proto se kombinace s lenalidomidem nepovažují za optimální léčbu první linie s výjimkou případů AL-amyloidózy s kontraindikací bortezomibu (těžká neuropatie způsobená AL-amyloidózou). Bylo prokázáno, že lenalidomid v kombinaci s dalšími léky může navodit remisi u pacientů, jejichž onemocnění bylo rezistentní k iniciální léčbě bortezomibem. Proto lze lenalidomid (a případně i thalidomid) použít jako léčbu druhé linie v případě neúspěchu léčby bortezomibem s nadějí na dosažení kompletní remise. Zvýšení počtu kompletních remisí, které přinesly léčebné postupy obsahující bortezomib pacientům s AL-amyloidózou, navozuje otázku, zda pro mladší pacienty s nevelkým poškozením organizmu použít vysokodávkovanou chemoterapii s autologní transplantací krvetvorných buněk, či kombinovanou léčbu s bortezomibem. K zodpovězení této otázky je zapotřebí dalších srovnávacích studií, které nám odpoví na otázku, která z těchto léčebných modalit je optimální. Zda zvýšení počtu kompletních remisí díky bortezomibu přinese automaticky také delší přežití, srovnatelné s výsledky léčby vysokodávkovanou chemoterapií s autologní transplantací krvetvorných buněk.
Klíčová slova:
AL – amyloidóza – bortezomib – lenalidomid – thalidomid – vysokodávkovaná chemoterapie s autologní transplantací
Základní informace o AL-amyloidóze
Biologie a klasifikace AL-amyloidózy
Amyloidóza z lehkých řetězců (immunoglobulin light chain amyloidosis) – akronymem AL-amyloidóza – je choroba, která vzniká důsledkem tvorby monoklonálních lehkých řetězců se specifickou mutací. Tato mutace způsobuje odlišnou (lineární) prostorovou konfiguraci volných lehkých řetězců na rozdíl od fyziologické α helikální. Díky této odchylné struktuře amyloidogenní lehké řetězce spontánně agregují, vznikají tak oligomery, a ty pak vytvářejí depozita amyloidových fibril ve stabilní lineární struktuře. Podstatně méně často se ukládají ve formě amorfních hmot (light chain deposition disease) [1,2].
Amyloidogenní lehké řetězce jsou produkovány monoklonálními plazmatickými buňkami, lokalizovanými v kostní dřeni, případně ve slezině. Počet těchto klonálních plazmocytů obvykle není nijak velký. S pomocí krevního oběhu se pak amyloidogenní lehké řetězce imunoglobulinů dostávající k cílovým orgánům, v nichž se usazují díky zatím neznámým interakcím s matrix jednotlivých tkání (s glykosaminoglykany a s molekulami buněčných membrán). Tyto interakce mohou potencovat tvorbu oligomerů klonálních lehkých řetězců a konečné formování amyloidového depozita. To, v kterém orgánu se vytvoří nejmasivnější depozitum, a tedy největší poškození, je determinováno geny pro variabilní část lehkého řetězce, tedy strukturou amyloidogenního lehkého řetězce [3–5].
Klinické projevy mohou být velmi pestré a jsou popsány ve vícero českých publikacích [6–11] a připomínáme je na obr. 1–10.
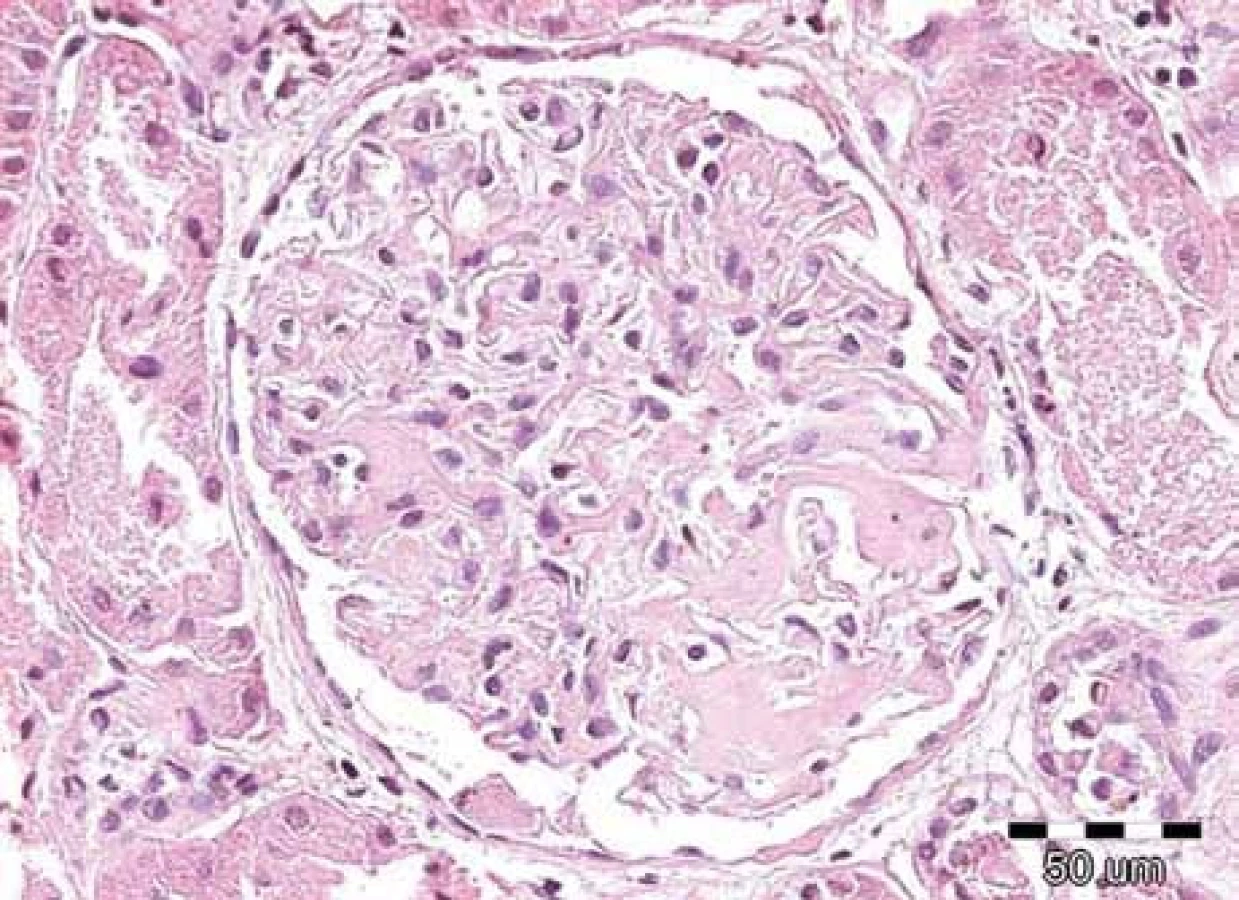
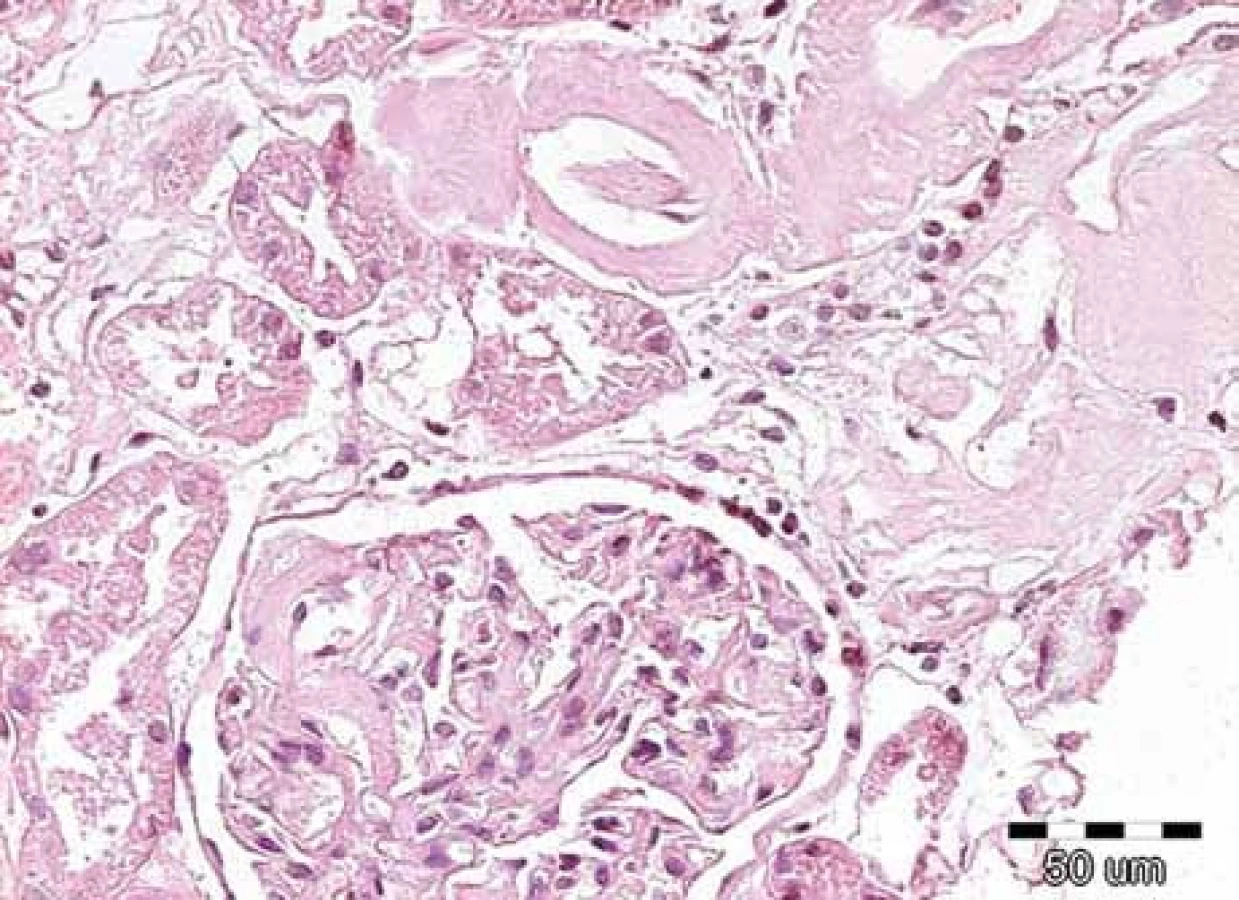
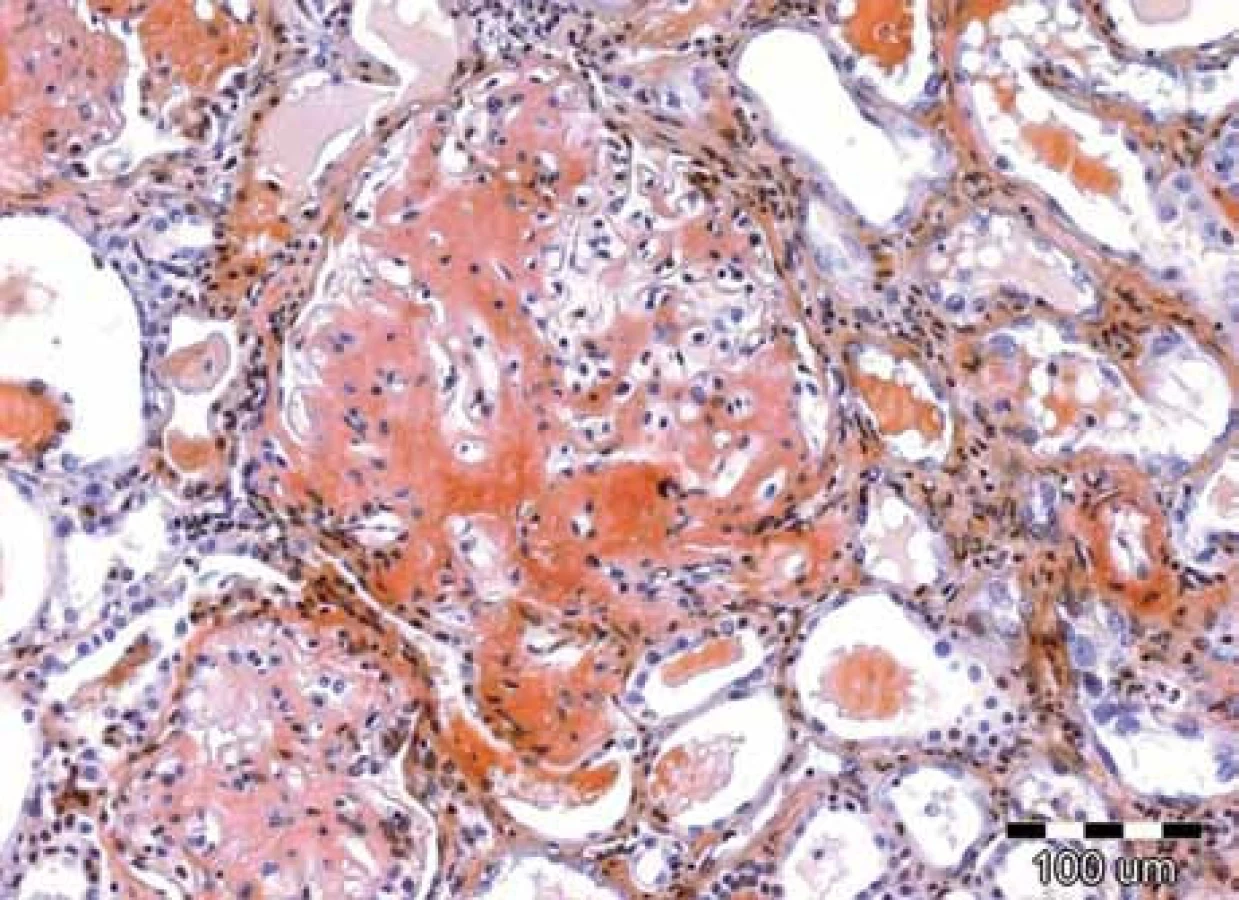
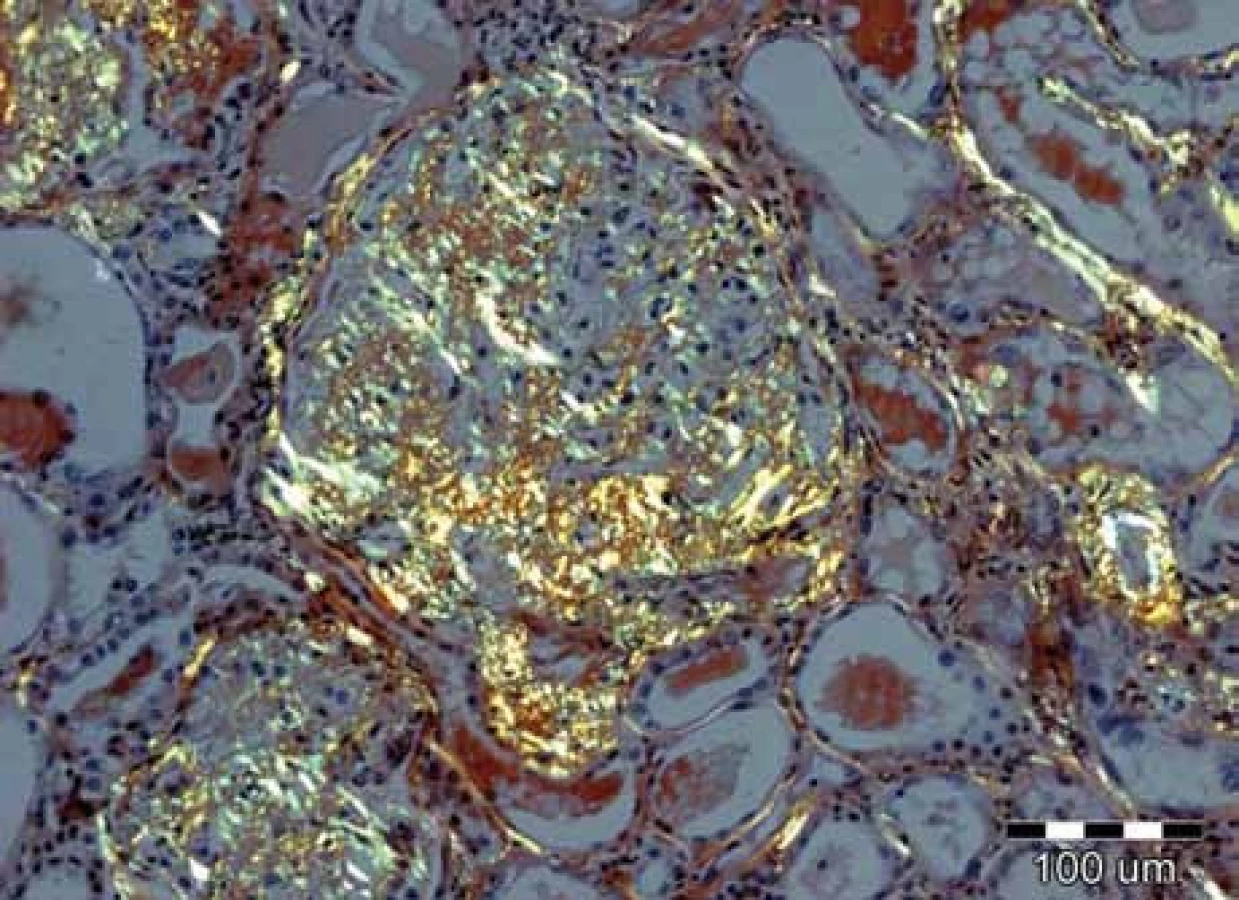
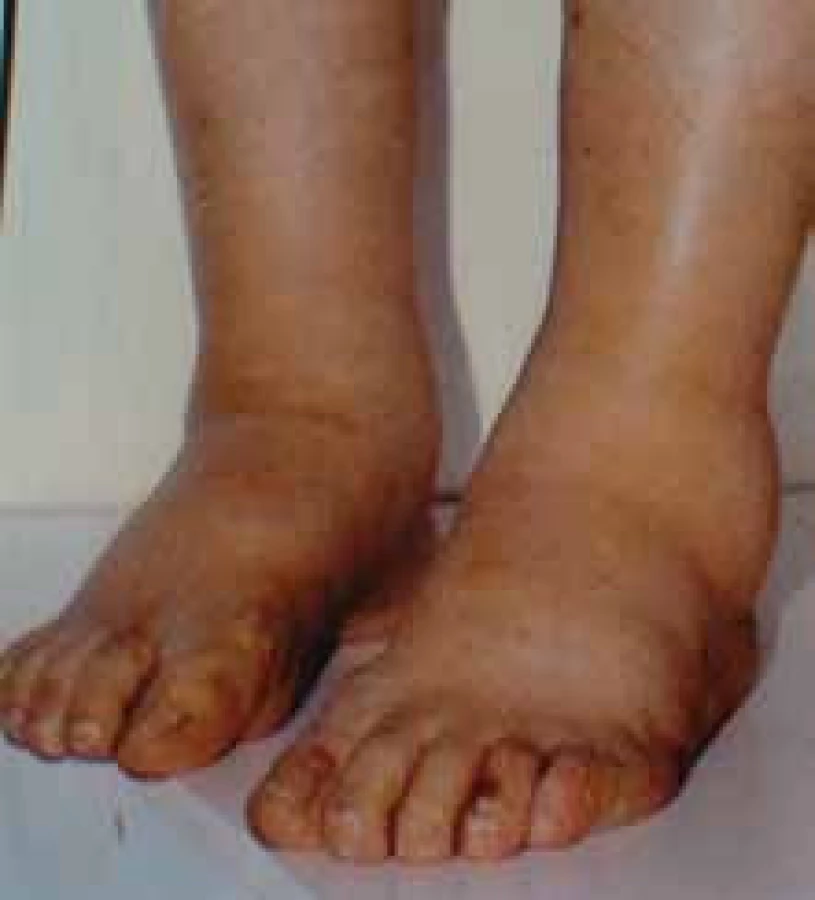


Zda a kde vznikne amyloidové depozitum, je dáno mutací genetické informace pro lehký řetězec imunoglobulinu v klonálním plazmocytu [2,3].
Podle rozsahu postižení tkání se rozlišuje:
- systémová AL-amyloidóza (je postižen difuzně jeden či více orgánů),
- ložisková AL-amyloidóza (jen jedno ložisko amyloidu v orgánu); je podstatně vzácnější než systémová AL-amyloidóza.
Podle počtu plazmocytů v kostní dřeni se pak formálně rozlišuje:
- systémová AL-amyloidóza provázející mnohočetný myelom (jde o systémovou AL-amyloidózu a jsou splněna všeobecně platná kritéria mnohočetného myelomu),
- primární systémová AL-amyloidóza (jde o systémovou AL-amyloidózu a nejsou splněna kritéria mnohočetného myelomu).
Problémy s rozpoznáním proteinu, z něhož je amyloid vytvořen
Obecnou histochemickou vlastností depozit patologických proteinů v lineární amyloidové struktuře je charakteristické zelené zbarvení po barvení konžskou červení. Jako další možné barvení se používá barvení thioflavinem a někdy také sulfátovou alciánovou modrou. V elektronovém mikroskopu jsou zřetelné nevětvené fibrily průměru 7,5–10 nm. Ale nejen molekuly monoklonálních lehkých řetězců mohou tvořit amyloidová depozita. V roce 2012 bylo popsáno 28 dalších molekul, které mohou také tvořit amyloidová depozita, obtížně odlišitelná od amyloidových depozit, tvořených amyloidogenními lehkými řetězci imunoglobulinu [12]. Pro terapeutický plán je zásadní správná typizace amyloidogenních proteinů, protože omyl může mít tragické následky. Např. provedení vysokodávkované chemoterapie s autologní transplantací kostní dřeně u pacienta s amyloidózou, tvořenou mutovaným transthyretinem, nebude mít žádný terapeutický efekt a naopak může pacienta poškodit, protože u nemocného s transthyretinovou amyloidózou je léčbou volby transplantace jater. Proto je nutné provést veškerá dostupná vyšetření s cílem přesně stanovit typ amyloidu a v nejasných případech před zahájením vlastní léčby konzultovat i specializovaná pracoviště [13,14].
Průkaz monoklonální gamapatie také neznamená jednoznačný důkaz toho, že se jedná o AL-amyloidózu. Asi 3 % starších osob mají monoklonální gamapatii nejistého významu (MGUS). Proto je možný současný výskyt hereditárního typu amyloidového depozita a monoklonální gamapatie. V roce 2002 byla uveřejněna analýza 350 pacientů s nálezem amyloidu a monoklonální gamapatie, u nichž se po prvním vyšetření předpokládala souvislost těchto 2 jevů. Vyšetřili u nich přítomnost dalších amyloidogenních bílkovin, či přítomnost amyloidogenní mutace genů (transthyretinu, apolipoproteinu A-1, lysozymu a α-řetězce fibrinogenu). Tato vyšetření odhalila hereditární amyloidózu u 9,7 % nemocných, u nichž se pro přítomnost monoklonální gamapatie předpokládala AL-amyloidóza. V 5 % se jednalo o amyloidózu z α-řetězce fibrinogenu, ve 4 % o amyloidózu způsobenou nerozpoznanou mutací transthyretinu. Amyloidóza z aberantního fibrinogenu poškozuje ledviny podobně jako AL-amyloidóza [15].
Imunohistochemická analýza depozit amyloidu má své limity, i když je prováděna zkušenými odborníky. Proto se v posledních letech ve světě dostává do popředí zájmu rozpoznávání bílkoviny, z níž je složeno amyloidové depozitum, pomocí hmotnostní spektrometrie [16–26]. Jak je zřejmé z citované literatury, je diagnostika hmotnostní spektrometrií mezi odborníky velmi aktuálním tématem.
Doufáme, že v brzké budoucnosti bude diagnostika amyloidogenních proteinů metodou hmotnostní spektrometrie dostupná i v České republice.
Incidence AL-amyloidózy
Výskyt amyloidózy není dostatečně zmapován. Autoři z Mayo Clinic uvedli 0,5–1,2 případu na 100 000 obyvatel, tedy 5–12 případů na milión obyvatel [27]. Incidenci podle francouzských autorů [28] uvádí tab. 1, tedy 2,4/1 000 000 obyvatel. Další údaje o incidenci amyloidózy v Evropě jsme nenalezli. Merlini v ústním sdělení potvrdil data z Mayo Clinic. Uvedl, že v jeho regionu (Toskánsko) dosahuje incidence amyloidózy až 10 případů na milión obyvatel, z toho 3/4 představuje AL-amyloidóza.
![Výskyt amyloidóz ve francouzském regionu Magy-Bertrand 2008 [28].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/22541d3f494770285e541c4801283dda.png)
Četnost AL-amyloidózy v ČR nebyla doposud publikována. Podle našich zkušeností je podstatně menší četnost pacientů s nově diagnostikovanou AL-amyloidózou oproti pacientům s nově diagnostikovaným mnohočetným myelomem (incidence myelomu je 4/100 000 obyvatel). Na naše pracoviště přichází přibližně 1 pacient s AL-amyloidózou na 10–20 pacientů s mnohočetným myelomem. To by odpovídalo spíše incidenci uvedené ve francouzské studii.
Rozdíly mezi nízkou incidencí z Francie a vysokou incidencí uváděnou z Mayo Clinic a také prof. Merlinim odvisí od způsobu sběru dat. Ve Francii byla hlášena jen klinicky manifestní onemocnění, zatímco data v regionu Mayo Clinic z Rochesteru a Merliniho data z Toskánska byla získána při realizaci plošného programu časného záchytu monoklonálních gamapatií. Podle prof. Merliniho tak byly odhaleny i oligosymptomatické formy, které by bez usilovného pátrání nebyly rozpoznány.
Podle incidence ve Francii lze tedy odhadovat, že incidence manifestních případů v ČR bude podobná, tedy asi 20–30 nových případů v celé ČR ročně. Při cílevědomějším pátrání po této nemoci by se mohla incidence zdvojnásobit či ztrojnásobit, ale zřejmě nepřekročí 100 nových případů v celé ČR za rok.
Jedná se tedy o relativně malou skupinu pacientů v celé České republice. Ale i tito pacienti by měli dostávat tu nejlepší léčbu podle současných poznatků lékařské vědy.
Poslední přehled léčebných postupů byl zveřejněn v roce 2009 [1]. Od té doby došlo k ověření přínosu nových léků (bortezomibu, thalidomidu, lenalidomidu) pro nemocné s touto chorobou, a proto se k tématu léčba AL-amyloidózy vracíme.
Léčba AL-amyloidózy
Cíle léčby a hodnocení léčebné odpovědi
Cílem léčby je rychlá eliminace tvorby amyloidogenních lehkých řetězců, což je podmínkou, aby došlo ke zlepšení funkce amyloidem poškozených orgánů [29,30]. V klinické studii bylo histologicky prokázáno, že po dokončení chemoterapie dochází k postupnému odbourávání a odstraňování amyloidu v místech jeho depozit. Při opakovaném vyšetřování podkožního tuku břišní stěny na přítomnost AL-amyloidu u pacientů po dokončení léčby bylo zjištěno, že depozita amyloidu vymizela u 80 % pacientů, kteří po 3,2 roku zůstávali v trvalé kompletní remisi, zatímco u pacientů v parciální remisi došlo k vymizení amyloidových depozit jen u velmi malé části nemocných [31].
Tato práce mění dosavadní pohled na léčbu této nemoci. Jasně dokazuje, že cílem léčby u AL-amyloidózy musí být dosažení kompletní hematologické remise. Dosažení parciální hematologické remise se dnes nepovažuje za dostatečné, protože neumožní reparační pochody v amyloidem poškozených tkáních a orgánech!
Při vyhodnocování léčby AL-amyloidózy se běžně udávají počty hematologických léčebných odpovědí, které jsou definovány na základně vyšetření přítomnosti amyloidotvorných lehkých řetězců a na základě vyšetření kostní dřeně [32,33], ale také počet orgánových léčebných odpovědí, které jsou také definovány publikovanými kritérii [1,34,35]. Definici hematologických léčebných odpovědí uvádí tab. 2.
![Definice hematologických léčebných odpovědí u pacientů s AL-amyloidózou z roku 2005 [34,35].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/5cdf1f10ea4ca1abf73aa7f57678b1c4.png)
Klasická léčba založená na vysokých dávkách dexametazonu a alkylačních cytostatikách
Klasickým léčebným postupem v předchozích desetiletích byla léčba melfalanem a prednisonem [36], stejně jako u mnohočetného myelomu. Tato léčba však přinášela nevelký počet léčebných odpovědí, nevedla k zásadnímu prodloužení přežití [37]. Léčba kombinací více alkylačních cytostatik také nevedla k zásadnímu zlepšení [38]. Zvýšení počtu léčebných odpovědí přineslo až přidání vysokých dávek dexametazonu do léčebných schémat. Monoterapie dexametazonem a udržovací léčba dexametazon + interferon dosáhla 24 % kompletních remisí [39].
Podobně i další klinické studie, v nichž byly použity léčebné režimy, obsahující vysoké dávky dexametazonu, prokázaly vyšší počet léčebných odpovědí než dříve používaný alkeran a prednison (alkeran a dexametazon), VAD (vinkristin, adriamycin a dexametazon) a jemu podobné režimy, střední dávky melfalanu a dexametazonu [40–43].
Ve studii, zveřejněné roku 2007, dosáhla kombinace melfalan + dexametazon léčebné odpovědi v 67 % případů, z toho u 33 % pacientů se jednalo o kompletní remisi (CR). Orgánové léčebné odpovědi bylo dosaženo ve 48 %. Dosažené léčebné odpovědi trvaly v 70 % nejméně 3 roky [44].
Nejaktuálnější hodnocení dat z italského centra uvádí v souboru 126 pacientů, léčených kombinací vysokých dávek dexametazonu a alkylačního cytostatika, dosažení kompletní remise u 26 % nemocných a parciální remise (PR) u 36 %. Tato léčba přinesla orgánové léčebné odpovědi u 33 % léčených [45].
Na rozdíl od výše uvedené italské studie uvádí studie z USA nižší výskyt léčebných odpovědí. Celkem 70 pacientů, nevhodných k transplantační léčbě, bylo léčeno kombinací dexametazonu a alkylačního cytostatika. U 22 z nich nebyla hodnocena léčba pro časné úmrtí, takže pouze u 48 ze 70 bylo možné provést závěrečné vyhodnocení účinnosti léčby. Jenom 6 pacientů (13 %) dosáhlo kompletní remise a dalších 12 (25 %) parciální remise [46].
Výsledky velkých studií, v nichž byly použity dexametazon a alkylační cytostatikum, jsou shrnuty v tab. 3. Je zřetelné, že léčba AL-amyloidózy kombinací alkylačního cytostatika a dexametazonu dosahuje kompletní remise přibližně u 20 % léčených, při značném rozptylu počtu kompletních remisí v jednotlivých studiích (13–33 %).
Bortezomib v léčbě AL-amyloidózy
Mechanizmus účinku bortezomibu u AL-amyloidózy
Úlohou proteasomu je regulovaná degradace proteinů. Degradovány jsou nesprávně prostorově složené proteiny, aberantní proteiny nebo nepotřebné proteiny, které již splnily svoji regulační úlohu v buněčném cyklu či v apoptóze. Proteasom rozkládá proteiny určené k degradaci, které jsou deponovány v lumen endoplazmatického retikula (ER) [48,49].
Pokud je proces degradace bílkovin v proteasomu zastaven, pak se v endoplazmatickém retikulu (ER) proteiny hromadí. To způsobuje takzvaný „stres ER“, tedy stav, při kterém je porušena metabolická a redoxní rovnováha a buňka se stává citlivější a náchylnější k apoptóze (plánované buněčné smrti) v důsledku stabilizace některých proapoptotických proteinů. Dlouhodobý stres ER pak přivodí apoptózu patologického plazmocytu.
V případě amyloidózy je v ER klonálních plazmocytů velké množství aberantních (amyloidogenních) lehkých řetězců. Inhibice proteasomu působením bortezomibu vede k další akumulaci proteinů v ER, způsobí tak stres ER, který následně indukuje apoptotickou smrt klonálního plazmocytu [50,51].
V některých případech bylo popsáno, že amyloidogenní proteiny přímo inhibují aktivitu proteasomu [52,53].
Na základě těchto skutečností byla vytvořena hypotéza, že klonální plazmatické buňky, tvořící amyloidogenní volné lehké řetězce, jsou citlivější k inhibici proteasomu, než jsou klonální plazmatické buňky u mnohočetného myelomu, který netvoří amyloidogenní bílkoviny. Tato hypotéza vysvětluje vyšší počet léčebných odpovědí po bortezomibu u AL-amyloidózy, než je dosahován u mnohočetného myelomu [54,55].
Monoterapie bortezomibem
Nejdůležitější jsou informace z velké multicentrické studie, která hodnotila účinnost různých dávek bortezomibu v monoterapii [58,59]. Do studie bylo zařazeno celkem 70 nemocných, u nichž již předcházela nejméně jedna linie léčby. Pacienti byli rozděleni do skupin léčených bortezomibem v dávce 0,7 mg/m2; 1,0 mg/m2 a 1,3 mg/m2. Tyto dávky byly aplikovány 2krát týdně. Dále byla u další skupiny použita dávka 1,6 mg/m2, aplikovaná 1krát týdně. Ze 70 zařazených pacientů bylo možné léčebnou odpověď vyhodnotit u 67 z nich.
Hodnocení léčebné odpovědi bortezomibu v monoterapii považujeme za natolik důležité, že je uvádíme v samostatné tab. 4. Bortezomib v monoterapii dosáhl u pacientů již dříve léčených jinou léčbou 24–37 % kompletních remisí. Žádný další lék zatím nedosáhl v monoterapii takového úspěchu.
![Léčebné odpovědi u pacientů s AL-amyloidózou po bortezomibu v monoterapii [56,57].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/699bd812773414816a07e3a4853adfcd.png)
Bortezomib v kombinaci s dalšími léky
První klinické zkušenosti s bortezomibem přinesly pozitivní překvapení. Kastritis léčil skupinu 18 pacientů s AL-amyloidózou (z toho 7 bylo relabujících) bortezomibem a dexametazonem a dosáhl 94 % léčebných odpovědí, z toho 44 % kompletních remisí. Medián intervalu do dosažení léčebné odpovědi byl jen 0,9 měsíce. Při mediánu sledování 11,2 měsíce zaznamenali již 28 % orgánových léčebných odpovědí [58,59]. Tak vysoký počet léčebných odpovědí po klasické léčbě zatím nebyl zaznamenán. Efektivita bortezomibu u AL-amyloidózy předčila v této studii efektivitu bortezomibu u mnohočetného myelomu. Proto bylo nutno ověřit vysokou účinnost bortezomibu u AL-amyloidózy dalšími studiemi, které uvádíme v přehledné tab. 5. Uvádíme pouze počet hematologických odpovědí, nikoliv počet orgánových léčebných odpovědí. To proto, že orgánové léčebné odpovědi vyžadují další interval sledování, neboť k reparaci orgánového poškození dochází později, až po dosažení hematologické léčebné odpovědi, neboli po vymizení či zásadním snížení koncentrace amyloidotvorných lehkých řetězců. Proto také orgánové léčebné odpovědi nejsou uvedeny ve všech citovaných pracích.


Z tab. 5, sumarizující výsledky studií s bortezomibem aplikovaným většinou v kombinaci s dalšími léky, je zřetelné, že počet léčebných odpovědí zásadně závisí na tom, zda léčebný protokol obsahující bortezomib je zahájen u nemocného s nově diagnostikovým onemocněním, nebo u nemocného, u něhož již předcházely jiné léčebné linie, které nevedly k dlouhodobější léčebné odpovědi. V poslední době je pro léčbu těchto nemocných často používána trojkombinace bortezomibu, dexametazonu a alkylačního cytostatika (cyklofosfamidu nebo melfalanu) [80].
V případě podání kombinovaného léčebného režimu obsahujícího bortezomib v rámci první léčebné linie, se počet léčebných odpovědí pohybuje kolem 40 % (od 20 % do 67 %). Při srovnání výsledků léčby bortezomibovými režimy s výsledky klasické léčby (melfalan a dexametazon) je zřetelné zlepšení výsledků léčby vlivem bortezomibu nejméně o 10–20 %.
Do konce roku 2012 nebyly zveřejněny výsledky žádné velké prospektivní srovnávací studie. Formou abstraktů na konferenci Americké hematologické společnosti byla zveřejněna retrospektivní srovnání. Pro ilustraci jsme vybrali retrospektivní srovnání léčebných režimů obsahujících cyklofosfamid, dexametazon a thalidomid oproti cyklofosfamidu, dexametazonu a bortezomibu. Celkem tato britská studie zhodnotila 78 pacientů, 39 v každé skupině. V rámci „intent to treat analysis“ bylo ve skupině léčené režimem obsahujícím bortezomib 28,2 % kompletních remisí, zatímco ve skupině léčené režimem obsahujícím thalidomid to bylo jen 14,7 % (p = 0,1). Roční přežití dosáhlo 94 % pacientů léčených režimem s bortezomibem a pouze 62,1 % pacientů léčených režimem s thalidomidem (p = 0,01) [81].
Konzolidační léčba bortezomibovým režimem po vysokodávkované chemoterapii
Bortezomibové režimy jsou dále přínosné i u pacientů, kteří se po vysokodávkované chemoterapii nedostanou do kompletní remise [78]. To testovala klinická studie (fáze II) v Bostonu. Celkem do ní bylo zahrnuto 37 pacientů. Devatenáct pacientů se po vysokodávkované chemoterapii nedostalo do kompletní remise a byli v rámci druhé linie léčeni bortezomibem a dexametazonem; z této skupiny po léčbě bortezomibem a dexametazonem se 67 % dostalo do CR [82].
Tolerance bortezomibu pacienty s AL-amyloidózou
O pacientech s AL-amyloidózou obecně platí, že díky poškození orgánů depozity amyloidu jsou celkově křehčí a zranitelnější, a tedy hůře tolerují léčbu než pacienti stejného věku s mnohočetným myelomem [1]. Proto je vždy zásadní otázkou, jak pacienti s AL-amyloidózou tolerují jednotlivé léčebné postupy.
Nejpodrobněji byla tato otázka analyzována v rámci prospektivní evropsko-americké multicentrické studie, která testovala v podskupinách pacientů bortezomib v monoterapii v dávce 0,7 mg/m2, 1,0 mg/m2 a 1,3 mg/m2, aplikované 2krát týdně a dále pak bortezomib v dávce 1,6 mg/m2, aplikovaný 1krát týdně [55,56].
Autoři této studie konstatovali, že je možné u pacientů s AL-amyloidózou zahájit léčbu stejnými dávkami jako u pacientů s mnohočetným myelomem, tedy dávkou 1,3 mg/m2 aplikovanou 2krát týdně. Konstatovali, že není nutné u pacientů s AL-amyloidózou používat nižší dávky, než se běžně používají u mnohočetného myelomu, neboť ve frekvenci nežádoucích účinků není zásadní rozdíl [56,57].
Dále v rámci této studie srovnávali toleranci a účinnost aplikace bortezomibu v dávce 1,3 mg/m2 i.v. 2krát týdně anebo 1,6 mg/m2 1krát týdně.
V obou skupinách bylo mírně rozdílné spektrum nežádoucích účinků bortezomibu. Gastrointestinální problémy a infekce byly častější ve skupině léčené vyšší dávkou bortezomibu, aplikovanou 1krát týdně. Neuropatie a ortostatická hypotenze byly častější ve skupině s bortezomibem aplikovaným 2krát týdně. Tento rozdíl autoři vysvětlují tím, že některé typy nežádoucích účinků (gastrointestinální obtíže) souvisejí s maximální dosaženou plazmatickou koncentrací, zatímco jiné nežádoucí účinky (periferní neuropatie, hypotenze a případně trombocytopenie) závisejí na celkové kumulativní dávce podané za určitý čas, čili na intenzitě léčby („dose density“) [56,57]. Frekvenci komplikací v průběhu léčby v této studii shrnuje tab. 6.
![Počet hlášených komplikací v průběhu monoterapie AL-amyloidózy bortezomibem [57].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/1272d04552765b77e2345a716b315c87.png)
Na nežádoucí účinky v oblasti kardiovaskulární se zaměřila i další práce, navazující na výše zmíněnou studii. Celkem 31 pacientů bylo randomizováno do podskupiny léčených podle výše uvedeného schématu bortezomibem v monoterapii. Všichni pacienti byli echokardiograficky sledováni a měli opakovaně prováděno sledování srdečního rytmu metodou holterovského monitorování. V průběhu léčby se neobjevily závažné supraventrikulární či ventrikulární arytmie vyvolané léčbou.
Pouze v jednom případě Dubrey v roce 2011 popsal klinicky signifikantní zhoršení srdeční zdatnosti. Z komplikací, které se v průběhu léčby vyskytly a které by mohly souviset s léčbou, uvádějí periferní edémy (23 %), ortostatickou hypotenzi (13 %) a hypotenzi u 10 %. U 2 pacientů se v průběhu léčby objevila kongestivní srdeční slabost, která odezněla po přerušení léčby [70].
Stejně vyznívá hodnocení tolerance tohoto léku i dalšími autory, nepřekvapující je, že nejčastějším nežádoucím účinkem je periferní senzorická neuropatie. Kastritis v roce 2010 popisuje v průběhu léčby u některých nemocných zhoršení ortostatické hypotenze, periferní edémy a také zácpu anebo průjem [64].
Jako zcela výjimečný stav popsal Ghose v roce 2011 případ akutní dušnosti v souvislosti s aplikací bortezomibu [83]. Lamm v roce 2011 popisuje častější výskyt hyponatremie, hypokalemie a 2 výjimečné případy arytmie [79].
Souhrnně lze tedy konstatovat, že tolerance této léčby je dobrá, srovnatelná s tolerancí této léčby u mnohočetného myelomu.
Thalidomid v léčbě AL-amyloidózy
Thalidomid v monoterapii
Thalidomid podaný v rámci monoterapie v nízkých dávkách je v případě AL-amyloidózy málo účinný. Vyšší dávky thalidomidu jsou pacienty s AL-amyloidózou špatně tolerovány [84–86].
Palladini, který používal maximální tolerovatelné dávky, až 400 mg, uvádí symptomatickou bradykardii u 26 % léčených [87].
Thalidomid v kombinaci s dalšími léky
Při kombinaci s dexametazonem se však účinnost léčby zvyšuje a počet léčebných odpovědí dosáhl 48 % [87]. Kombinovaná léčba (cyklofosfamid, thalidomid a dexametazon) v dávkách upravovaných podle předpokládané tolerance dosáhla celkem 74 % léčebných odpovědí [88]. Kombinace melfalanu, dexametazonu a thalidomidu pomohla i v případech závažné kardiální formy amyloidózy, pokud srdeční postižení umožnilo absolvovat alespoň 3 cykly [89]. Největší popsaný soubor vyšel z britských ostrovů, celkem 202 pacientů bylo léčeno tímto režimem. Hematologickou léčebnou odpověď popisují u 62 % hodnocených pacientů, z toho bylo 25 % CR a 3 % nCR. Léčebná odpověď byla dosažena u 67 % nově léčených nemocných a u 54 % nemocných s relapsem nemoci [90]. Celkový počet orgánových léčebných odpovědí dosáhl 42 %. Frekvence dosažení orgánové léčebné odpovědi ovšem závisí na tom, který orgán je poškozen.
Orgánová léčebná odpověď byla dosažena při poškození ledvin u 46 % léčených, při poškození jater u 29 % léčených, při poškození nervů u 13 % léčených, při poškození srdce u 8 % léčených, při poškození gastrointestinálního traktu u 25 % [90].
V literatuře jsou zatím pouze 2 srovnávací studie klasické léčby kombinací melfalanu a dexametazonu (MD) s kombinací thalidomidu, dexametazonu a cyklofosfamidu (CTD). Po 12 měsících léčby byla orgánová léčebná odpověď prokázána u 44 ze 113 (tedy u 39 %) pacientů, léčených režimem CTD a u 12 z 56 (tedy u 21 %) pacientů léčených MD (p = 0,03). Autoři uvádějí, že mezi oběma režimy nebyl statisticky signifikantní rozdíl v počtu hematologických kompletních remisí, celkovém počtu hematologických odpovědí, toxicitě a délce celkového přežití. Rozdílný byl pouze medián nástupu léčebné odpovědi, podle hodnot FLC byl 3 měsíce po MD a 2 měsíce po CTD [55,90–92]. Ani v dalším menším souboru, který srovnával tyto 2 léčebné režimy (12 MD + 12 CTD), nebyl mezi jednotlivými režimy nalezen signifikantní rozdíl [93].
Publikované zkušenosti lze shrnout do konstatování, že přidání thalidomidu k základním kamenům léčby AL-amyloidózy nepřineslo zásadní zlepšení.
Nicméně režim cyklofosfamid, thalidomid a dexametazon (CTD) je použitelný před sběrem periferních kmenových buněk, zatímco o režimu melfalan a dexametazon je známo, že snižuje pravděpodobnost úspěšného sběru kmenových buněk krvetvorby z periferní krve. Zkušenosti s léčbou AL-amyloidózy thalidomidem shrnuje tab. 7.
Lenalidomid
Lenalidomid v monoterapii
K dispozici je pouze jedna studie zveřejněná in extenso, v níž byl lenalidomid testován první 3 měsíce v monoterapii a teprve po 3 měsících byl k lenalidomidu přidán dexametazon. Monoterapie lenalidomidem byla zahájena u 23 pacientů. Po 3 měsících však pouze u 1 z nich došlo k léčebné odpovědi. Nutno však říci, že z těchto 23 nemocných 10 nedokončilo plánované 3 cykly. Pouze 11 pacientů, kteří dokončili první 3 měsíce monoterapie lenalidomidem, bylo schopno pokračovat v kombinované léčbě. Lenalidomid s dexametazonem vedl u 9 z nich k léčebné odpovědi.
Tato studie tedy prokázala nízkou účinnost lenalidomidu v monoterapii AL-amyloidózy, poukázala na jeho toxicitu a prokázala jeho účinnost v kombinaci s dexametazonem [95]. V analýze souboru z roku 2008 autoři uvádějí, že lenalidomid v monoterapii není vhodný pro léčbu AL-amyloidózy, ale v kombinaci může být pro tyto nemocné užitečný [96,97].
Lenalidomid v kombinaci s dalšími léky
V rámci dalších klinických studií již byla testována léčba lenalidomidem v kombinaci s dexametazonem a případně s dalšími léky. Zároveň se hledala optimální dávka lenalidomidu.
Standardní dávka 25 mg/den se u většiny pacientů s AL-amyloidózou ukázala jako velmi toxická. Jako optimální dávka se pro tyto nemocné jevila dávka 15 mg při kombinaci s dexametazonem. Při použití lenalidomidu v dávce 15 mg/den a dexametazonu bylo dosaženo 29 % kompletních hematologických léčebných odpovědí, 38 % dosáhlo PR [96].
Lenalidomid v kombinaci s dalšími léky představuje pro pacienty s AL-amyloidózou účinný lék [96–109]. Důležité je, že lenalidomid v kombinaci může docílit léčebnou odpověď i tam, kde léčba obsahující bortezomib nevedla ke kompletní remisi, jak prokázala jedna klinická studie [106]. Tento jev můžeme potvrdit z vlastní zkušenosti. U nemocného s relapsem po vysokodávkované chemoterapii, nereagujícím na režim cyklofofamid, dexametazon a bortezomib, dosáhla léčba lenalidomidem a dexametazonem kompletní remise.
Nejvýznamnější klinické studie shrnuje tab. 8. Je zřetelné, že lenalidomid v kombinaci s dexametazonem a případně s dalšími léky dosahuje počet léčebných odpovědí až nad 50 %. Počty kompletních remisí, uvedené v této tabulce, jsou o něco nižší než při použití bortezomibu.

V roce 2012 nemáme žádnou přímou srovnávací studii léčby založené na lenalidomidu s jinými léčebnými postupy. Při pohledu na tabulky sumarizující přehled léčebných odpovědí u lenalidomidu a u bortezomibu je zřetelné, že počty kompletních remisí jsou zřejmě u lenalidomidu nižší, než je tomu u režimů obsahujících bortezomib. Léčebné odpovědi jsou dosahovány léčebnými režimy obsahujícími lenalidomid podstatně pomaleji než u léčebných režimů obsahujících bortezomib.
Důležitým momentem je zjištění, že v některých případech má lenalidomid potenciál dosáhnout kompletní remise u pacienta s AL-amyloidózou, která nereagovala na léčebný režim obsahující bortezomib [106].
Informace o účinnosti lenalidomidu u AL-amyloidózy, dostupné v roce 2012, dokazují přínos tohoto léku pro některé nemocné s AL-amyloidózou, a proto lze tento lék považovat za vhodný pro pacienty, nereagující na iniciální léčbu jiným režimem. Lenalidomid představuje rovněž možnou alternativu pro nemocné s AL-amyloidózou a s těžkou neuropatií, která znemožňuje podání léčebného režimu s bortezomibem.
Tolerance lenalidomidu pacienty s AL-amyloidózou
U lenalidomidu se neuvádí nefrotoxicita. Ledviny s depozity amyloidu jsou však výjimkou. Specter et al v roce 2011 z myelomového centra v Bostonu popsali soubor 41 pacientů léčených lenalidomidem, z nichž u 27 (66 %) došlo v průběhu léčby lenalidomidem ke zhoršení funkce ledvin, které bylo definováno jako vzestup kreatininu o 50 % a výše. Medián intervalu od zahájení léčby do klinicky signifikantního zhoršení funkce ledvin byl pouze 44 dnů [110]. Poněvadž i u pacientů s mnohočetným myelomem může být ledvina poškozena amyloidovými depozity, o nichž lékaři nevědí, je nutno myslet na tento zatím patofyziologicky neobjasněný jev i v případě mnohočetného myelomu, podáváme-li lenalidomid pacientovi se signifikantní proteinurií a s přítomností volných lehkých řetězců v séru a v moči. U těchto pacientů bychom měli sledovat vývoj kreatininu v 1–2týdenních intervalech a při vzestupu kreatininu změnit léčebný postup.
Batts v roce 2008 popsal poškození funkce ledvin při léčbě lenalidomidem i u pacientů s jinými monoklonální migamapatiemi [111]. Je pravděpodobné, že to může být způsobeno nerozpoznaným depozitem amyloidu v ledvině.
V průběhu léčby AL-amyloidózy lenalidomidem byl dále popsán vzestup hodnot BNP (brain natriuretic peptide) [112] a náhlá smrt u pacienta se srdeční formou AL-amyloidózy [113].
Vysokodávkovaná chemoterapie
Léčebná účinnost vysokodávkované chemoterapie
Léčba vysokodávkovaným melfalanem s transplantací autologních krvetvorných buněk dosahuje vysokého počtu kompletních hematologických remisí, což s sebou podle některých studií přináší zlepšení kvality života a prodloužení přežití [114]. Nicméně tento postup je spojen s dosti vysokou toxicitou. To je při zvýšené fragilitě pacientů, způsobené depozity amyloidu, spojeno se zvýšenou mortalitou, která se pohybuje od 11 % [115] do 27 % [116]. Tuto vysokou mortalitu je možné zmenšit selekcí nemocných, vhodných pro tuto léčbu, založenou na pečlivém vyhodnocení míry srdeční dysfunkce a tíže postižení dalších důležitých orgánů [117–123].
Další možností, jak snížit mortalitu, je snížení dávky melfalanu na 100–140 mg/m2. Redukce dávky však snížila počet hematologických odpovědí (53 %), při zachování nezanedbatelné mortality [124].
V retrospektivní studii měli pacienti s vysokodávkovanou chemoterapií delší přežití než pacienti léčení melfalanem a prednisonem [125].
První prospektivní multicentrickou randomizovanou studií, která měla za cíl srovnat klasickou chemoterapii, melfalan a dexametazon, s vysokodávkovanou chemoterapií, byla studie francouzské myelomové skupiny. Do každé skupiny bylo zařazeno 50 pacientů a analýza byla provedena formou „intent to treat analysis“. Medián přežití ve skupině léčené vysokodávkovanou chemoterapií byl 22,2 měsíce, medián přežití ve skupině léčené klasickou chemoterapií byl 56 měsíců. Z pacientů, randomizovaných do skupiny vysokodávkované chemoterapie, zemřelo před podáním vysokodávkované chemoterapie celkem 13 osob, jeden pacient odmítl léčbu, 2 pacienti měli insuficietní sběr kmenových krvetvorných buněk, 8 pacientů náhle zemřelo, 1 pacient zemřel na progresivní jaterní amyloidózu a 1 na sepsi. Takže z 50 randomizovaných do skupiny s vysokodávkovanou chemoterapií ji podstoupilo pouze 37 nemocných. Z nich 9 (24 %) zemřelo do 100. dne po transplantaci. Tato prospektivní randomizovaná klinická studie tedy nepotvrdila lepší výsledky díky vysokodávkované chemoterapii [126], takže melfalan a dexametazon z ní vyšel jako zlatý standard.
Kritické hlasy ovšem vyčítají této studii nevhodný výběr pacientů pro vysokodávkovanou chemoterapii, což vedlo k vysoké mortalitě před vlastní vysokodávkovanou chemoterapií (12 z 50 = 24 %) a 24% peritransplantační mortalitě, což je vyšší mortalita spojená s léčbou než ve studiích ze specializovaných center. Tato provokativní francouzská studie iniciovala další analýzy.
Retrospektivní srovnání z Mayo Clinic prokázalo, že vysokodávkovaná chemoterapie je jednoznačně lepší než kombinace melfalanu a dexametazonu pro pacienty s počtem plazmocytů v kostní dřeni nad 20 % [127]. Tato práce ukazuje, jak různorodý může být pohled na to, co je to AL-amyloidóza. Jiní by tuto jednotku klasifikovali jako mnohočetný myelom s amyloidózou. Nicméně z této práce vychází, že pro nemocné s vyšší masou patologických plazmocytů je vyšší agresivita léčby přínosnější.
Důležitost dosažení kompletní remise vysokou dávkou melfalanu pro prognózu nemocných zdůrazňuje analýza 80 pacientů, kteří podstoupili vysokodávkovanou chemoterapii s autologní transplantací před 10 lety. Sice 22 % léčených zemřelo v průběhu 1. roku (14 % důsledkem toxicity léčby a 8 % důsledkem progrese), ale z 63 pacientů, kteří přežili 1. rok po transplantaci, se 51 dostalo do kompletní remise nemoci. Pro celou skupinu 80 pacientů byl medián přežití 4,75 roku. Medián přežití pacientů, kteří dosáhli kompletní remisi, přesahoval 10 roků, zatímco medián přežití těch, kteří se nedostali do kompletní remise, byl pouze 50 měsíců. Tato studie prokázala, že dosažení kompletní remise vysokou dávkou melfalanu navodí u AL-amyloidózy dlouhodobé přežití [128].
Postižení srdce (pokud není klinicky závažné) není kontraindikací. V souboru 194 pacientů s kardiálním poškozením a vysokodávkovanou chemoterapií byla dosažena hematologická léčebná odpověď u 69 % nemocných a orgánová léčebná odpověď u 47 %, a to za cenu 16,5% mortality do dne 100 po transplantaci [129]. Efektivitu a mortalitu největších publikovaných studií shrnuje tab. 9.
Rizika vysokodávkované chemoterapie u pacientů s AL-amyloidózou
Problémem vysokodávkované chemoterapie u AL-amyloidózy je kumulace toxicit, a proto je spojena s mnohem vyšším rizikem úmrtí než u mnohočetného myelomu. Již sběr periferních kmenových buněk byl ojediněle komplikován nekardiálním plicním edémem s hypoxií. U tohoto pacienta byla při sekci rozpoznána depozita AL-amyloidu v plicích [137]. Ve výše citované francouzské randomizované studii zemřeli 4 pacienti v průběhu aplikace G-CSF [126]. Také ruptura sleziny v průběhu aplikace leukocytárních růstových faktorů byla popsána.
V případě postižení zažívacího traktu depozity amyloidu může následná mukozitida po chemoterapii způsobit značné krvácení a ohrozit život (toxické megakolon) [138–141].
Další závažnou komplikací vysokodávkované chemoterapie je zhoršení funkce ledvin. Vzestup kreatininu nejméně o 44 μmol/l (0,5 mg/dl) byl v jedné studii zaznamenán u 19 % pacientů [141,142]. V další studii byl vzestup kreatininu nejméně o 88 μmol/l v potransplantačním období (melfalan 100–200 mg/m2) zjištěn dokonce u 23 % nemocných [143].
Proč vysokodávkovaný melfalan vede u pacientů s AL-amyloidózou k poškození funkce ledvin, to stále není jasné. Nicméně je nutné tento fakt respektovat a raději podávat vyšší dávky melfalanu rozděleně ve 2 dnech po sobě [143].
Vysokodávkovaná chemoterapie má u pacientů s mnohočetným myelomem nízkou mortalitu (1–3 %). Pokud se ale stejná léčba použila u nemocných s AL-amyloidózou, pak byla mortalita podstatně vyšší. Analýza výsledků ze 48 transplantačních center (107 pacientů) prokázala transplantační mortalitu 18 % do 30. dne od provedení vysokodávkované chemoterapie s autologní transplantací, při mediánu přežití 4 roky. Výsledná časná transplantační mortalita (18 %) je vyšší, než je obvykle publikováno v analýzách velkých center pro tuto léčbu, která uvádějí 10–15% mortalitu spojenou s vysokodávkovanou chemoterapií s autologní transplantací [116]. To souvisí s většími zkušenostmi s výběrem pacientů s AL-amyloidózou, vhodných pro transplantační léčbu a se zkušenostmi se zvládáním komplikací u takto léčených osob ve specializovaných centrech.
Kteří pacienti s AL-amyloidózou jsou vhodní pro vysokodávkovanou chemoterapii?
Vysokodávkovaná chemoterapie s autologní transplantací se doporučuje pro osoby s amyloidovým postižením jednoho nebo maximálně 2 orgánů. Nesmí ale předcházet krvácení do zažívacího traktu způsobené depozity amyloidu [145] a nesmí být přítomno klinicky závažné postižení srdce anebo ledvin [146,147].
Bostonské centrum například podmínky pro podání vysokodávkované chemoterapie uvádí následovně: dobrá celková tělesná zdatnost (performance status SWOG 0-2), přiměřená srdeční funkce (LV EF > 40 %), saturace krve kyslíkem > 95 % a hemodynamická stabilita, čímž je míněn systolický krevní tlak > 90 mm Hg. Léčba hemodialýzou není kontraindikací.
Pro pacienty mladší 65 let s ejekční frakcí levé komory nad 45 % použili dávku melfalanu 200 mg/m2. Pacientům se sníženou ejekční frakcí na 40–45 % podávali 140 mg/m2, při ejekční frakci pod 40 % nepřipadá provedení vysokodávkované chemoterapie v úvahu. Aplikaci melfalanu vždy rozdělili do 2 dávek a aplikovali je 2. a 1. den před transplantací. U takto vybrané skupiny nemocných měli v kategorii do 65 let 10,4% mortalitu a v kategorii nad 65 let 12,3%. Počet kompletních remisí byl u mladší skupiny 43 % a u starší 32 % a medián přežití byl 4 roky [116]. Kritéria italské pracovní skupiny pro výběr pacientů vhodných k vysokodávkované chemoterapii uvádí tab. 10.
![Výběrová kritéria italské skupiny pro studium amyloidózy pro výběr pacientů k vysokodávkované chemoterapii s autologní transplantací [148].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/71558dfbc0cc364803cda4d5296ad6ae.png)
Vysokodávkovaná chemoterapie následovaná konzolidační chemoterapií
Pro pacienty, kteří po vysokodávkované chemoterapii nedosáhnou kompletní remise, jsou vhodné další léčebné linie. Kombinace thalidomidu a dexametazonu byla v této indikaci zkoušena u 31 nemocných, u 42 % z nich prohloubila léčebnou odpověď. Tolerance této léčby však nebyla bezproblémová [94].
V následující klinické studii (27 pacientů) zaměnili thalidomid za bortezomib a u 7/8 pacientů došlo ke zlepšení léčebné odpovědi [78,82,131].
Časná konzolidační léčba, navazující na vysokodávkovanou chemoterapii s autologní transplantací, může prohloubit léčebnou odpověď.
Role vysokodávkované chemoterapie s autologní transplantací v roce 2012
V roce 2012 lze nalézt více než 50 studií, které potvrdily účinnost vysokodávkované chemoterapie u pacientů s AL-amyloidózou. Tato léčba je však doporučována obvykle pouze pro nemocné mladší 65 let, u nichž jsou závažně postiženy pouze 2 orgány či systémy a kteří nemají klinicky signifikantní poškození srdce depozity amyloidu. Týká se tedy pouze malé podskupiny pacientů s AL-amyloidózou.
V době, kdy se do léčebné strategie dostávají kombinované léčebné postupy s bortezomibem, které dosahují vysokého počtu léčebných odpovědí s menší toxicitou, je indikace vysokodávkované chemoterapie méně jasná než v období, kdy nebyly bortezomib a lenalidomid dostupné. Role vysokodávkované chemoterapie s autologní transplantací pro pacienty s AL-amyloidózou bude teprve krystalizovat ze srovnávacích klinických studií.
K dispozici je zatím jedna velká metaanalýza, která neprokázala rozdíly mezi klasickou a vysokodávkovanou chemoterapií, a několik přehledů problematiky [149–153].
Zatím neznáme odpověď na otázku, zda kompletní remise, navozená vysokou dávkou melfalanu anebo režimem obsahujícím bortezomib, budou mít stejně dlouhé trvání.
Pokud je pacient s AL-amyloidózou vhodný k provedení vysokodávkované chemoterapie (splňuje kritéria podle tab. 8), považujeme za správné mu ji nabídnout, ale zároveň jej seznámit s potenciálními riziky i dalšími léčebnými možnostmi.
Podávat indukční chemoterapii před vysokodávkovanou chemoterapií?
Vysokodávkovaná chemoterapie u AL-amyloidózy se obvykle provádí bez předchozí indukční chemoterapie. Faktem totiž je, že pacienti s AL-amyloidózou mají obvykle nízký počet plazmatických buněk v kostní dřeni a nevelkou koncentraci monoklonálního imunoglobulinu, takže masa patologických plazmocytů u nich není velká. A faktem je, že orgánové poškození kontinuálně progreduje, pokud se rychle nesníží tvorba volných lehkých řetězců. V roce 2004 byla zveřejněna randomizovaná studie, která srovnávala vysokodávkovanou chemoterapii s předchozí indukční léčbou a bez předchozí indukční léčby. V obou skupinách nebylo rozdílu mezi počtem hematologických a orgánových léčebných odpovědí. Nicméně ve skupině s indukční chemoterapií došlo v jejím průběhu ke zhoršení orgánového poškození, a proto nakonec nebylo u některých z těchto pacientů přistoupeno k vysokodávkované chemoterapii. Takže aplikace popisované indukční chemoterapie nebyla přínosná. Pacienti s kardiálním postižením, u nichž díky indukční chemoterapii došlo k oddálení vysokodávkované chemoterapie, měli horší přežití [154].
Transplantace srdce, případně jiných orgánů, následovaná vysokodávkovanou chemoterapií s autologní transplantací krvetvorných buněk
Těžce poškozené srdce je kontraindikací vysokodávkované chemoterapie. Ale i konvenční léčba, obsahující vysoké dávky dexametazonu, která obvykle způsobuje retenci tekutin, nemusí být proveditelná při závažném poškození srdce. Pokud jde o mladšího člověka a ostatní orgány nejsou kriticky poškozeny, je možné provést napřed transplantaci srdce. Tím se výrazně zlepší celková zdatnost, a pak je možno použít vysokodávkovanou chemoterapii s podporou transplantace vlastních krvetvorných buněk s cílem eradikovat amyloidogenní klon plazmocytů. V literatuře lze nalézt četné popisy menších skupin nemocných, které dokládají přínos provedení orgánové transplantace nejvíce poškozeného orgánu (tedy nejen srdce) následované léčbou amyloidózy [155–162]. Podstatné je, aby pacient byl v celkově dobrém stavu a měl amyloidózou závažně poškozen pouze jeden orgán, který je nahrazen orgánem transplantovaným. Zřejmě i do budoucna půjde o výjimečnou léčbu, její častější použití bude váznout v důsledku nedostatku vhodných lidských srdcí pro transplantační program.
Prognóza pacientů s AL amyloidózou
Prognóza pacientů s AL-amyloidózou je dána, stejně jako je tomu u všech onkologických chorob, rozsahem nemoci, tedy mírou pokročilosti poškození orgánů amyloidovými depozity.
Nejvíce ze všeho má vliv na prognózu poškození srdce. Proto četné práce dávají do souvislosti prognózu a funkci srdce dokumentovanou laboratorně či sonografickým a MR vyšetřením [163–166]. Tento fakt se odráží v analýzách jednotlivých prognostických faktorů a je zohledněn při tvorbě prognostických indexů.
Nejnovější index byl stanoven analýzou v souboru 810 pacientů s nově stanovenou diagnózou AL-amyloidóza [166]. V multivariantní analýze byly jako nezávislé prognostické faktory prokázány následující:
- kardiální biomarker troponin-T (cTnT);
- kardiální biomarker N-terminální pro-B-typ natriuretického peptidu (NT-proBNP). V ČR je možné stanovit jak NT-pro BNP, tak BNP. Výpovědní hodnotu mají stejnou, ale numerické výsledky nejsou srovnatelné. U akutního srdečního selhání je jasně patologická hodnota NT-proBNP > 1 800 pg/ml a BNP > 500 pg/ml. Hodnocení BNP je: BNP < 100 pg/ml = srdeční selhání nepravděpodobné (< 2 %), BNP 100–500 pg/ml = srdeční selhání možné, BNP > 500 pg/ml = sr-deční selhání potvrzeno s 95% jistotou, BNP > 800 pg/ml = srdeční selhání s vážnou prognózou. Pokud bychom uvedený index aplikovali v českých poměrech, doporučuje místo NT-proBNP ≥ 1 800 pg/ml použít hodnotu BNP > 500 pg/ml;
- hodnota FLC, konkrétně diference mezi klonálním typem lehkého řetězce a neklonálním typem (FLC-diff).
Z těchto tří faktorů bylo vytvořeno skóre 0–3 poměrně robustně odlišující 4 prognostické skupiny (p < 0,001), které je uvedeno v tab. 11.
![Prognostický skórovací systém pro všechny pacienty s AL-amyloidózou z roku 2012 [14].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0cc17b82c930bd435ccef5213fbd86d2.png)
V tab. 12 je uveden index založený pouze na měření hladiny troponinu a NT-proBNP [167]. V našich poměrech lze použít místo NT-proPNP hodnotu BNP.
![Prognostický index založený pouze na troponinu a NT-proBNP Dispenzieri, 2004 [167].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f6d35af513f52a1369b3f0236a89b7c9.png)
Závěry pro praxi
- Výsledky léčby AL-amyloidózy, a tedy osud nemocného, závisí na míře poškození organizmu v době zahájení léčby. Proto je důležité na tuto diagnózu myslet a časně ji stanovit.
- Cílem léčby je dosáhnout poklesu koncentrace amyloidogenních volných lehkých řetězců pod měřitelnou koncentraci, neboli dosáhnout kompletní hematologické léčebné odpovědi, což je předpokladem orgánové léčebné odpovědi.
- Zlatým standardem léčby pro pacienty s AL-amyloidózou byla až do roku 2011 kombinace alkylačního cytostatika (melfalanu či cyklofosfosfamidu) s dexametazonem.
- Pro vybrané nemocné mladší 65 let, s nevelkým poškozením organizmu amyloidem a s dobrou srdeční funkcí (EF > 40 %), byla do roku 2011 považována za optimální vysokodávkovaná chemoterapie s autologní transplantací krvetvorných buněk. Při nízkém počtu plazmocytů se prováděl sběr krvetvorných buněk a následná vysokodávkovaná chemoterapie bez předchozích cyklů indukční chemoterapie.
- V roce 2012 byly zveřejněny rozsáhlé zkušenosti s novými léky, které byly použity pro léčbu pacientů s AL-amyloidózou v rámci klinických studií fáze I a II a ojediněle fáze III. Vyplývá z nich, že z dostupných nových léků (bortezomib, thalidomid a lenalidomid) má bortezomib nejvyšší účinnost v monoterapii. Nejvyšší počet léčebných odpovědí přináší léčba kombinací bortezomibu s cyklofosfamidem nebo melfalanem a s dexametazonem.
- Léčba kombinací bortezomibu, alkylačního cytostatika a dexametazonu přináší podstatně vyšší počet léčebných odpovědí a kompletních remisí, pokud je použita v rámci léčby první linie, než při použití v rámci léčby další linie.
- Klinické studie s lenalidomidem v kombinaci s dalšími léky přinesly menší počet léčebných odpovědí, než je popisováno při léčbě bortezomibem v kombinaci s dalšími léky. Proto se kombinace s lenalidomidem nepovažují za optimální léčbu první linie s výjimkou případů s kontraindikací bortezomibu (těžká neuropatie způsobená AL-amyloidózou).
- Bylo prokázáno, že lenalidomid v kombinaci s dalšími léky může navodit remisi u pacientů, jejichž onemocnění bylo rezistentní k iniciální léčbě bortezomibem. Proto lze lenalidomid (a případně i thalidomid) použít jako léčbu druhé linie v případě neúspěchu bortezomibové léčby s nadějí na dosažení kompletní remise.
- Zlepšení léčebných výsledků, které přinesl bortezomib pacientům s AL-amyloidózou, přinesl i otázku, zda pro mladší pacienty s nevelkým poškozením organizmu použít vysokodávkovanou chemoterapii s autologní transplantací či kombinovanou léčbu s bortezomibem. V roce 2012 neznáme odpověď na otázku, zda výrazné zvýšení počtu kompletních remisí při léčbě bortezomibem bude znamenat stejně významné prodloužení přežití. Nevíme, zda kompletní remise, navozené u pacientů s AL-amyloidózou bortezomibem budou mít stejně dlouhé trvání jako kompletní remise navozené vysokodávkovanou chemoterapií s autologní transplantací krvetvorných buněk. Proto má vysokodávkovaná chemoterapie s autologní transplantací krvetvorných buněk stále svoji indikaci u podskupiny mladších pacientů bez závažného poškození srdce či dalších orgánů.
- Zcela novým trendem, prováděným jen vybranými pracovišti, je nahrazení kriticky poškozeného orgánu vhodným transplantátem, např. srdce, a pak teprve léčba AL-amyloidózy pomocí vysokodávkované chemoterapie či jiné léčby. U mladších pacientů s AL-amyloidózou a závažným poškozením srdce lze v případě izolovaného či dominantního postižení srdce zvážit zařazení do programu transplantace srdce. Předřazení transplantace srdce před léčbu AL-amyloidózy umožní nemocnému absolvovat účinnou léčbou AL-amyloidózy, kterou by bez transplantace srdce nebyl schopen tolerovat. Tento postup vede k podstatnému prodloužení života pacientů s těžkým poškozením srdce depozity amyloidu.
Tato práce byla podpořena granty IGA Ministerstva zdravotnictví NT12215, NT12130 a NT13190, ale také i institucionální podpory výzkumné organizace poskytnuté Ministerstvem zdravotnictví ČR v roce 2012.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 26. 9. 2012
Přijato po recenzi: 26. 11. 2012
Sources
1. Ščudla V, Pika. T Současné možnosti léčby systémové AL amyloidózy. Vnitř Lék 2009; 55: (Suppl. 1): 77–87.
2. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349 : 583–596.
3. Abraham RS, Geyer SM, Price-Troska L et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome of light chain-associated amyloidosis. Blood 2003; 101 : 3801–3808.
4. 1Comenzo RL, Zhang Y, Martinez C et al. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig VL germ line use and clonal plasma cell burden. Blood 2001; 98 : 714–720.
5. Bellotti V, Chiti F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein mis-folding diseases. Curr Opin Struct Biol 2008; 18 : 771–779.
6. Adam Z, Ščudla V. Klinické projevy a diagnostika AL-amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 36–45.
7. Tichý M. Primární amyloidóza. Lék Zpr UK Hradec Králové 1999; 44 : 99–107.
8. Kroupa R, Dastych M, Šenkyřík M et al. Systémové amyloidóza s dominující klinickou manifestací v trávicím traktu. Vnitř Lék 2005; 51 : 588–592.
9. Ryšavá R Amyloidóza ledvin. Postgrad Med 2006; 8 : 207–212.
10. Ryšavá R. Léčba paraproteinemické nefropatie a primární amyloidózy ledvin. Aktual v Nefrol 2005; 11 : 62–65.
11. Linhartová K, Daum O. Srdeční amyloidóza. Cor et Vasa 2005; 47 : 328.
12. Sideras K, Gertz MA. Amyloidosis. Adv Clin Chem 2009; 47 : 1–44.
13. Lavatelli F, Perlman DH, Spencer B. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics 2008; 7 : 1570–1583.
14. Anesi E, Palladini G, Perfetti V et al. Therapeutic advances demand accurate typing of amyloid deposits. Am J Med 2001; 111 : 243–244.
15. Lachman HJ, Booth DR, Booth SE et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002;346; 23 : 1786–1791.
16. Vrana JA, Zeldenrust SR, Theis JD et al. Diagnosis and classification of amyloidosis in abdominal subcutaneous fat specimens using mass spectrometry based proteomics. Blood 2008; 112 : 937, abstr. 2710.
17. Linke RP. On typing amyloidosis using immunohistochemistry. Detailled illustrations, review and a note on mass spectrometry. Prog Histochem Cytochem 2012; 47 : 61–132.
18. Sethi S, Vrana JA, Theis JD et al. Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int 2012; 82 : 226–234.
19. Ribeiro-Silva C, Gilberto S, Gomes RA et al. The relative amounts of plasma transthyretin forms in familial transthyretin amyloidosis: a quantitative analysis by Fourier transform ion-cyclotron resonance mass spectrometry. Amyloid 2011; 18 : 191–199.
20. Lavatelli F, Vrana JA. Proteomic typing of amyloid deposits in systemic amyloidoses. Amyloid 2011; 18 : 177–782.
21. Westermark P. Amyloid diagnosis, subcutaneous adipose tissue, immunohistochemistry and mass spectrometry. Amyloid 2011; 18 : 175–176.
22. Brambilla F, Lavatelli F, Di Silvestre D et al. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012; 119 : 1844–1847.
23. Lavatelli F, Valentini V, Palladini G et al. Mass spectrometry-based proteomics as a diagnostic tool when immunoelectron microscopy fails in typing amyloid deposits. Amyloid 2011; 18: (Suppl. 1): 59–61.
24. Klein CJ, Vrana JA, Theis JD et al. Mass spectrometric-based proteomic analysis of amyloid neuropathy type in nerve tissue. Arch Neurol 2011; 68 : 195–199.
25. Sethi S, Theis JD, Leung N et al. Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 2010; 5 : 2180–2187.
26. Vrana JA, Gamez JD, Madden BJ et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 2009; 114 : 4957–4959.
27. Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County Minnesota. Blood 1992; 78 : 1817–1822.
28. Magy-Bertrand N, Dupond JL, Mauny F et al. Incidence of amyloidosis over 3 years. The Amypro study. Clin Experiment Rheumatology 2008; 26 : 139–142.
29. Merlini G, Palladini G. Amyloidosis: is a cure possible? Ann Oncol 2008; 19: (Suppl. 4): 63–66.
30. Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108 : 2520–2530.
31. Van Gameren I, van Rijswijk MH, Bijzet J et al. Histological regression of amyloid in AL amyloidosis is exclusively seen after normalization of serum free light chain. Haematologica 2009; 94 : 1094–1100.
32. International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies, multiple myelom and releated disorders: a report of the International Myeloma Working Group. Brit J Haematol 2003; 121 : 749–457.
33. Kyle RA. Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23 : 3–9.
34. Gertz MA, Comenzo R, Falk RH. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis. A consensus opinion from the 10th international symposium on amyloid and amyloidosis. Amer J Hematol 2005; 79 : 319–328.
35. Gertz MA, Merlini G. Definition of organ involvement and response to treatment in AL amyloidosis: an updated consensus opinion. Amyloid 2010; 17: (Suppl. 1): 48–49.
36. Kyle RA, Gertz MA, Greipp PR et al. A trial of three regimens for primary amyloidosis: Colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336 : 1202–1207.
37. Gertz MA, Kyla RA, Greip PP. Response rates, and survival in primary systemic amyloidosis. Blood 1991; 77 : 257–262.
38. Gertz MA, Lacy MQ, Lust JA et al. Prospective randomized trial of melphalan and prednisone versus vincristine, carmustine, melpahalan, cylophosphamide and prednisone in the treatment of primary systemic amyloidosis. J Clin Oncol 1999; 17 : 262–267.
39. Dhodapkar MV, Hussein MA, Rasmussen E et al. Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood 2004; 104 : 3520–3526.
40. Palladini G, Perfetti V, Obici L et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood 2004; 103 : 2936–2938.
41. Goodman H, Lachmann H, Bradsell AR et al. Intermediate dose melpahalan and dexamethasone treatment in 144 patients with systemic AL-amyloidosis. Blood 2004; 104 : 755–759.
42. Goodman H, Wechalekar A, Lachmann H et al. Clonal disease response and clinical outcome in 229 patients with AL-amyloidosis treated with VAD-like chemotherapy Hematologica 2008; 90 : 201–203.
43. Palladini G, Anesi E, Perfetti V et al. A modified high-dose dexamethasone regimen for primary systemic (AL) amyloidosis. Br J Haematol 2001; 113 : 1044–1046.
44. Palladini G, Russo P, Nuvolone M et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood 2007; 110 : 787–788.
45. Palladini G, Russo P, Foli A et al. Treatment with oral melphalan and dexamethasone in an extended population with AL amyloidosis. Blood 2009; 114: (Suppl. N 22): 1496, abstr. 3889.
46. Sanchowarala V, Seldin DC, Sloan JM et al. Oral cyclic melphalan and dexametasone treatment in the treatment of patients with high dose amyloidosis, ineligible for high dose melphalan and stem cell transplantation. Blood 2009; 114: (Suppl. N 22): 748, abstr. 1883.
47. Sanchorawala V, Sedlin CD, Berk JL et al. Oral cyclic melphalan and dexametasone for pacient with AL amyloidosis. Clin Lymphoma Myeloma Leukemia 2010; 10 : 469–472.
48. Sitia R, Braakman I. Duality control in the endoplasmatic reticulum protein factory. Nature 2003; 426 : 891–894.
49. Breckenridge DG, Germain M, Mathai JP et al. Regulation of apoptosis by endoplasmatic reticulum pathway Oncogene 2003; 22 : 8608–8618.
50. Teixeira PR, Cerca F, Santos SD et al. Endoplasmatic reticulum stress associated with extracelullar aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. J Biol Chem 2006; 281 : 21998–22003.
51. Huang CJ, Haataje L, Gurlo T et al. Induction of endoplasmatic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated protein by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab 2007; 293: E1656–E1662.
52. Keller JN, Hanni KB Markesbery WR. Impaired proteasome function in Alzheimer´s disease. J Neurochem 2000; 75 : 436–439.
53. Casas S, Gomis R, Gribble FM et al. Impairment of the ubiquitin-proteasome pathway is a dowstream endoplasmatic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 2007; 56 : 2284–2294.
54. Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyloidosis: targeted therapy? Haematologica 2007; 92 : 1302–1307.
55. Bianchi G, Oliva L, Casctio P et al. The proteasome load versus capaticy balance determinates apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009; 113 : 3040–3049.
56. Reece D, Hegenbart U, Merlini G et al. Weekly and twice-weekly bortezomib in patients with systemic AL-amyloidosis: results of a phase 1 dose-escalation study. Blood 2009; 114 : 1489–1497.
57. Reece DE, Hegenbart U, Sanchorawala V et al. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood 2011; 118 : 865–873.
58. Kastritis E, Anagnostopoulos A, Rousnou M et al. Treatment of ligh chain deposition disease with the combination of bortezomibe and dexamethasone. Blood 2007; 110: (N.11 Suppl.): abstr. 64.
59. Kastritis E, Anagnostopoulos A, Roussou M et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica 2007; 92 : 1351–1358.
60. Wechalekar AD, Lachmann HJ, Offer M et al. Efficacy of bortezomib in systemic AL amyloidosis with relapsed/refractory clonal disease. Haematologica 2008; 93 : 295–298.
61. Kastritis E, Wechalekar AD, Dimopoulos MA et al. Significant activity of bortezomib--based therapy in patients with primary systemic AL-amyloidosis. Blood 2008; 112 : 321, abstr. 869.
62. Zonder JA, Sanchorawala, V, Snyder RM et al. Melphalan, dexamethosone plus bortezomib induces hematologic and organ response in AL-amylolidosis with tolerable neurotoxicity. Blood 2009; 114: (Suppl. N 22): 310, abstr. 746.
63. Singh V, Saad A, Palmer J et al. Response to bortezomib based induction therapy in newly diagnosed light chain (AL) amyloidosis. Blood 2009; 114: (Suppl. N 22): 740, abstr. 1867.
64. Kastritis E, Wechalekar AD, Dimopoulos MA et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol 2010; 28 : 1031–1037.
65. Lamm W, Willenbacher W, Zojer N et al. Efficacy of the combination of bortezomib and dexamethasone in systemic AL amyloidosis. Blood 2009; 114: (Suppl. N 22): 1121, abstr. 2871.
66. Palladini G, Foli A, Russo P et al. Treatment of IgM-associated AL amyloidosis with the combination of rituximab, bortezomib, and dexamethasone. Clin Lymphoma Myeloma Leuk 2011; 11 : 3–5.
67. Moscetti A, Saltarelli F, Bianchi MP et al. Quick response to bortezomib plus dexamethasone in a patient with AL amyloidosis in first relapse. Amyloid 2011; 18: (Suppl 1): 147–149.
68. Abonour R, Kramer G, Suvannasankha A et al. Bortezomib (Velcade) treatment of AL amyloidosis: Indiana University experience. Amyloid 2011; 18: (Suppl. 1): 146.
69. Coriu D, Badelita S, Talmaci R et al. Bortezomib in systemic AL amyloidosis: a single center experience. Amyloid 2011; 18: (Suppl. 1): 143–145.
70. Dubrey SW, Reece DE, Sanchorawala V et al. Velcade Can 2007 Study Group. Bortezomib in a phase 1 trial for patients with relapsed AL amyloidosis: cardiac responses and overall effects. QJM 2011; 104 : 957–970.
71. Maramattom LV. Bortezomib Based Therapy for Newly Diagnosed Patients with Advanced Multisystem Light Chain Amyloidosis (AL). Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 1880.
72. Palladini G. Treatment of AL Amyloidosis with Bortezomib Combined with Alkylating Agents: Results From a Prospective Series of Unselected Patients. Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 3977.
73. Re A. Bortezomib-Containing Regimens in the Treatment of AL Amyloidosis: A Single Center Experience. Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 4614.
74. Cossor FI. Weekly Combination Chemotherapy with Cyclophosphamide, Bortezomib and Dexamethasone (CyBorD) for Newly Diagnosed Patients with Advanced Cardiac Disease Due to Systemic ALAmyloidosis. Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 5139.
75. Jaccard A. Efficacy of Bortezomib/Cyclophosphamide/Dexamethasone (VCD) Chemotherapy in Naive Patients with High Risk Cardiac Light Chain Amyloidosis (Mayo Clinic stage III). Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 5126.
76. Mikhael JR, Schuster SR, Jimenez-Zepeda VH et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 2012; 119 : 4391–4394.
77. Venner CP, Lane T, Foard D et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood 2012; 119 : 4387–4390.
78. Dimopoulos MA, Kastritis E. Bortezomib for AL amyloidosis: moving forward. Blood 2011; 118 : 827–828.
79. Lamm W, Willenbacher W, Lang A et al. Efficacy of the combination of bortezomib and dexamethasone in systemic AL amyloidosis. Ann Hematol 2011; 90 : 201–206.
80. Jimenez-Zepeda VH, Reeder CB, Mikhael JR et al. Cyclophophamide, bortezomib and dexamethasone induces rapid and complete response in patients with amyloidosis non eligible for peripheral stem cell transplant. Blood 2009; 114: (Suppl. N 22): 737, abstr. 1857.
81. Venner ChP, Lane T, Foard D et al. A matchen comparison of cyclophosphamide, bortzomib and dexamethasone versus cyclophosphamide, thalidomide and dexamethasone in the treatment of Mayo cardiac stage II patients with AL-amyloidosis. Blood 2012; Suppl. Abstracts from the 54th ASH annual meeting December 8–11, Atlanta 2012, Abstr. N 2966.
82. Landau H, Hassoun H, Bello C et al. Consolidation with bortezomib and dexamethasone following risk-adapted melphalan and stem cell transplant in systemic AL amyloidosis. Amyloid 2011; 18 (Suppl. 1): 1300–1301.
83. Ghose A, Tariq Z, Taj A et al. Acute dyspnea from treatment of AL amyloidisis with bortezomib. Am J Ther 2011; 18: e123–e125.
84. Dispenzieri A, Lacy MQ, Rajkumar SV et al. Poor tolerance to high dose thalidomide in patients with light chain associated (AL) amyloidosis. Amyloid 2003; 10 : 257–261.
85. Dispenziery A, Lacy MQ, Zeyer SM et al. Low dose single agent thalidomid is tolerated in patients with primary amyloidosis, but responses are limited. Blood 2004; 104 : 4920–4922.
86. Seldin DC, Choufani EB, Dember LM et al. Tolerability and efficacy of thalidomide for the treatment of patients with light chain-associated (AL) amyloidosis. Clin Lymphoma 2003; 3 : 241–246.
87. Palladinin G, Perfetti V, Perlini S et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis Blood 2005; 105 : 2949–2951.
88. Wechalekar AD, Goodman HJ, Lachmann HJ et al. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood 2007; 109 : 457–464.
89. Palladini G, Russo P, Lavatelli F et al. Treatment of patients with cardiac AL-amyloidosis with oral melphalan, dexamethasone and thalidomide. Ann Hematol 2009; 88 : 347–350.
90. Gibbs SDJ, Sattianayagam PT, Lachmann H et al. Risk adapted cyclophosphamide, thalidomide and dexamethasone (CTD) for treatment of systemic AL-amyloidosis: long term outcome of 202 Patients. Blood 2008; 112 : 611, abstr. 1733.
91. Gibbs SDJ, Gillmore J, Satttianayagam PT et al. in AL-amyloidosis both oral melphalan and dexametzasone and risk adapted cyclophosphamide, thalidomide, and dexamethasone (CTD) have similar efficacy as upfront treatment Blood 2009; 114: (Suppl. N 22): 310, abstr. 745.
92. Gibbs SDJ, Gillmore JD, Sattianayagam PT et al. CTD versus Mel-Dex as upfront treatment in AL-amyloidosis.: a matched case-control study. Amyloid 2010; 17 : 4922–4924.
93. Gillmore J, Cocks K, Gibbs DJB et al. Cyclophosphamide, thalidomide and dexamethasone (CTD) versus melphalan and dexamethasone (MD) for newly diagnosed systemic AL amyloidosis. Result from the UK amyloidosis treatment trial. Blood 2009; 114: (Suppl. N 22): 1120, abstr. 2869.
94. Cohen AD, Zhou P, Chou J et al. Risk-adapted autologous stem cell transplantation with adjuvant dexamethasone ± thalidomide for systemic light-chain amyloidosis: results of a phase II trial. Br J Haematol 2007; 139 : 224–233.
95. Dispenzieri A, Lacy MQ, Zeldenrust S et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood 2007; 109 : 465–470.
96. Sanchorawala V, Wright DG, Rosenzweig M et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood 2007; 109 : 492–496.
97. Dispenzieri L, Lacy M, Zeldenrust SR et al. Long term follow up of patients with immunoglobulin light chain amyloidosis treated with lenalidomide and dexamethasone. Blood 2008; 112 : 612, abstr. 1737.
98. Kastritis E, Zahoura F, Rousnou M et al. Phase I/II study of lenalidomid, intermediate dose of dexamethasone and low dose cyclophosphamide for the treatment of AL-amyloidosis. Blood 2008; 112 : 611, abstr. 1734.
99. Schoenland SO, Bochler T, Ditrich S et al. Single center experience of lenalidomide/dexametazone treatment in 40 patients with light chain amyloidosis. High toxicity in patients with impaired renal and cardiac function. Blood 2008; 112 : 612, abstr. 1736.
100. Moreau P, Jaccard A, Benboubker L et at. Lenalidomide with melphalan and dexamethasone in patients with newly diagnosed light chain amyloidosis. A multicenter phase I/II dose escalation study. Blood 2009; 114: (Suppl. N 22): 177, abstr. 427.
101. Kastritis E, Roussou M, Migkou M et al. A phase I/II study of lenalidomide with low dose dexamethasone and cyclophosphamide for patients with primary systemic light chain amyloidosis. Blood 2009; 114: (Suppl. N 22): 177, abstr. 428.
102. Sloan JM, Sanchowarala V, Girnius S et al. Melphalan, lenalidomide and dexamethasone combination therapy in patients with AL amyloidosis. Blood 2009; 114: (Suppl. N 22): 737, abstr. 1859.
103. Palladini G, Russo P, Bragotti LZ et al. A phase II trial of cyclophosphamide, lenalidomide and dexamethasone in previously treated patients with AL-amyloidosis. Blood 2009; 114: (Suppl. N 22): 1117, abstr. 2863.
104. Kumar S, Hayman SR, Buaudi F et al. A phase II trial of lenalidomide, cyclophosphamide and dexamethasone in patients with light chain amyloidosis. Blood 2009; 114: (Suppl N 22): 1482, abstr. 3853.
105. Sanchorawala V, Finn KT, Fennessey S et al. Durable hematologic complete responses can be achieved with lenalidomide in AL amyloidosis. Blood 2010; 116 : 1990–1991.
106. Palladini G, Russo P, Foli A et al. Salvage therapy with lenalidomide and dexamethasone in patients with advanced AL amyloidosis refractory to melphalan, bortezomib, and thalidomide. Ann Hematol 2012; 91 : 89–92.
107. Kastritis E, Terpos E, Roussou M et al. A phase 1/2 study of lenalidomide with low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL amyloidosis. Blood 2012; 119 : 5384–5390.
108. Kumar SK, Hayman SR, Buadi FK et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long--term results from a phase 2 trial. Blood 2012; 119 : 4860–4867.
109. Moreau P, Jaccard A, Benboubker L et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood 2010; 116 : 4777–4782.
110. Specter R, Sanchorawala V, Seldin DC et al. Kidney dysfunction during lenalidomide treatment for AL amyloidosis. Nephrol Dial Transplant 2011; 26 : 881–886.
111. Batts ED, Sanchorawala V, Hegerfeldt Y et al. Azotemia associated with use of lenalidomide in plasma cell dyscrasias. Leuk Lymphoma 2008; 49 : 1108–1115.
112. Tapan U, Seldin DC, Finn KT et al. Increases in B-type natriuretic peptide (BNP) during treatment with lenalidomide in AL amyloidosis. Blood 2010; 116 : 5071–5072.
113. Finsterer J, Höftberger R, Stöllberger C et al. Sudden death possibly related to lenalidomide given for cardiac and muscle AL amyloidosis secondary to light chain deposition disease. J Oncol Pharm Pract 2012; Apr 13, 112–115. [Epub ahead of print].
114. Sanchorawala V, Skinner M, Quillen K et al. Long-term outcome of patients with AL amyloidosis treated with melphalan and stem cell transpalntation. Blood 2007; 110 : 3561–3563.
115. Gertz MA, Lacy MQ, Dispenzieri A et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica 2007; 92 : 1415–1418.
116. Vesole DH, Perez WS, Akasheh M et al. High-dose therapy and autologous hematopoietic stem cell transplantation for patients with primary systemic amyloidosis: A Center for International Blood and Marrow Transplant Research Study. Mayo Clin Proc 2006; 81 : 880–888.
117. Dispenzieri A, Gertz MA, Kyle RA et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2004; 104 : 1881–1887.
118. Dispenzieri A, Kyle RA, Gertz MA et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet 2003; 361 : 1787–1789.
119. Gertz MA, Lacy M, Dispenzieri A et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk Lymphoma 2008; 49 : 36–041.
120. Skinner M, Sanchorawala V, Seldin DC et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med 2004; 140 : 85–93.
121. Yagi S, Akaike M, Ozaki S. Improvement of cardiac diastolic function and prognosis after autologous peripheral blood stem cell transplantation in AL cardiac amyloidosis. Intern Med. 2007; 46 : 1705–1710.
122. Perfetti V, Siena S, Palladini G et al. Long--term results of risk adapted approach to melphalan conditioning in autologous peripheral blood stem cell transplantation for primary AL amyloidosis. Hematologica 2006; 91 : 1635–1643.
123. Dispenzieri A, Kyle RA, Lacy MQ et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral stem cell transplantation. A case control study. Blood 2004; 103 : 2949–2951.
124. Gertz MA, Lacy MQ, Dispenzieri A et al. Risk-adjusted manipulation of melphalan dose before stem cell transplantation in patients with amyloidosis is associated with a lower response rate. Bone Marrow Transplant 2004; 34 : 1025–1031.
125. Dispenzieri A, Kyle RA, Lacy MQ et al. Superior survival in primary systemic amyloidosis patients udergoing peripheral stem cell transplantation: a case control study Blood 2004; 103 : 3960–3963.
126. Jaccard A, Moreau P, Leblond V et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007; 357 : 83–93.
127. Leung N, Gunderson H, Tan TS et al. Melphalan and dexamethason is less effective for patients with immunoglobulin light chain amyloidosis with high bone marrow plasmocytosis. Blood 2008; 112 : 612, abstr. 1735.
128. Sanchorawala V, Seldin DC, Magnani B et al. Serum free light-chain responses after high-dose intravenous melphalan and autologous stem cell transplantation for AL (primary) amyloidosis. Bone Marrow Transplant 2005; 36 : 597–600.
129. Madan S, Kumar S, Dispenzieri A et al. Outcomes with high dose therapy and peripheral blood stem cell transplantation for light chain amyloidosis with cardiac involvement. Blood 2009; 114: (Suppl. N 22): 223, abstr. 534.
130. Landau H, Hassoun H, Cohen AD et al. Adjuvant bortezomib and dexamethasone following risk adapted melphalan and stem cell transplant in systemic light chain amyloidosis. Blood 2009; 114: (Suppl. N 22): 222, abstr. 533.
131. Landau HJ, Hoffman J, Hassoun H et al. Adjuvant bortezomib and dexamethasone following risk-adapted melphalan and stem cell transplant in patients with light-chain amyloidosis (AL). J Clin Oncol 2009; 27 : 15, abstr. 8540.
132. Hoffman JE, Hassoun H, Landau H et al. Early adjuvant treatment after risk adapted autologou stem cell transplant for systemic light chain amyloidosis. Increased hospitalization and impaired immune recovery but improved responses and overall survival. Blood 2008; 112 : 1143, abstr. 3329.
133. Cibeira MT, Sanchorawala V, Seldin DC et al. Outcome of AL amyloidosis after high--dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood 2011; 118 : 4346–4352.
134. Goodman HJ, Gillmore JD, Lachmann HJ et al. Outcome of autologous stem cell transplantation for AL amyloidosis in the UK. Br J Haematol 2006; 134 : 417–425.
135. Madan S, Kumar SK, Dispenzieri A et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood 2012; 119 : 1117–1122.
136. Schonland SO, Bochtler T, Perz J et al: Results of two consecutive phase II trials of patients with systemic AL amyloidosis treated with high--dose melphalan after induction and mobilization chemotherapy. 12th international symposium on amyloidosis abstracts. Amyloid 2010; 17 : 80–81.
137. Gerz MA, Lacy MA, Bjornsson J et al. Fatal pulmonary toxicity releated to the administration of granulocyte colony stimulation factor in amyloidosis. A report and review of growth factor pulmonary toxicity. J. Hematotherapy and stem cell research 2000; 9 : 635–643.
138. Haynes-Lattin BM, Kurtin PT, Fleming WH. Toxic megacolon, a life threteating complication of high dose therapy and autologous transplantation among patients with AL-amyloidosis. Bone Marrow Transplantation 2002; 30 : 279–285.
139. Kumar S, Dispenzieri A, Lacy MQ et al. High incidence of gastrointestinal tract bleeding after autologous stem cell transplant for primary systemic amyloidosis. Bone Marrow Transplant 2001; 28 : 381–385.
140. Kyle RA, Gertz MA, Lacy MQ et al. Localized AL amyloidosis of the colon: an unrecognized entity. Amyloid. 2003; 10 : 36–41.
141. Gerz, MA, Lacy MQ, Dispenzieri A et al. Amyloidosis. Best Practice Research Clinical Hematology 2005; 18 : 709–727.
142. Leung N, Slezak JM Bertstralh EJ. Acute renal insufficiency after high dose melphalan in patients with primary systemic amyloidosis during stem cell transplantation. Amer J Kidney Dis 2005; 45 : 102–111.
143. Fadia A, Casserly LF, Sanchorawala V et al. Incidence and outcome of acute renal failure complicating autologous stem cell transplantation for AL-amyloidosis. Kidney International 2003; 63 : 1868–1878.
144. Vesole D, Waleska S, Peréz S et al. High dose therapy and autologout hematopoietic stem cell transplantation for patients with primary systemic amyloidosis. A center for International blood and marrow transplant research center. Mayo Clin Proc 2006; 81 : 880–888.
145. Spier BJ, Einstein M, Johnson EA et al. Amyloidosis presenting as lower gastrointestinal hemorrhage. WMJ 2008; 107 : 40–43.
146. Röcken C, Erns J, Hund E et al. Interdisciplinary guidelines on the diagnosis and therapy for systemic amyloidosis – issued by German society of amyloid disease (www.amyloid.de) Deutsche Med Wochenschrift 2006; 131 : 1–28.
147. United Kingdom Myeloma Forum: Guidelines on the diagnosis and management of AL-amyloidosis. Brit J. Hematology 2004; 125 : 681–700.
148. Perfetti V, Siena S, Palladii G et al. Long term results of a risk adapted aproach to melphalan conditioning in autologous peripheral blood stem cell transplantation for primary AL amyloidosis. Hematologica 2006; 91 : 1635–1643.
149. Mhaskar R, Kumar A, Behera M et al. Role of high-dose chemotherapy and autologous hematopoietic cell transplantation in primary systemic amyloidosis: a systematic review. Biol Blood Marrow Transplant 2009; 15 : 893–902.
150. Sanchorawala V. Role of high-dose melphalan and autologous peripheral blood stem cell transplantation in AL amyloidosis. Am J Blood Res 2012; 2 : 9–17.
151. Schönland SO, Dreger P, de Witte T et al. Current status of hematopoietic cell transplantation in the treatment of systemic amyloid light--chain amyloidosis. Bone Marrow Transplant 2012; 47 : 895–905.
152. Palladini G, Merlini G. Transplantation vs. conventional-dose therapy for amyloidosis. Curr Opin Oncol 2011; 23 : 214–220.
153. Mehta J, Dispenzieri A, Gertz MA. High--dose chemotherapy with autotransplantation in AL amyloidosis: a flawed meta-analysis. Biol Blood Marrow Transplant 2010; 16 : 138–140; author reply 140–141.
154. Sanchorawala V, Wright DG, Seldin DC et al. High-dose intravenous melphalan and autologous stem cell transplantation as initial therapy or following two cycles of oral chemotherapy for the treatment of AL amyloidosis: results of a prospective randomized trial. Bone Marrow Transplant 2004; 33 : 381–388.
155. Audard V, Matignon M, Weiss L et al. Successful long-term outcome of the first combined heart and kidney transplant in a patient with systemic Al amyloidosis. Am J Transplant 2009; 9 : 236–240.
156. Mignot A, Varnous S, Redonnet M et al. Heart transplantation in systemic (AL) amyloidosis: a retrospective study of eight French patients. Arch Cardiovasc Dis 2008; 101 : 523–532.
157. Lacy MQ, Dispenzieri A, Hayman SR et al. Autologous stem cell transplant after heart transplant for light chain (Al) amyloid cardiomyopathy. J Heart Lung Transplant 2008; 27 : 823–829.
158. Mignot A, Bridoux F, Thierry A et al. Successful heart transplantation following melphalan plus dexamethasone therapy in systemic AL amyloidosis. Haematologica 2008; 93: e32–e35.
159. Sack FU, Kristen A, Goldschmidt H et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. Eur J Cardiothorac Surg 2008; 33 : 257–262.
160. Perz JB, Kristen AV, Rahemtulla A et al. Long-term survival in a patient with AL amyloidosis after cardiac transplantation followed by autologous stem cell transplantation. Clin Res Cardiol 2006; 95 : 671–674.
161. Gillmore JD, Goodman HJ, Lachmann HJ et al. Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood 2006; 107 : 1227–1229.
162. Dubrey SW, Burke MM, Hawkins PN et al. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant 2004; 23 : 1142–1153.
163. Kastritis E, Dimopoulos MA. Prognosis and risk assessment in AL amyloidosis – state of the art. Amyloid 2011; 18: (Suppl. 1): 84–86.
164. Mekinian A, Lions C, Leleu X et al. Amyloidosis Study Group. Prognosis assessment of cardiac involvement in systemic AL amyloidosis by magnetic resonance imaging. Am J Med 2010; 123 : 864–868.
165. Kristen AV, Giannitsis E, Lehrke S et al. Assessment of disease severity and outcome in patients with systemic light-chain amyloidosis by the high-sensitivity troponin T assay. Blood 2010; 116 : 2455–2461.
166. Kumar S, Dispenzieri A, Lacy MQ et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30 : 989–995.
167. Dispenzieri A, Gertz M, Kyle A et al. Serum cardiac troponins and N-terminal Pro-Brain Natriuretic Peptide: A staging system for primary AL-amyloidosis. J Clin Oncol 2004; 22 : 3751–3757.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2013 Issue 1
Most read in this issue
- Léky indukovaná osteoporóza
- Rizikové faktory rekurentního a těžkého průběhu kolitidy vyvolané infekcí Clostridium difficile
- Zvláštnosti diagnostiky a terapie exokrinní pankreatické nedostatečnosti
- Výsledky katetrové ablace fibrilace síní u pacientů nad 65 let