
Plicní hypertenze – patofyziologické mechanizmy
Pulmonary hypertension – disease mechanisms
Pulmonary hypertension (PH) is known for its variable etiology. PH pathophysiology is very complex and our therapeutic options are limited. Most of known underlying disease mechanisms play a role across all etiological groups of PH, and they are followed by the same morphological and functional changes of pulmonary vasculature. Mostly, we are not able to determine whether one particular mechanism works as a cause or consequence in the chain of events. An imbalance between vasoconstriction and vasodilation becomes the major functional change of pulmonary vasculature in PH. The main morphological changes (termed together as “remodeling”) include cell hyperplasia of pulmonary artery leading to its thickening and narrowing, and impaired regulation of extracellular matrix production leading to reduction in its elasticity. As a result of all these changes, the peripheral vascular resistance in pulmonary vascular bed rises, thus increasing afterload of the right ventricle and finally progressing to its failure. This review aims to summarize and explain the nature of the functional and histological changes in pulmonary arteries which occur in pulmonary hypertension, separately define the role of endothelium and pulmonary artery myocytes, and discuss the most important known pathophysiological mechanisms that lead to these changes.
Key words:
endothelium – intracellular calcium signaling – nitric oxide – pulmonary artery – pulmonary hypertension – remodeling – smooth muscle cell
Authors:
Martin Helán 1,2; Anna Konieczna 2,3; Martin Klabusay 2,3; Vladimír Šrámek 1
Authors‘ workplace:
Anesteziologickoresuscitační klinika LF MU a FN u sv. Anny Brno, přednosta doc. MUDr. Vladimír Šrámek, Ph. D.
1; Mezinárodní centrum klinického výzkumu FN u sv. Anny Brno, ředitel Gorazd B. Stokin, M. D., MSc., Ph. D.
2; LF UP Olomouc, děkan prof. MUDr. Milan Kolář, Ph. D.
3
Published in:
Vnitř Lék 2014; 60(10): 852-858
Category:
Reviews
Overview
Plicní hypertenze (PH) je onemocnění s bohatou etiologií. Také její patofyziologie je značně složitá a možnosti léčby zatím omezené. Většina známých patofyziologických mechanizmů se uplatňuje napříč všemi etiologickými skupinami PH a dochází u nich ke stejným histologickým a funkčním změnám plicní arterie. Většinou však stále nejsme schopni určit, zda příslušný mechanizmus je příčinou nebo následkem jiných. Hlavní funkční změnou plicního řečiště při PH je nerovnováha mezi vazokonstrikcí a vazodilatací. Mezi hlavní morfologické změny označované souhrnně jako remodeling patří hyperplazie buněk plicní arterie vedoucí k jejímu ztluštění a zúžení lumen a dále porušená regulace tvorby extracelulární matrix vedoucí ke snížení její elasticity. Důsledkem všech těchto změn je narůstající periferní vaskulární rezistence plicního řečiště, tedy zvyšování afterloadu pravé srdeční komory progredující až k jejímu selhání. Tento přehledový článek si klade za cíl shrnout a vysvětlit podstatu funkčních a histologických změn plicního řečiště, ke kterým dochází při plicní hypertenzi, odděleně definovat role endotelu a myocytů plicní arterie a představit nejdůležitější známé patofyziologické mechanizmy, které k těmto změnám vedou.
Klíčová slova:
endotel – myocyt – oxid dusnatý – plicní arterie – plicní hypertenze – regulace intracelulárního kalcia – remodeling
Úvod
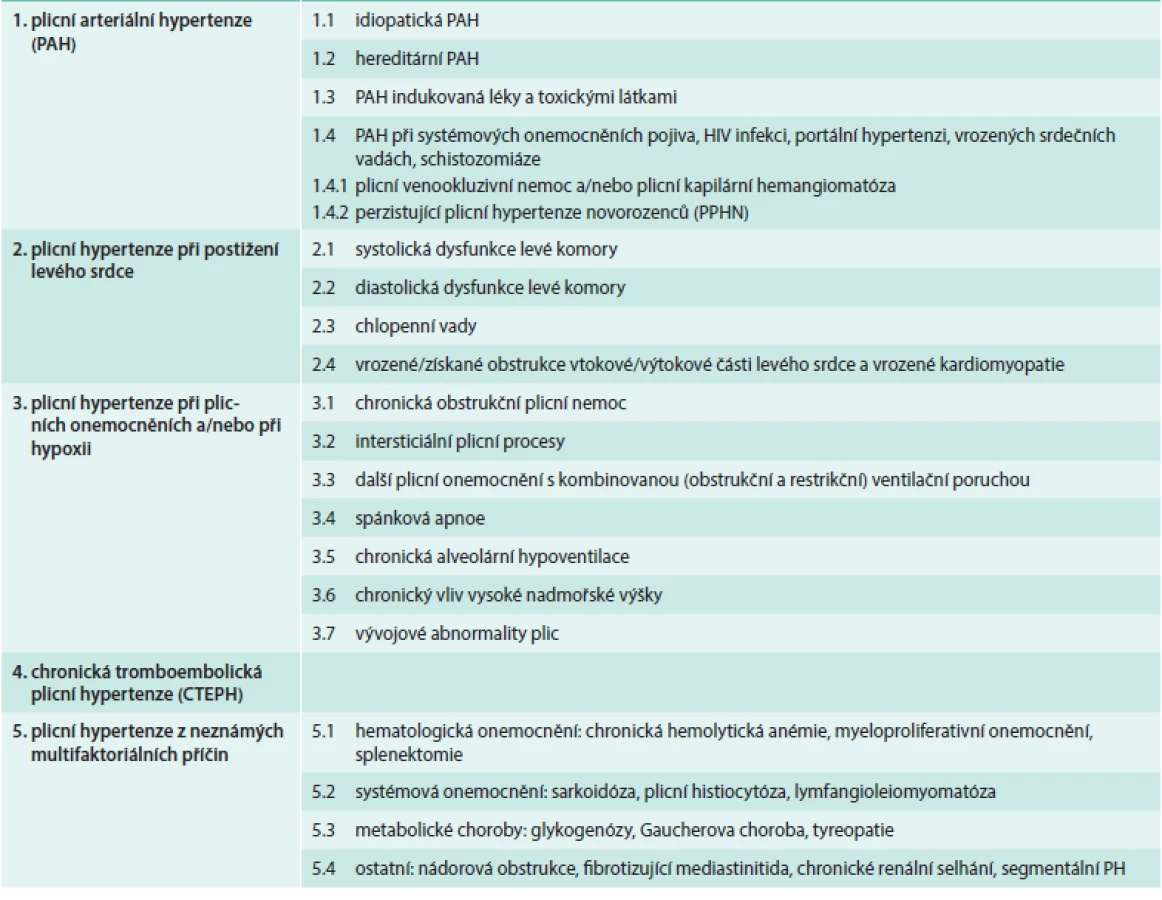
Plicní hypertenze (PH) je onemocnění se složitou etiologií. Podle vyvolávající příčiny jej dnes (dle poslední revize – 5th World Symposium, Nice, France, 2013) dělíme na několik základních etiologických skupin (tab):
- plicní arteriální hypertenze (PAH) zahrnující idiopatické, vrozené a léky či toxiny indukované formy a také PH související s některými jinými nemocemi (systémové nemoci pojiva, HIV atd)
- PH při onemocnění levého srdce
- PH při plicních onemocněních (astma, CHOPN, intersticiální plicní nemoci) a PH způsobená chronickým působením hypoxie (pobyt ve vysoké nadmořské výšce, syndrom spánkové apnoe)
- chronická tromboembolická plicní hypertenze (CTEPH)
- PH z jiné nebo neznámé příčiny (např. PH při sarkoidóze či nádorových onemocněních) [1]
Také patofyziologie a mechanizmy vedoucí k rozvoji a progresi PH jsou značně složité a nepřehledné, což je důvodem zatím bohužel omezených možností léčby. Bylo zjištěno, že většina známých mechanizmů se uplatňuje napříč všemi etiologickými skupinami PH a dochází u nich ke stejným histologickým a funkčním změnám plicní arterie.
Studium PH se kromě klinických dat opírá o poznatky získané použitím zvířecích modelů a izolovaných buněčných kultur. Oba přístupy však mají značné limitace. Každý z využívaných zvířecích modelů simuluje stav PH pouze z části a nezahrnuje všechny patofyziologické mechanizmy současně. Buněčné kultury pak představují in vitro prostředí vzdálené situaci in vivo a studuje se vždy izolovaně pouze vliv jednoho typu buňky, kdežto v patofyziologii PH se uplatňuje celá řada buněčných typů, které se navíc vzájemně bohatě ovlivňují (endotelové buňky, myocyty a fibroblasty plicní arterie, buňky imunitního systému, cirkulující progenitorové buňky atd).
Hlavní funkční změnou plicního řečiště při PH je nerovnováha mezi vazokonstrikcí a vazodilatací nastavená dlouhodobě ve prospěch konstrikce. Mezi hlavní morfologické změny pak patří hyperplazie buněk stěny plicní arterie (PA) vedoucí k jejímu ztluštění a zúžení lumen a porucha regulace tvorby extracelulární matrix vedoucí ke snížení její elasticity.
Hemodynamickým důsledkem těchto změn v plicním řečišti je postupný nárůst periferní plicní vaskulární rezistence, jež zvyšuje afterload pravé komory srdeční a vede při progresi onemocnění až k jejímu selhání.
Cílem tohoto přehledového článku je shrnout a vysvětlit podstatu funkčních a histologických změn plicního řečiště při plicní hypertenzi, oddělit patofyziologické role endotelu a myocytů PA a představit nejdůležitější patofyziologické faktory včetně genetických, které k těmto změnám vedou.
I. Funkční a histologické změny plicního řečiště při plicní hypertenzi
Vazokonstrikce
Stěžejní funkční změnou plicních arterií vedoucí k rozvoji a progresi PH je nerovnováha mezi vazokonstrikcí a vazodilatací. Tonus cév je složitě a jemně regulován mnoha mechanizmy, které reagují na aktuální potřeby tkání a udržují jejich dostatečnou perfuzi. V plicní cirkulaci je při PH tato rovnováha narušena trvale ve prospěch konstrikce. Některé studie poukazují na fakt, že sérové hladiny vazokonstrikčních látek (např. endotelin 1 – ET1, serotonin – 5HT) jsou u pacientů s plicní hypertenzí konstantně zvýšeny [2–6], zatímco produkce některých vazodilatačních faktorů (např. oxid dusnatý – NO, prostacyklin – PGI2) jsou naopak nižší [5,7]. Vazodilatační faktory stejně jako faktory vazokonstrikční jsou produkovány především endotelem PA, ale také myocyty, a mají autokrinní i parakrinní efekt. Síla kontrakce hladkých svalových buněk PA (PASMC) je regulována snížením nebo zvýšením hladiny intracelulárního vápníku.
Remodeling
Pojmem remodeling plicní arterie se označují histologické změny, ke kterým dochází při PH. Tyto povětšinou nevratné změny způsobují fixaci patologického stavu plicního řečiště a mají za následek progresi tíže onemocnění. Dochází k hyperplazii buněk tunica intima i tunica media, způsobené zvýšenou proliferací nebo sníženou úrovní apoptózy endotelových a hladkých svalových buněk. Dále dochází k jevu označovanému jako distální muskularizace, tedy k výskytu myocytům podobných buněk (nesoucích pouze některé jejich znaky) ve stěnách prekapilárních arteriol, tedy v cévách, které za normálního stavu svalové buňky neobsahují. Tento jev je způsoben migrací myocytů do distálnějších úseků PA a jejich proliferací, diferenciací cirkulujících progenitorových buněk [8] a také buněčnou transdiferenciací (tzv. endoteliálně-mezenchymovou transformací buněk) [9]. Buněčná hyperplazie může u některých typů PH vést až ke kompletní obturaci lumen plicních arteriol. Poněkud speciálním příkladem je tvorba tzv. plexiformních lézí, způsobených dezorganizovanou intraluminální proliferací endotelu za přispění mediátorů zánětu a růstových faktorů, která byla zjištěna hlavně u idiopatické PH [10].
Dalším mechanizmem podílejícím se na histologických změnách stěny PA je nadměrná produkce a kvalitativní změny extracelulární matrix. Látky jako kolagen, elastin, tenascin C, fibronektin, laminin a další jsou při PH produkovány zvýšeně [11–14].
Všechny popsané změny, souhrnně označované jako remodeling, vedou ke zvýšení tuhosti (a tedy snížené poddajnosti) stěny PA, což vede k nárůstu periferní vaskulární rezistence v plicním oběhu [15].
Trombóza arteriol
Kromě chronické tromboembolické plicní hypertenze (CTEPH), u níž je hlavním a primárním patofyziologickým mechanizmem, je trombóza pravidelně pozorována i u všech ostatních etiologických skupin. Obecně vzato, má-li dojít ke vzniku trombózy, musí být splněna alespoň jedna podmínka tzv. Virchowovy trias. Při PH se uplatňují všechny tři. Dochází k poškození endotelu, k poruše jeho integrity a permeability, vlivem vazokonstrikce a remodelace dochází ke změnám v průtoku a tlaku působícího na stěnu arterie a konečně je také narušena rovnováha mezi prokoagulačními/antikoagulačními mechanizmy, což vede k hyperkoagulačnímu stavu. U pacientů s PH dochází např. ke snížení produkce látek inhibujících agregaci krevních destiček (oxid dusnatý – NO a prostacyklin – PGI2) a naopak ke zvýšení produkce proagregačních látek (tromboxan A2 – TXA2) [5,16]. Dále je zvýšena aktivita trombinu a inhibována fibrinolýza. Antikoagulační léčba, jež je běžnou součástí léčby PH a zvyšuje přežívání pacientů, brání jednak tvorbě těchto lokálních trombóz, ale také snižuje riziko vzniku plicní embolie [17].
II. Role endotelu a myocytů plicní arterie při plicní hypertenzi
Role endotelu
Za normálního stavu spočívá funkce endotelu v produkci vazodilatačních (např. oxid dusnatý – NO, prostacyklin – PGI2) a vazokonstrikčních látek (např. tromboxan A2 – TXA2, endotelin 1 – ET1) a také faktorů ovlivňujících proliferaci, apoptózu a migraci buněk (např. vaskulární endoteliální růstový faktor/vascular endothelial growth factor – VEGF) [18]. Také vytváří mechanickou bariéru mezi intravaskulárním a intersticiálním prostorem, zároveň však umožňuje přestup některých mediátorů a buněk zánětu. V neposlední řadě má pak nepostradatelný vliv na regulaci krevního srážení. Za patologických stavů, plicní hypertenzi nevyjímaje, pak může dojít k porušení de facto všech jeho funkcí a nastává jev označovaný jako endoteliální dysfunkce. V PH je endoteliální dysfunkce vyjádřena především vzniklou nerovnováhou v regulaci cévního tonu, úrovně proliferace, apoptózy a migrace buněk PA, dále také dysregulací produkce protrombotických/antitrombotických látek a proimfalačních/antiinflamačních látek a vlastní permeability [19]. Také je změněna odezva samotného endotelu na hypoxii, zánět, protrombotické a angiogenní stimuly.
Jednou z hlavních vazodilatačních látek produkovaných endotelem je oxid dusnatý (NO). Jedná se o plynný radikál s velmi krátkým biologickým účinkem. Kromě jeho vazodilatačního působení byl popsán také jeho antiproliferační a proapoptotický účinek, chránící PA před nežádoucím remodelingem [20–22]. NO je produkován pomocí NOS (nitric oxide synthase) enzymů během oxidace substrátu L-argininu na L-citrulin [23]. V endotelových buňkách PA (PAEC) se uplatňuje především endoteliální izoforma (eNOS) zodpovědná za bazální produkci NO, a pak také inducibilní izoforma (iNOS) uplatňující se až při některých speciálních stavech (zánět, hypoxie) [24,25]. Funkce eNOS je složitě regulována, což napovídá, jak důležitou roli tento enzym a jeho produkt (NO) hrají. Aktivace eNOS je závislá na vzestupu intracelulárního Ca2+ (cestou aktivace PLC, IP3) a zahrnuje disociaci eNOS z dutinek (caveolae) buněčné membrány, spojení monomerů eNOS v dimery (tzv. eNOS coupling), vazbu na kalmodulin a fosforylaci na aktivní eNOS [26]. K inhibici eNOS pak vede např. kompetice asymetrického dimetylargininu (ADMA) s L-argininem, fosforylace eNOS pomocí proteinkinázy C (PKC), nedostatek nezbytných kofaktorů (např. tetrahydrobiopterin – BH4), kompetice eNOS s arginázou o L-arginin atd. Argináza např. využívá L-arginin k syntéze močoviny (místo NO) a L-ornitinu, jež je dále metabolizován na prolin a polyaminy, látky zvyšující proliferaci buněk a tvorbu extracelulární matrix [27]. Nerovnováha v produkci NO u pacientů s PH může být tedy částečně vysvětlena zvýšením aktivity arginázy [28]. O důležitosti NO v patofyziologii PH svědčí i klinické důkazy. Např. perorální suplementace L-argininu u pacientů s PAH snižuje tlak v plicnici a zvyšuje toleranci tělesné zátěže, přičemž současně dochází ke zvýšení hladin L-citrulinu, což dokazuje L-argininem navozené zvýšení produkce NO [29–31]. Inhalačně podávaný NO byl v minulosti testován jako slibná léčebná modalita PH, ale pro jeho nepohodlné použití, krátkodobý efekt a rebound fenomén nebyl uveden do běžné praxe.
Dalším velmi potentním vazodilatátorem produkovaným plicním endotelem je prostacyklin (PGI2). PGI2 je metabolit kyseliny arachidonové a je syntetizován cyklooxygenázami (COX). Jeho syntéza, konkrétně její první krok (uvolnění kyseliny arachidonové z membrány pomocí fosfolipázy A2), je závislá na zvýšení hladiny intracelulárního kalcia (podobně jako u eNOS). Stejně jako NO má kromě vazorelaxačního také antimitogenní a antitrombotický efekt [23]. Syntetická analoga prostacyklinu (iloprost, epoprostenol) jsou používána v terapii PH [32].
Kromě vazodilatačních látek produkuje endotel i vazokonstrikční a mitogenní faktory. Hlavním představitelem s oběma těmito účinky je endotelin 1 (ET1). Jeho autokrinní působení (na PAEC zprostředkované vazbou na ETB receptor) vede k produkci NO a tedy k vazodilataci, kdežto parakrinní účinek na PASMC (ETA i ETB receptor) vede k jejich vazokonstrikci, migraci a proliferaci [33–35]. Bylo ale zjištěno, že většina (80 %) endotelinu 1 je vylučována bazolaterálním směrem, tedy směrem od endotelu do hladké svaloviny, kdežto pouze 20 % intraluminálně, což vysvětluje jeho především nežádoucí efekt při PH [36]. ET1 také zvyšuje tvorbu proteinů mezibuněčné hmoty fibroblasty PA [37] a stimuluje sekreci proinflamačních cytokinů [38] a růstových faktorů [39] a potencuje jejich efekt. Za normálních podmínek je sérová hladina ET1 velmi nízká (dokonce pod farmakologickým prahem), což potvrzuje teorii o především autokrinním/parakrinním účinku ET1, avšak u pacientů s PH bylo zjištěno výrazné zvýšení sérové koncentrace. Navíc zvýšená hladina ET1 korelovala s vyšším tlakem v plicnici, vyšší plicní vaskulární rezistencí a vyšší mortalitou [40]. Také exprese ETA a ETB receptorů je vyšší u pacientů s PH [34]. Důležitost ET1 podtrhuje i fakt, že podávání antagonistů endotelinových receptorů (např. bosentan) je běžnou součástí léčby PH.
Kromě výše zmíněných působků je endotel při PH zdrojem i řady dalších látek: růstové faktory (např. VEGF, PDGF, BDNF) [25,41], mitogenně působící angiotenzin II [42], chemokiny (RANTES) [43] a další.
Kromě sekretorické role přispívá endotel rozvoji PH i vlastní proliferací (intimální hyperplazií), tvorbou okluzivních plexiformních lézí, zvýšenou trombogenní aktivitou, porušením své bariérové funkce a zvýšením permeability. Nově je také popisována role cirkulujících endotelových progenitorových buněk (EPC) [44] a slibně se jeví i pokusy o buněčnou terapii využívající autologní EPC [45].
Role hladkých svalových buněk
Hladké svalové buňky PA jsou efektorem vazokonstrikce a vazodilatace, mají vysoký proliferační a migrační potenciál, obzvláště za patologických podmínek, a jsou známé, i když v menší míře než PAEC, také pro svoji sekretorickou funkci.
Kontrakce myocytů, stejně jako jejich proliferace, je děj závislý na vzestupu hladiny intracelulárního kalcia ([Ca2+]i). Kontrakci myocytu vyvolá relativně rychlý vzestup [Ca2+]i, na který navazuje kaskáda dějů (vazba Ca2+ na kalmodulin, aktivace MLCK, fosforylace MLC) zakončená vazbou aktinu a myozinu. V případě proliferace stimuluje pomalý vzestup [Ca2+]i činnost transkripčních faktorů nezbytných pro buněčný růst (CREB, NFAT, AP-1, NFκB) a aktivuje proteiny účastnící se buněčného cyklu. Hladina [Ca2+]i a rychlost jejích změn mají tedy zásadní význam v regulaci cévního tonu i remodelace a jsou řízeny několika různými mechanizmy. Hladina [Ca2+]i a rychlosti jejích změn mají tedy zásadní význam v regulaci jak cévního tonu, tak i remodelace, která je řízena několika různými mechanizmy.
Změny polarizace plazmatické membrány, hlavně zajišťovány pomocí napěťově řízených K+ kanálů (Kv), regulují, zda dojde k vzestupu [Ca2+]i či nikoli. Pokud jsou Kv kanály uzavřeny, nepřestupují K+ ionty extracelulárně, a dochází tedy k depolarizaci, což vede k otevření napěťově řízených Ca2+ kanálů (VDCC), vzestupu [Ca2+]i a vazokonstrikci. Naopak, pokud jsou Kv kanály otevřeny, K+ ionty unikají extracelulárně, dochází k hyperpolarizaci, při níž zůstávají VDCC uzavřeny, k vzestupu [Ca2+]i nedochází a myocyt je relaxován.
Kromě VDCC je vzestup [Ca2+]i zajišťován dvojím mechanizmem: rozlišují se tzv. receptor-operated calcium entry (ROCE) a store-operated calcium entry (SOCE). V případě ROCE je vzestup [Ca2+]i navozen stimulací membránových receptorů, což vede k aktivaci fosfolipázy C, která štěpí PIP2 na IP3 a DAG. DAG se váže na receptor-operated Ca2+ channels (ROC) plazmatické membrány a umožňuje tak přestup Ca2+ z vnějšího prostředí do buňky, kdežto IP3 svojí vazbou na IP3 receptor (IP3R) způsobí uvolnění Ca2+ z ER. Při SOCE pak úbytek Ca2+ v ER způsobí otevření store-operated Ca2+ channels (SOC) plazmatické membrány a dojde k dalšímu přestupu Ca2+ do cytoplazmy, odkud je pumpováno zpět do ER pomocí Ca2+ ATPázy (SERCA) s cílem doplnit chybějící zásoby Ca2+ v ER [46].
V regulaci těchto mechanizmů se uplatňují i signální molekuly cGMP a cAMP. Oba tyto cyklické nukleotidy způsobují pokles [Ca2+]i, a tím relaxaci a inhibici proliferace PASMC. Např. NO je aktivátorem guanylylcyklázy, enzymu, který přeměňuje GTP na cGMP, kdežto PGI2 naopak účinkuje prostřednictvím cAMP. Za deaktivaci cGMP v buňce je mimo jiné zodpovědná fosfodiesteráza 5 (PDE5). Její farmakologická inhibice (např. sildenafilem) zvyšuje dostupnost cGMP, a tím i účinek NO a je využívána v terapii PH [47].
III. Některé další faktory uplatňující se při vzniku a progresi PH
Hypoxie
Za fyziologických podmínek způsobuje akutně působící hypoxie v plicním řečišti vazokonstrikci. Jedná se o adaptační mechanizmus redukující perfuzi neventilovaných (hypoxických) oblastí plicní tkáně. Chronicky působící hypoxie však vyvolává strukturální změny vedoucí k PH. Důkazem, že sama hypoxie způsobuje PH, je fakt, že lidé žijící ve vysoké nadmořské výšce mají signifikantně vyšší tlak v plicnici. U zvířecích modelů s hypoxií indukovanou PH dochází ke stejným strukturálním změnám PA jako u PH jiných etiologických skupin.
Hlavním regulátorem buněčné odezvy na hypoxii je HIF1. Jedná se o transkripční faktor, který za přítomnosti O2 podléhá rychlé degradaci a je tedy aktivní pouze v hypoxii a za speciálních stavů, jako je např. oxidační stres [48]. Činností HIF1 je regulováno více než 100 známých proteinů, včetně těch podílejících se na regulaci cévního tonu PA a její remodelaci [49]. Studie využívající farmakologickou inhibici HIF1 poukazují na slibnou terapeutickou možnost léčby PH [25,50].
Velmi zajímavé je zjištění, že původní obyvatelé Tibetu mají lepší adaptační schopnost pro život ve vysoké nadmořské výšce a tedy na chronicky působící hypoxii. Ve srovnání s jinými populacemi (včetně i jiných vysokohorských populací) mají Tibeťané ve vysoké nadmořské výšce vyšší hladiny NO, vyšší úroveň klidové ventilace, nižší hladiny hemoglobinu a nižší incidenci vysokohorské nemoci [51]. Studiem jejich genomu byly zjištěny polymorfizmy genů zodpovědných za adaptaci na hypoxii (mimo jiné i genů regulovaných pomocí HIF systému) [52,53].
Oxidační stres
Nadprodukce reaktivních forem kyslíku (ROS; hlavně superoxidu a peroxidu vodíku), tedy oxidační stres, se uplatňuje v patofyziologii mnoha onemocnění včetně PH. ROS způsobují přímé poškození tkání oxidací (lipidová peroxidace, poškození proteinů a DNA), ale také fungují jako signální molekuly a ovlivňují řadu důležitých signálních drah: mění funkci transkripčních faktorů (HIF1, NFκB) [48], aktivují proteinkinázy (PKC, ROCK), regulují hladinu [Ca2+]i [54] a těmito mechanizmy ovlivňují tonus PA a její remodelaci.
ROS vznikají několika mechanizmy. Především je to enzymový komplex NADPH-oxidáza, jehož NOX-podjednotky (NADPH oxidase systems) jsou schopné přesunout elektron z NADPH na molekulu kyslíku, a tím produkovat superoxid. V PA jsou nejvíce zastoupeny izoformy NOX2 a NOX4 a mohou být aktivovány řadou látek známých v patofyziologii PH (endotelin 1, angiotenzin II, tromboxan A2, cytokiny) a také změnami v dostupnosti O2 (hypoxie, hyperoxie). Dalším důležitým zdrojem ROS jsou mitochondrie. Za fyziologického stavu jsou 2–3 % O2 vstupujícího do respiračního řetězce nedostatečně redukovány za vzniku superoxidu. Při PH se produkce ROS mitochondriemi výrazně zvyšuje např. vlivem hypoxie, zánětu, angiotenzinu II. Také je popisován pozitivní zpětnovazebný mechanizmus ROS-induced ROS release. Na produkci ROS se také podílejí enzymy cyklooxygenáza, xantin oxidáza a také uncoupled eNOS [55]. Za patologických stavů spojených se zvýšenou aktivací iNOS (hypoxie, zánět, oxidační stres) dochází interakcí superoxidu a NO ke vzniku peroxynitritu (reactive nitrogen species – RNS), který poškozuje proteiny přeměnou tyrozinu na 3-nitrotyrozin [56]. Kromě zvýšené produkce ROS dochází při PH i ke snížení přirozených antioxidačních mechanizmů (superoxid dismutáza – SOD, glutathion peroxidáza – GPX) [57]. O důležitosti oxidačního stresu v PH svědčí i slibné výsledky antioxidační terapie na zvířecích modelech [58–60].
Zánět
Jednou z dalších histologických a funkčních změn PA při PH je chronicky působící zánět. Jsou popisovány infiltráty zánětlivých buněk v plexiformních lézích a perivaskulárně [10], zvýšená exprese adhezních molekul (ICAM1, ELAM1) endotelem [61], up-regulace chemokinů (RANTES, fraktalkine, MCP1, osteopontin) [43,62,63], zvýšené sérové hladiny řady cytokinů a růstových faktorů [64,65]. Také bylo zjištěno, že PASMC izolované od pacientů s PH reagují na zánětlivou stimulaci výraznějším zvýšením proliferace a migrace než kontrolní buňky [63]. U pacientů trpících systémovým zánětlivým onemocněním (SLE, sklerodermie) je rozvoj PH častým jevem, histologické změny jejich plicní tkáně jsou srovnatelné se změnami při IPAH a cílená imunosupresivní terapie snižuje závažnost přidružené PH [66].
Shear stress
Zvýšení tlaku a průtoku plicním řečištěm zvyšuje i tzv. shear stress (česky překládáno jako „střižné napětí“) působící na endotel PA. Studie ukazují, že zvyšující se shear stress má na PA další nežádoucí účinky a napomáhá tedy progresi PH. Vyvolává zánětlivou odpověď (zvyšuje expresi adhezních molekul, cytokinů, aktivuje NFκB) [67,68], stimuluje produkci ROS [69], zvyšuje hypertrofii PASMC a expresi kontraktilních molekul [70] a ovlivňuje produkci růstových faktorů a vazoaktivních látek endotelem s výsledným vazokonstrikčním efektem [71].
Genetické faktory
Již dlouho je známo, že v některých rodinách má PAH familiární výskyt. Genealogické studie u vybraných rodin potvrdily autozomálně dominantní typ dědičnosti. Dnes je tato forma onemocnění označována jako hereditární plicní arteriální hypertenze (HPAH). Následně v roce 2000 byl objeven gen kódující BMPR2 (bone morphogenetic protein receptor, type II), jakožto gen, jehož mutace jsou zodpovědné za naprostou většinu případů HPAH [72,73]. Dále byly zmapovány ještě mutace genů pro ALK1 (activin receptor-like kinase) [74] a endoglin [75] zodpovědné za PAH spojenou s hereditární hemoragickou teleangiektazií. Všechny 3 zmíněné proteiny jsou úzce spojeny se signální dráhou TGFβ.
Shrnutí
Známé patofyziologické mechanizmy způsobující vznik a progresi plicní hypertenze a jejich složité vzájemné vztahy představují komplikovanou a zdánlivě nepřehlednou síť, v níž působení jednoho faktoru, nebo aktivace jedné signální dráhy, vedou k mnoha dalším příčinným mechanizmům. Ve většině případů nejsme schopni určit, který mechanizmus je příčinou a který je následkem. U každé etiologické skupiny PH je zřejmě vyvolávající příčina jiná, ale její následky v podobě aktivovaných signálních drah a výsledné funkční a strukturální změny stejně jako klinické příznaky jsou stejné. Úspěchem dosavadního výzkumu PH je využití důležitých mechanizmů v běžně dostupné terapii. Je pravděpodobné, že klinicky úspěšná terapie může být pouze ta, která ovlivňuje současně více důležitých patofyziologických mechanizmů.
Podpořeno z Evropského regionálního rozvojového fondu – Projekt FNUSA-ICRC (CZ.1.05/1.1.00/02.0123), Evropského sociálního fondu a státního rozpočtu České republiky – Project ICRC Human Bridge – Support of Study Stays of Czech Researchers Abroad III: Young Talent Incubator (CZ.1.07/2.3.00/20.0239).
MUDr. Martin Helán
martin.helan@fnusa.cz
Anesteziologicko-resuscitační klinika LF MU a FN u sv. Anny, Brno
www.fnusa.cz
Doručeno do redakce 11. 5. 2014
Přijato po recenzi 30. 6. 2014
Sources
1. Simonneau G, Gatzoulis MA, Adatia I et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol 2013; 62(25 Suppl): D34-D41.
2. Stewart DJ, Levy RD, Cernacek P et al. Increased Plasma Endothelin-1 in Pulmonary Hypertension: Marker or Mediator of Disease? Ann Intern Med 1991; 114(6): 464–469.
3. Dupuis J, Cernacek P, Tardif JC et al. Reduced pulmonary clearance of endothelin-1 in pulmonary hypertension. Am Heart J 1998; 135(4): 614–620.
4. Kéreveur A, Callebert J, Humbert M et al. High Plasma Serotonin Levels in Primary Pulmonary Hypertension Effect of Long-Term Epoprostenol (Prostacyclin) Therapy. Arterioscler Thromb Vasc Biol 2000; 20(10): 2233–2239.
5. Christman BW, McPherson CD, Newman JH et al. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary Hypertension. N Engl J Med 1992; 327(2): 70–75.
6. Yoshibayashi M, Nishioka K, Nakao K et al. Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Evidence for increased production of endothelin in pulmonary circulation. Circulation 1991; 84(6): 2280–2285.
7. Giaid A, Saleh D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N Engl J Med 1995; 333(4): 214–221.
8. Frid MG, Brunetti JA, Burke DL et al. Hypoxia-Induced Pulmonary Vascular Remodeling Requires Recruitment of Circulating Mesenchymal Precursors of a Monocyte/Macrophage Lineage. Am J Pathol 2006; 168(2): 659–669.
9. Frid MG, Kale VA, Stenmark KR. Mature Vascular Endothelium Can Give Rise to Smooth Muscle Cells via Endothelial-Mesenchymal Transdifferentiation In Vitro Analysis. Circ Res 2002; 90(11): 1189–1196.
10. Tuder RM, Groves B, Badesch DB et al. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994; 144(2): 275–285.
11. Crouch EC, Parks WC, Rosenbaum JL et al. Regulation of collagen production by medial smooth muscle cells in hypoxic pulmonary hypertension. Am Rev Respir Dis 1989; 140(4): 1045–1051.
12. Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol 1997; 150(4): 1349–1360.
13. Botney MD, Kaiser LR, Cooper JD et al. Extracellular matrix protein gene expression in atherosclerotic hypertensive pulmonary arteries. Am J Pathol 1992; 140(2): 357–364.
14. Novotná J, Herget J. Exposure to chronic hypoxia induces qualitative changes of collagen in the walls of peripheral pulmonary arteries. Life Sci 1998; 62(1): 1–12.
15. Chesler N, Wang Z. Pulmonary vascular wall stiffness: An important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulm Circ 2011; 1(2): 212.
16. Chaouat A, Weitzenblum E, Higenbottam T. The role of thrombosis in severe pulmonary hypertension. Eur Respir J 1996; 9(2): 356–363.
17. Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J 2006; 28(5): 999–1004.
18. Mirzapoiazova T, Kolosova I, Usatyuk PV et al. Diverse effects of vascular endothelial growth factor on human pulmonary endothelial barrier and migration. Am J Physiol Lung Cell Mol Physiol 2006; 291(4): L718-L724.
19. Morrell NW, Adnot S, Archer SL et al. Cellular and Molecular Basis of Pulmonary Arterial Hypertension. J Am Coll Cardiol 2009; 54(1 Suppl): S20-S31.
20. Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 2000; 80(4): 1337–1372.
21. Smith JD, McLean SD, Nakayama DK. Nitric Oxide Causes Apoptosis in Pulmonary Vascular Smooth Muscle Cells. J Surg Res 1998; 79(2): 121–127.
22. Tanner FC, Meier P, Greutert H et al. Nitric Oxide Modulates Expression of Cell Cycle Regulatory Proteins A Cytostatic Strategy for Inhibition of Human Vascular Smooth Muscle Cell Proliferation. Circulation 2000; 101(16): 1982–1989.
23. Mitchell JA, Ali F, Bailey L et al. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp Physiol 2008; 93(1): 141–147.
24. Jung F, Palmer LA, Zhou N et al. Hypoxic Regulation of Inducible Nitric Oxide Synthase via Hypoxia Inducible Factor-1 in Cardiac Myocytes. Circ Res 2000; 86(3): 319–325.
25. Helan M, Aravamudan B, Hartman WR et al. BDNF secretion by human pulmonary artery endothelial cells in response to hypoxia. J Mol Cell Cardiol 2014; 68 : 89–97.
26. Dimmeler S, Fleming I, Fisslthaler B et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999; 399(6736): 601–605.
27. Durante W, Johnson FK, Johnson RA. Arginase: A Critical Regulator of Nitric Oxide Synthesis and Vascular Function. Clin Exp Pharmacol Physiol 2007; 34(9): 906–911.
28. Xu W, Kaneko FT, Zheng S et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004; 18(14): 1746–1748.
29. Morris CR, Morris SM, Hagar W et al. Arginine Therapy: A New Treatment for Pulmonary Hypertension in Sickle Cell Disease? Am J Respir Crit Care Med 2003; 168(1): 63–69.
30. Nagaya N, Uematsu M, Oya H et al. Short-term Oral Administration of L-Arginine Improves Hemodynamics and Exercise Capacity in Patients with Precapillary Pulmonary Hypertension. Am J Respir Crit Care Med 2001; 163(4): 887–891.
31. Al-Hiti H, Chovanec M, Melenovský V et al. L-arginine in combination with sildenafil potentiates the attenuation of hypoxic pulmonary hypertension in rats. Physiol Res 2013; 62(6): 589–595.
32. Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J 2008; 31(4): 891–901.
33. Janakidevi K, Fisher MA, Del Vecchio PJ et al. Endothelin-1 stimulates DNA synthesis and proliferation of pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 1992; 263(6 Pt 1): C1295-C1301.
34. Davie N, Haleen SJ, Upton PD et al. ETA and ETB Receptors Modulate the Proliferation of Human Pulmonary Artery Smooth Muscle Cells. Am J Respir Crit Care Med 2002; 165(3): 398–405.
35. Meoli DF, White RJ. Endothelin-1 induces pulmonary but not aortic smooth muscle cell migration by activating ERK1/2 MAP kinase. Can J Physiol Pharmacol 2010; 88(8): 830–839.
36. Wagner OF, Christ G, Wojta J et al. Polar secretion of endothelin-1 by cultured endothelial cells. J Biol Chem 1992; 267(23): 16066–16068.
37. Shi-Wen X, Renzoni EA, Kennedy L et al. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol 2007; 26(8): 625–632.
38. Gallelli L, Pelaia G, D’Agostino B et al. Endothelin-1 induces proliferation of human lung fibroblasts and IL-11 secretion through an ETA receptor-dependent activation of map kinases. J Cell Biochem 2005; 96(4): 858–868.
39. Okuda Y, Tsurumaru K, Suzuki S et al. Hypoxia and endothelin-1 induce VEGF production in human vascular smooth muscle cells. Life Sci 1998; 63(6): 477–484.
40. Nootens M, Kaufmann E, Rector T et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: Relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol 1995; 26(7): 1581–1585.
41. Gerber HP, Dixit V, Ferrara N. Vascular Endothelial Growth Factor Induces Expression of the Antiapoptotic Proteins Bcl-2 and A1 in Vascular Endothelial Cells. J Biol Chem 1998; 273(21): 13313–13316.
42. Schuster DP, Crouch EC, Parks WC et al. Angiotensin converting enzyme expression in primary pulmonary hypertension. Am J Respir Crit Care Med 1996; 154(4 Pt 1): 1087–1091.
43. Dorfmüller P, Zarka V, Durand-Gasselin I et al. Chemokine RANTES in Severe Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2002; 165(4): 534–539.
44. Diller GP, Thum T, Wilkins MR et al. Endothelial progenitor cells in pulmonary arterial hypertension. Trends Cardiovasc Med 2010; 20(1): 22–29.
45. Wang XX, Zhang FR, Shang YP et al. Transplantation of autologous endothelial progenitor cells may be beneficial in patients with idiopathic pulmonary arterial hypertension: a pilot randomized controlled trial. J Am Coll Cardiol 2007; 49(14): 1566–1571.
46. Kuhr FK, Smith KA, Song MY et al. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 2012; 302(8): H1546-H1562.
47. Zhao L, Mason NA, Morrell NW et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation 2001; 104(4): 424–428.
48. BelAiba RS, Djordjevic T, Bonello S et al. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem 2004; 385(3–4): 249–257.
49. Wang J, Weigand L, Lu W et al. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 2006; 98(12): 1528–1537.
50. Abud EM, Maylor J, Undem C et al. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 2012; 109(4): 1239–1244.
51. West JB. High-altitude medicine. Am J Respir Crit Care Med 2012; 186(12): 1229–1237.
52. Simonson TS, Yang Y, Huff CD et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010; 329(5987): 72–75.
53. Beall CM, Cavalleri GL, Deng L et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA 2010; 107(25): 11459–11464.
54. Ko EA, Wan J, Yamamura A et al. Functional characterization of voltage-dependent Ca(2+) channels in mouse pulmonary arterial smooth muscle cells: divergent effect of ROS. Am J Physiol Cell Physiol 2013; 304(11): C1042-C1052.
55. Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol. Neurobiol 2010; 174(3): 212–220.
56. Chovanec M. Role of reactive oxygen species and nitric oxide in development of the hypoxic pulmonary hypertension. Česk Fysiol 2013; 62(1): 4–9.
57. Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA 1997; 94(13): 6954–6958.
58. Masri FA, Comhair SAA, Dostanic-Larson I et al. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin Transl Sci 2008; 1(2): 99–106.
59. Lai YL, Wu HD, Chen CF. Antioxidants attenuate chronic hypoxic pulmonary hypertension. J Cardiovasc Pharmacol 1998; 32(5): 714–720.
60. Lachmanová V, Hnilicková O, Povýsilová V et al. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sci 2005; 77(2): 175–182.
61. Okawa-Takatsuji M, Aotsuka S, Fujinami M et al. Up-regulation of intercellular adhesion molecule-1 (ICAM-1), endothelial leucocyte adhesion molecule-1 (ELAM-1) and class II MHC molecules on pulmonary artery endothelial cells by antibodies against U1-ribonucleoprotein. Clin Exp Immunol 1999; 116(1): 174–180.
62. Lorenzen JM, Nickel N, Krämer R et al. Osteopontin in patients with idiopathic pulmonary hypertension. CHEST J 2011; 139(5): 1010–1017.
63. Sanchez O, Marcos E, Perros F et al. Role of Endothelium-derived CC Chemokine Ligand 2 in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2007; 176(10): 1041–1047.
64. Selimovic N, Bergh C-H, Andersson B et al. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J 2009; 34(3): 662–668.
65. Soon E, Holmes AM, Treacy CM et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010; 122(9): 920–927.
66. Sanchez O, Sitbon O, Jaïs X et al. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. CHEST J 2006; 130(1): 182–189.
67. Li M, Scott DE, Shandas R et al. High pulsatility flow induces adhesion molecule and cytokine mRNA expression in distal pulmonary artery endothelial cells. Ann Biomed Eng 2009; 37(6): 1082–1092.
68. Li M, Tan Y, Stenmark KR et al. High pulsatility flow induces acute endothelial inflammation through over polarizing cells to activate NF-κB. Cardiovasc Eng Technol 2013; 4(1): 26–38.
69. Hsieh HJ, Cheng CC, Wu ST et al. Increase of reactive oxygen species (ROS) in endothelial cells by shear flow and involvement of ROS in shear-induced c-fos expression. J Cell Physiol 1998; 175(2): 156–162.
70. Scott D, Tan Y, Shandas R et al. High pulsatility flow stimulates smooth muscle cell hypertrophy and contractile protein expression. Am J Physiol Lung Cell Mol Physiol 2013; 304(1): L70-L81.
71. Li M, Stenmark KR, Shandas R et al. Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J Vasc Res 2009; 46(6): 561–571.
72. Deng Z, Morse JH, Slager SL et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67(3): 737–744.
73. International PPH Consortium, Lane KB, Machado RD et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26(1): 81–84.
74. Johnson DW, Berg JN, Baldwin MA et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13(2): 189–195.
75. McAllister KA, Grogg KM, Johnson DW et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994; 8(4): 345–351.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 10
Most read in this issue
- Terapeutické monitorování vankomycinu v rutinní klinické praxi
- Optimální způsob podání vysokodávkového i.v. furosemidu – kontinuálně nebo bolusově?
- Monoklonální gamapatie nejistého významu a asymptomatický mnohočetný myelom z pohledu roku 2014
- Dna a kardiovaskulární riziko