
Lymfomatoidní granulomatóza – minulost a současnost
Lymphomatoid granulomatosis – the past and present
Background:
Lymphomatoid granulomatosis (LyG) is a rare multisystemic angiocentric and angiodestructive B lymphoproliferative disease that was first described by Liebow in 1972. Disease was then in the “gray zone” between vasculitis and lymphoproliferative disease. LyG is currently categorized as a primary B lymphoproliferative disease associated with Epstein-Barr (EB) virus according to the World Health Organization (WHO) classification of tumours.
Epidemiology, clinical course and treatment:
Lymphomatoid granulomatosis is a rare disease with unknown prevalence. It occurs more often in males (male : female ratio 2 : 1) between the 5th to 6th decade of life and is more frequent in Europe than in Asia. Lungs are typically the predominantly affected organ; the disease spreads predominantly by extralymphatic manner. Spleen and lymph nodes are affected at an advanced stage. The clinical features are often nonspecific. Dyspnea, cough, hemoptysis, chest pain are the most common features with/without B symptoms (fever, night sweats, weight loss) in the pulmonary involvement. The radiographic finding of the lung is very diverse, but when there are multiple bilateral nodular lesions with basal predominance in perilymphatic distribution, we should think of this disease, although LyG rarely occurs. The histopathologic examination of affected tissue (most commonly the lung) is necessary to confirm the diagnosis. The thoracoscopy is used mainly. When the pulmonary findings are without any response to antibiotics, the autoimmune cause and other granulomatous inflammations (tuberculosis, sarcoidosis, etc.) are excluded, this diagnostic performance is indicated. Prognosis is variable – from spontaneous remission to progressive disease, often with aggressive behavior. Median survival is 14 months from diagnosis and mortality rate is 60% in the first year – despite the treatment. Treatment strategy is chosen depending on the histological grade. The therapy is not yet standardized. Interferon α, rituximab, glucocorticoids, cyclophosphamide and combined immunochemotherapy have been used for the treatment. The disease may lead to pulmonary failure, fatal CNS (central nervous system) involvement and sometimes develops into progressive EB virus positive lymphoproliferative disorder.
Conclusion:
Improvements in understanding of the biology of LyG, especially in determining the precise role of EB virus infection in its pathogenesis may lead to optimization of treatment strategies for this disease. Novel treatment modalities are urgently needed due to unfavourable prognosis. Adoptive immunotherapy appeals to be a promising approach.
Key words:
epidemiology – Epstein-Barr virus – lymphomatoid granulomatosis
Authors:
Alice Sýkorová 1; Vít Campr 2; Petra Kašparová 3; Eva Kočová 4; David Belada 1; Marek Trněný 5; Pavel Žák 1
Authors‘ workplace:
IV. interní hematologická klinika LF UK a FN Hradec Králové, přednosta doc. MUDr. Pavel Žák, Ph. D.
1; Ústav patologie a molekulární medicíny 2. LF UK a FN Motol Praha, přednosta prof. MUDr. Roman Kodet, CSc.
2; Fingerlandův ústav patologie LF UK a FN Hradec Králové, přednosta prof. MUDr. Aleš Ryška, Ph. D.
3; Radiologická klinika LF UK a FN Hradec Králové, přednosta prof. MUDr. Antonín Krajina, CSc.
4; I. interní klinika – klinika hematologie 1. LF UK a VFN Praha, přednosta prof. MUDr. Marek Trněný, CSc.
5
Published in:
Vnitř Lék 2014; 60(3): 225-238
Category:
Reviews
Overview
Úvod:
Lymfomatoidní granulomatóza (LyG) je vzácné angiocentrické a angiodestruktivní multisystémové B-lymfoproliferativní onemocnění, které bylo poprvé popsáno v roce 1972 (A. A. Liebow). Tehdy onemocnění představovalo „šedou zónu“ mezi vaskulitidou a lymfoproliferací. V současnosti se dle klasifikace Světové zdravotnické organizace (World Health Organization – WHO) řadí mezi primární B lymfoproliferace asociované s virovou infekcí Epsteina-Barrové (EB) a je často provázené imunodeficitním stavem.
Epidemiologie, klinický průběh a léčba:
Toto raritní onemocnění s neznámou prevalencí se častěji objevuje u mužů mezi 50.–60. rokem života; častěji v západních zemích než v Asii. Predilekčně bývají postiženy plíce a onemocnění se šíří převážně extranodálně. Slezina a uzliny bývají postiženy až v pokročilém stadiu choroby. Klinické projevy jsou často nespecifické. Plicní postižení se může projevit dušností, kašlem, hemoptýzou, svíravým pocitem na hrudi s možností přítomnosti B-symptomů (febrilie, noční pocení, úbytek váhy). Radiografický obraz plicní LyG je velice rozličný, ale pokud se jedná o mnohočetné bilaterální nodulární léze s bazální predominancí v perilymfatické distribuci, měli bychom na toto onemocnění, i přes jeho vzácný výskyt, myslet. K potvrzení diagnózy je nutné histologické vyšetření postižené tkáně (nejčastěji plic), především pomocí torakoskopie. Tento diagnostický výkon je indikován nejčastěji při neregredujícím plicním nálezu po antibiotické terapii, po vyloučení autoimunitní příčiny procesu či vyloučení jiných granulomatózních zánětů (tuberkulóza, sarkoidóza atp). Prognóza je variabilní – od spontánní remise k progresivní chorobě, častěji s agresivním chováním. Medián přežití dosahuje 14 měsíců od stanovení diagnózy a mortalita představuje 60 % v 1. roce, a to navzdory terapii. V závislosti na histologickém „grade“ onemocnění se volí léčebná strategie, která dosud není standardizována. Léčebně se uplatňuje interferon α, rituximab, glukokortikoidy, cyklofosfamid a kombinovaná imunochemoterapie. Onemocnění může vést k plicnímu selhání, k fatálnímu postižení centrálního nervového systému (CNS) nebo se může rozvinout progresivní EBV pozitivní lymfoproliferace.
Závěr:
Zdokonalení v porozumění biologickému chování, zvláště v určení přesné role EBV infekce v patogenezi LyG, slibuje optimalizaci léčebné strategie u tohoto onemocnění. Pro neuspokojivou prognózu onemocnění je nutné hledat stále nové léčebné přístupy. Nadějně se jeví uplatnění adoptivní imunoterapie v praxi.
Klíčová slova:
epidemiologie – Epsteinův-Barrové virus – lymfomatoidní granulomatóza
Úvod – od minulosti k současnosti
Lymfomatoidní granulomatóza (LyG) je vzácné angiocentrické a angiodestruktivní multisystémové B-lymfoproliferativní onemocnění asociované s virovou infekcí Epsteina-Barrové (EB) postihující nejčastěji plíce [1]. Řadí se mezi primární plicní lymfoproliferace, které představují 0,5 % všech primárních plicních tumorů [2]. Poprvé bylo onemocnění popsáno v roce 1972 A. A. Liebowem et al jako triáda polymorfních lymfoidních infiltrátů, angiitidy a granulomatózy [3]. V práci Liebowa et al, která zahrnovala 40 pacientů, je pravděpodobné, že některé případy diagnosticky nesplňovaly současná histopatologická kritéria LyG. Onemocnění se řadilo mezi benigní choroby. Otázkou tehdy bylo, zda je LyG zánětlivé onemocnění typu vaskulitidy podobné např. Wegenerově granulomatóze, nebo zda se jedná o lymfoproliferaci [3–5]. LyG se od charakteristického průběhu lymfomů lišila častou absencí lymfadenopatie, splenomegalie a naopak přítomností vaskulitidy [3]. Katzensteinová et al přiblížili ve své práci příbuznost LyG s lymfoproliferací prostřednictvím prezentace 12 % případů transformací do nehodgkinského lymfomu (NHL) a přítomností uzlinového postižení [4]. V minulosti vůbec bývalo chápání LyG nejednotné a při pohledu do literatury zjistíme, že co autor, to většinou jiná definice onemocnění. Dokud nebyla dána jasná diagnostická imunohistologická kritéria s podpůrnými klinickými daty, obraz LyG byl v minulosti klasifikován jako vaskulitida, některé podjednotky B - či T-NHL nebo jako posttransplantační lymfoproliferace (post-transplant lymphoproliferative disease – PTLD) [6–9]. Převažující zastoupení T-lymfocytů na pozadí histologického obrazu často vedlo k domněnce, že se jedná o T-lymfom [8]. Postupem času byla LyG zařazena k B-lymfoproliferativním stavům proto, že ačkoliv je v imunohistologickém obraze nalézána převaha T-lymfocytů, tak maligní populaci tvoří monoklonální B-lymfocyty [10,11]. Od roku 2001 je LyG součástí WHO klasifikace jako B lymfoproliferativní onemocnění. V této minulé klasifikaci bylo onemocnění (společně s polymorfním PTLD) uvedeno v podkapitole „B lymfoproliferativní onemocnění nejistého maligního potenciálu“ [12]. Toto bylo ve WHO klasifikaci hematopoetických a lymfatických orgánů z roku 2008 zrušeno, takže se v současné době řadí mezi B nehodgkinské lymfomy, ale stále nikoliv jako lymfom, nýbrž pouze jako lymfoproliferativní onemocnění – tomu odpovídá i kód lomený jedničkou (M-9766/1) [1].
Vnímání tohoto onemocnění je však stále nejednotné a v současnosti se objevují názory, že dojde k rozpadu této jednotky a jako LyG bude nadále vnímáno onemocnění grade 3 (dle současné klasifikace). U LyG grade 1 a 2 je předpoklad, že se vzhledem k morfologickému nálezu a odlišnému klinickému chování jedná spíše o zánětlivá onemocnění, podobně jako případy nemoci spojené s imunoglobuliny IgG4, Castlemanovy choroby či jiných obdobných chorob.
Etiopatogeneze LyG
Ačkoliv Liebow et al ve své původní práci zvažovali virovou patogenezi LyG, teprve v roce 1990 Katzensteinová, Peiper et al identifikovali EB virový (EBV) genom ve velkých B-buňkách [13]. Následně Guinee et al v roce 1994 a Myers et al v roce 1995 prokázali přítomnost EBV RNA ve velkých B-buňkách pomocí metody in situ hybridizace. Tento výsledek potvrdily následné studie, a tak byla zjištěna významná role EBV infekce ve vzniku tohoto onemocnění [10,11,13,14].
Epsteinův-Barrové virus
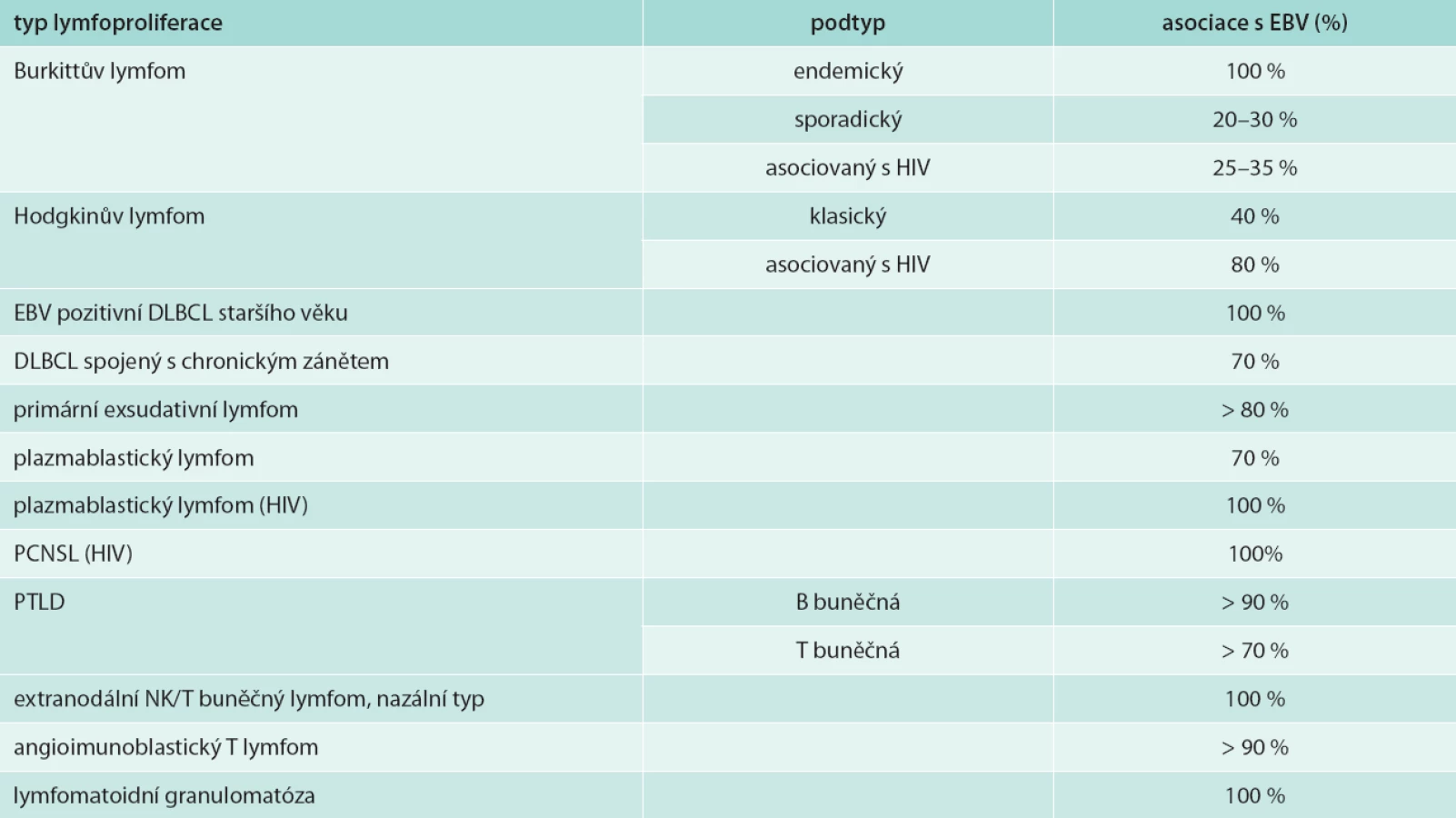
EBV byl poprvé popsán Epsteinem a Barrovou v roce 1964 v histologickém materiálu Burkittova lymfomu [15]. Jedná se o herpetický DNA virus vyskytující se ve 2 typech: 1 a 2. Klinický průběh je u obou typů shodný, liší se pouze geny pro nukleární proteiny. Až 90 % dospělé populace je během života infikováno EBV, který infikuje především B-lymfocyty. Primoinfekce probíhá buď inaparentně, nespecifickým horečnatým onemocněním, nebo se projevuje onemocněním – infekční mononukleózou. Organizmus se brání proti viru nespecifickými mechanizmy, např. NK buňky (natural killer), fagocytóza, interferony, komplementový systém, a specifickými obrannými mechanizmy (protilátková imunita IgG a IgM s neutralizací viru protilátkami proti povrchovým antigenům viru, vznik imunokomplexů, opsonizace) s cílem destrukce virů a lýzy infikovaných buněk [16]. Virus je schopen perzistovat v organizmu člověka a vyvolávat latentní infekci (latentní vylučování viru slinami, přítomnost antigenu viru v postižených buňkách) s možností symptomatické či asymptomatické reakce [16]. V době latence dochází k expresi některých nestrukturálních virem kódovaných antigenů. Existuje 6 EB nukleárních antigenů (EB-nuclear antigen – EBNA) – 1, 2, 3a, 3b, 3c, LP a 3 latentní membránové proteiny (LMP) – 1, 2a a 2b. K tomu jsou ještě v jádru přítomny netranslatované řetězce RNA EBER – 1 a 2 (EB virus – encoded RNA) [17–19]. Tři různé typy latence (typ I, II, III) EBV, lišící se kombinací exprimovaných znaků, jsou charakteristické pro jednotlivé lymfoproliferace asociované s EBV [19]. Virus Epsteinův-Barrové má řadu vlastností společných s ostatními herpetickými viry, ale odlišný je v tom, že má onkogenní potenciál uplatňující se především u imunokompromitovaných nemocných. Porucha buněčné imunity hostitele může vést k EBV indukované proliferaci B buněk, a tak ke vzniku maligního lymfoproliferativního stavu [16,20]. Epsteinův-Barrové virus může vyvolat celou řadu lymfoproliferací s převahou B buněčných neoplazií včetně LyG [17–20]. EBV u LyG vykazuje latenci typu II (LMP – 1, 2, EBER) [20]. Přehled lymfoproliferací asociovaných s EBV uvádí tab. 1 [20].
Lymfomatoidní granulomatóza a porucha imunity
Při vzniku LyG se uplatňuje nepřiměřená reakce na virovou infekci EB v důsledku snížené imunitní kontroly T-lymfocyty (deficit CD4 pozitivních pomocných lymfocytů, CD8 pozitivních cytotoxických lymfocytů) [21] a podobá se vzniku lymfoproliferativních chorob spojených s poruchami imunity (např. PTLD) [22,23]. Predisponujícím faktorem vzniku LyG je tedy přítomnost primárního či sekundárního imunodeficitního stavu [1,3,4,24,25].
Tab. 2 ukazuje nejčastější primární imunodeficitní stavy se zvýšeným rizikem vzniku lymfoproliferace a sekundární poruchy imunity [25,26]. Nemocní při diagnóze LyG nemusí trpět žádnou klinicky manifestní poruchou imunity, ale často u nich bývá přítomna imunitní deregulace na EBV. I když etiologie procesu není detailně prozkoumána, zdá se, že by LyG mohla vznikat při globálním deficitu CD8 pozitivních cytotoxických lymfocytů s přidruženými imunitními poruchami [26].
![Přehled nejčastějších primárních a sekundárních imunodeficitů asociovaných s lymfoproliferací [25]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/cc0bcc6292c0a5a52f78d10b30f4fa61.png)
Epidemiologie
LyG je vzácné onemocnění bez známé prevalence. Častěji se vyskytuje v západních zemích než v Asii. Objevuje se více u mužů než u žen (2 : 1) mezi 50.–60. rokem života [1]. Byly popsány ale i případy s výskytem nemoci u dětí mladších 4 let [21,27,28].
Klinická manifestace
Příznaky, pro které nemocný vyhledá lékaře, jsou často nespecifické a odvíjejí se od tíže orgánového postižení, které je celkem variabilní. Predominantně jsou postiženy plíce (často bilaterálně) [4]. Onemocnění se šíří většinou extranodálně – nejčastěji do CNS, ledvin, kůže a jater [3,4,8]. Postižení CNS se objevuje až u 1/3 nemocných a je spojeno s nepříznivou prognózou [3,4]. Literárně byly popsány případy s postižením orbit, varlat či očí [29–31]. Extranodální postižení může být pouze izolované na jeden orgán [29,31]. Přítomnost lymfadenopatie se splenomegalií bývá neobvyklá, nicméně byla popsána. Častěji se objevuje až v pokročilém stadiu onemocnění nebo se již jedná o velkobuněčný lymfom či transformaci do difuzního velkobuněčného B-lymfomu (diffuse large B-cell lymphoma – DLBCL) asociovaného s EBV infekcí [3,4].
Projevy orgánového postižení
Plicní postižení se může projevovat kašlem, dušností, hemoptýzou, svíravým pocitem na hrudi [3,4,32]. Kožní postižení se objevuje především na končetinách, projevuje se heterogenně v podobě kožních a podkožních nodulů podobného vzhledu jako v plicích, makulopapulárních erupcí, erytému. Podkožní noduly a makulopapulární erupce se objevují u 40–45 % pacientů [33]. Indurované kožní plaky a léze s různým stupněm ulcerace jsou spíše vzácností. CNS postižení může způsobit mentální změny, ataxii, diplopii, vertigo, parézy, záchvaty a objevuje se asi v 30 % případů. Distální senzorické neuropatie, mononeuritidy, bývají u postižení periferních nervů (do 10 % případů) [34]. Postižení jater se projevuje klinicky velmi vzácně, hepatomegalie je přítomna asi jen u 12 % nemocných a představuje horší prognózu [4]. Vzácně se LyG může projevit i fatální hemoptýzou (jako projev kavitací) nebo plicním selháním. Ojediněle může probíhat naopak zcela asymptomaticky [4,36]. Prevalenci orgánového postižení ukazuje tab. 3.
![Prevalence orgánového postižení u LyG [3,4]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/324ce3fc83fedc9f38c36030916e5509.png)
Fyzikální nález
Na plicích je většinou normální nebo lehce patologický nález navzdory velkému plicnímu postižení. V práci Liebowa et al měla pouze méně než polovina pacientů s plicním postižením patologický fyzikální nález [3]. Fyzikální vyšetření může častěji odhalit kožní či neurologické postižení.
Celkové příznaky
B-příznaky (febrilie, noční pocení a úbytek hmotnosti až o 10 % za 6 měsíců) bývají většinou přítomny [3,14,30,37]. Někdy anamnesticky zjišťujeme rekurentní a perzistující infekce na podkladě imunodeficitního stavu.
Laboratorní změny
Charakteristické laboratorní ukazatele nejsou přítomny [3,4]. Kvantita imunoglobulinů je většinou v normě [4]. Řada pacientů má dysfunkční subpopulaci T-lymfoctyů (CD4 pozitivních a CD8 pozitivních T-lymfocytů) [14]. Sedimentace erytrocytů, hodnota laktátdehydrogenázy, C-reaktivního proteinu či β2-mikroglobulinu mohou být v normě či zvýšeny. Testy na autoimunitu (revmatoidní faktor, antinukleární faktor) jsou obvykle negativní, výsledky sérologie pro EBV nejsou spolehlivé, ale pozitivita může být přítomna.
Radiologické nálezy
Radiografický obraz slouží především k monitoraci léčebné odezvy. Stanovení diagnózy na základě zobrazovacích metod není možné vzhledem k rozličnému a nespecifickému obrazu. Popisovány jsou mnohočetné (až v 75 %) bilaterální (v 60–80 %) nodulární léze [4,32,38]. Většina těchto lézí je velikosti do 1 cm, jsou však popisovány i nodularity velké 6–8 cm [32,39]. Výjimečně se vyskytuje hilová a mediastinální lymfadenopatie [32,36].
RTG vyšetření
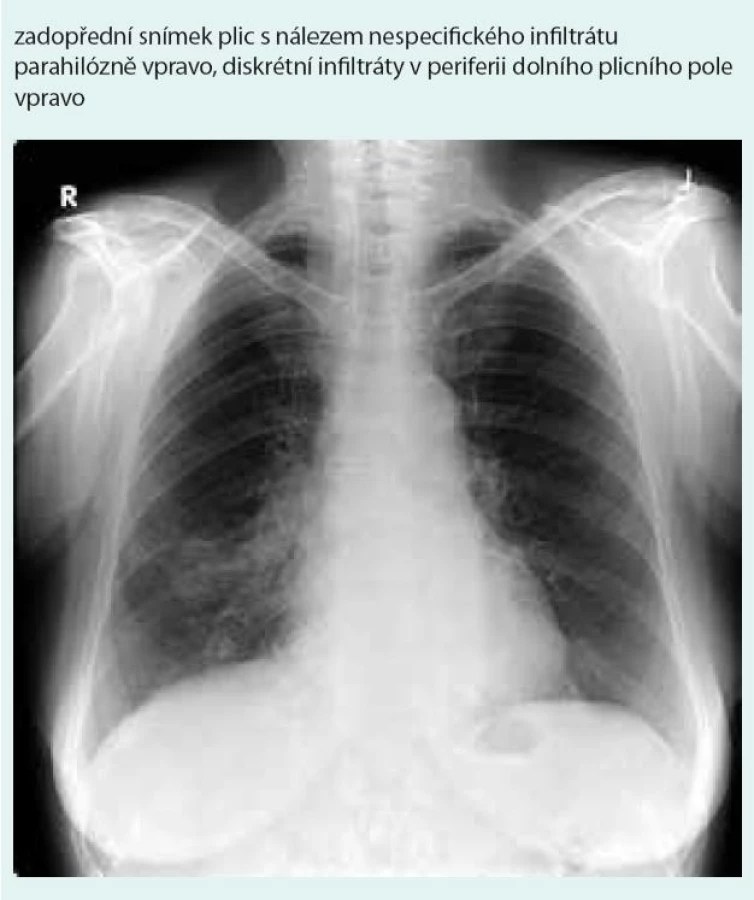
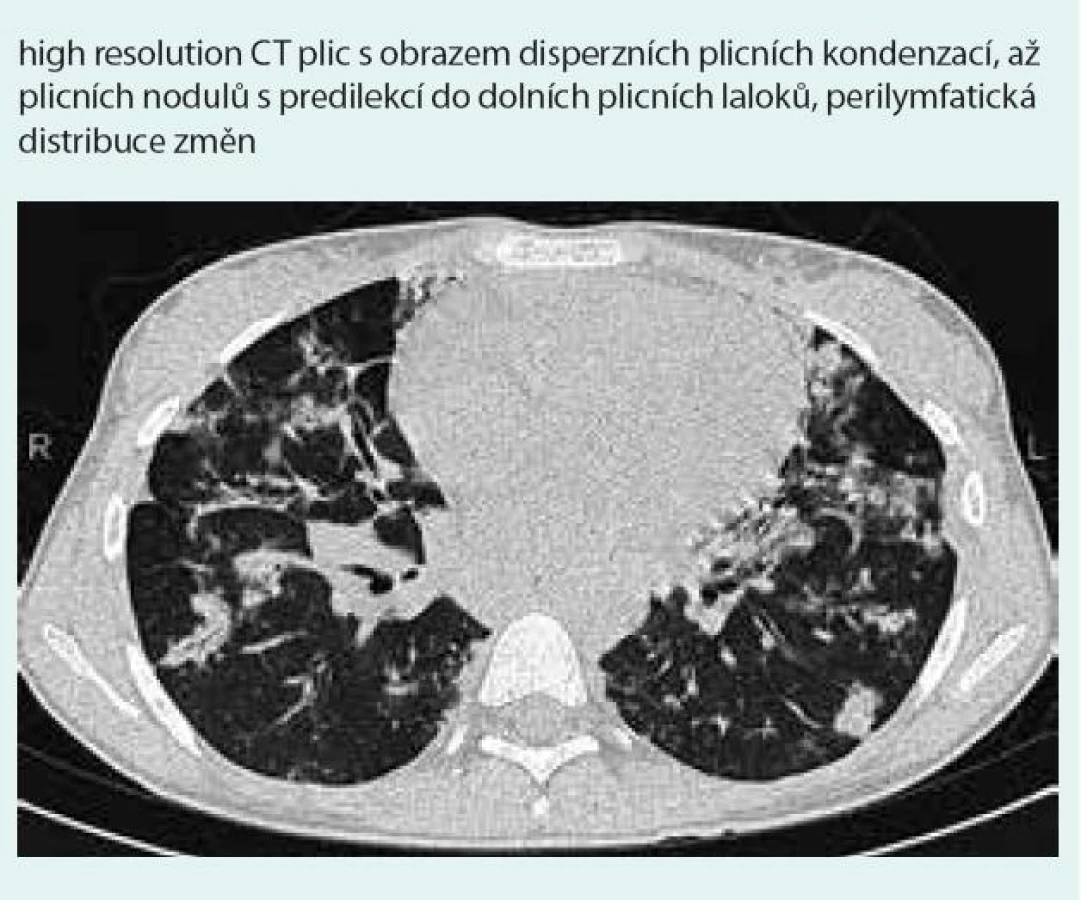
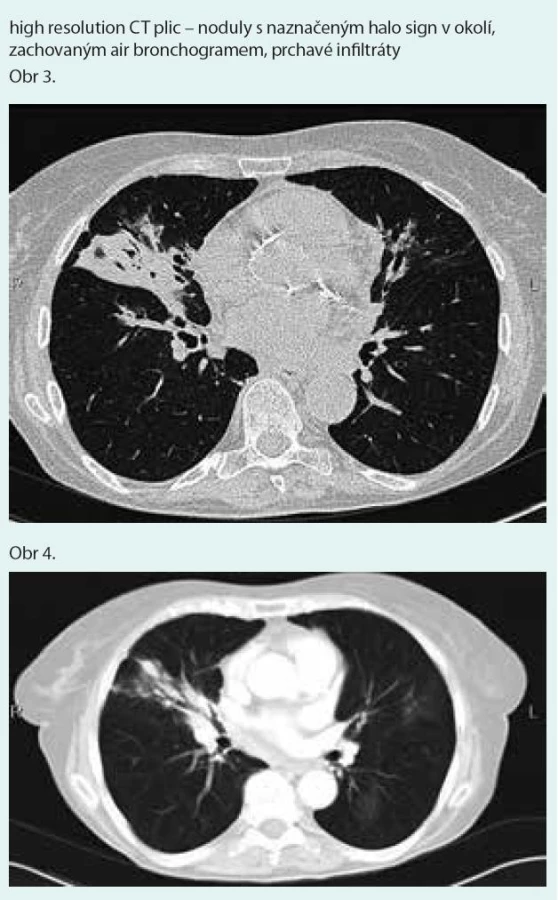
Prostý snímek plic zobrazuje nespecifické infiltrativní změny s predilekcí do periferie dolních plicních polí s širokou diferenciální diagnostikou (obr. 1). Výpočetní tomografie s vysokým prostorovým rozlišením (HRCT – high-resolution computerized tomography): nejčastěji jsou popisovány nodulární léze s bazální predominancí v perilymfatické distribuci (obr. 2). Maximum jich je v oblasti peribronchovaskulárního intersticia a interlobulárních sept. Noduly mohou rychle progredovat, kavitovat či spontánně mizet [2,4]. Typicky jsou tyto léze dobře ohraničené, jsou však popsány případy s jasně zřetelným „halo sign“ (denzity mléčného skla) v okolí vyskytující se vzhledem k angioinvazi (obr. 3 a 4) [38]. V literatuře se však objevuje i popis denzit mléčného skla uvnitř uzlu jako „reverse halo sign“ [2]. Noduly mají zachovaný air bronchogram a jasně zřetelné tepny procházející nodulem (obr. 5 a 6). Bronchy v nodulu mohou být ektatické. Pozitronová emisní tomografie s 18F deoxyglukózou (FDG-PET): literární zkušenosti s FDG-PET vyšetřením u LyG jsou malé, představují odkazy na jednotlivé kazuistiky s převážně pozitivním záchytem FDG v plicních nodulech [40–43]. Problém představuje nespecificita vyšetření. U low grade 1–2 LyG byly popsány kazuistické případy jak se zvýšením záchytu FDG [36,41,42], tak i bez záchytu [44]. Přehled FDG-PET nálezů na některých literárně zpracovaných kazuistických sděleních ukazují ve své práci Arai et al [41].
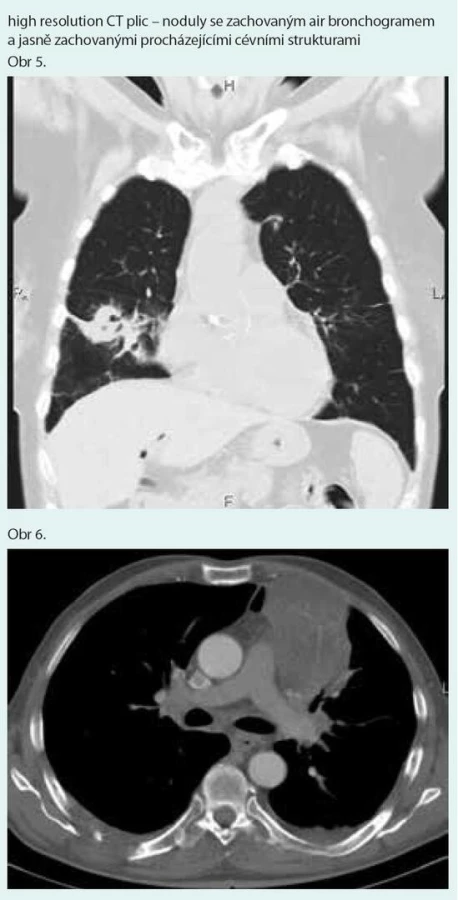
Diagnostika
Diagnóza LyG se stanovuje histologicky odběrem biopsie z postiženého místa. Jelikož je predominantně postižena plíce (asi v 90 % případů), jedná se většinou o odběr vzorku z plicní tkáně. Nejčastěji používanou metodou je videotorakoskopie s bioptickým odběrem a plicní transbronchiální biopsie. Protože se LyG nevyskytuje bronchocentricky, bývá transbronchiální biopsie často nevýtěžná. Pro přítomnost nekróz bývá nediagnostický i odběr punkční jehlou pod CT kontrolou. Druhotně je pak nezbytné přistoupit k videotorakoskopii. V práci Pisani et al byla otevřená plicní biopsie diagnosticky úspěšná v 70 % případů, transbronchiální plicní biopsie v 15 % a mimoplicní biopsie v 15 % případů [45].
Základní diagnóza LyG se provádí v korelaci morfologického nálezu, imunohistochemické typizace nádorových buněk a metod na průkaz EBV infekce.
Histologická diagnostická kritéria [1,5,6,46–48] (obr. 7 a 8)
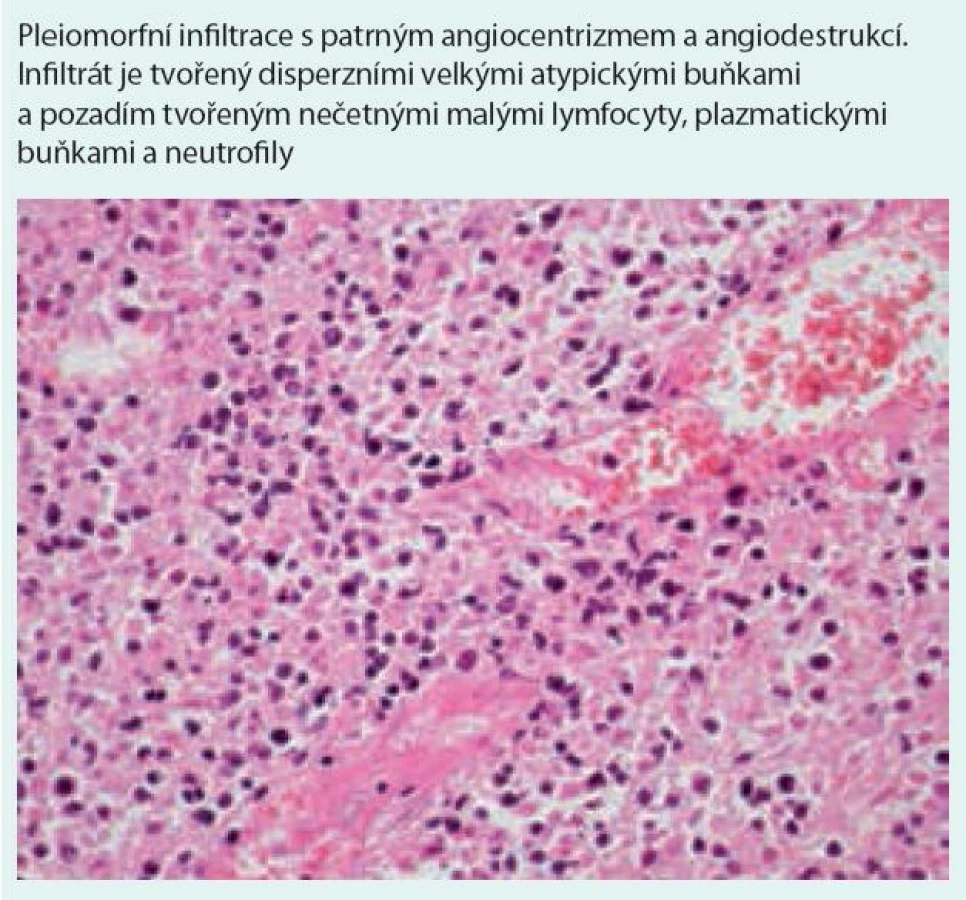
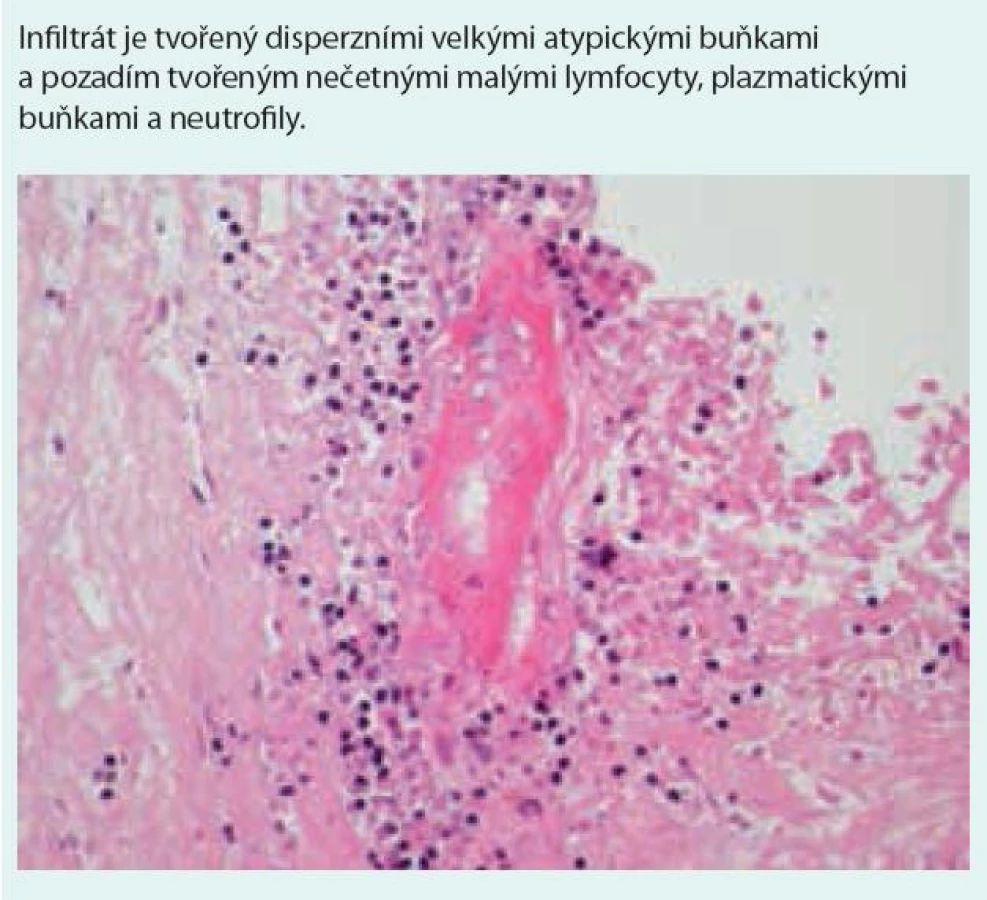
Nodulární polymorfní lymfoidní infiltráty tvořené z malých lymfocytů, imunoblastů, histiocytů, plazmatických buněk a různého počtu velkých atypických mononukleárních buněk, B-lymfocytů s pozitivitou CD20, CD79 a variabilní pozitivitou CD30 (při negativitě CD15 antigenu). Neutrofilní segmenty a eozinofily jsou přítomny jen ojediněle, ve velkém početním zastoupení na pozadí histologického obrazu jsou reaktivní CD3 pozitivní, CD4 pozitivní a CD8 pozitivní T-lymfocyty. Lymfoidní infiltrace je vázána na cévy, nejedná se však o intravaskulární postižení.
Transmurální vaskulitida arterie nebo vény – infiltrace lymfocyty (proces odlišný od klasické vaskulitidy), preference postižení intimy s následným zúžením cévy a vznikem nekrózy. Poznámka: Jedním z mechanizmů je infiltrace T-lymfocyty bránící se proti EBV infekci. Dalším mechanizmem je postižení cévy chemokiny jako IP-10 a M-ig EBV s následkem destrukce cévní stěny.
„Granulomatóza“ – v lymfoidních uzlech se vyskytují nekrózy. Nejedná se o klasické granulomy, LyG je klasický „misnomer“ na podkladě obdobného zobrazovacího nálezu LyG a klasických granulomatózních lézí (vaskulitidy atp) v plicích. Typickou granulomatózní reakci možno ale vidět při postižení kůže (v podkoží). Poznámka: Tomuto původně makroskopickému označení – granulomatóza – odpovídá v mikroskopii solidní hnízdo epiteloidně transformovaných histiocytů s variabilní příměsí drobných lymfocytů. Tyto klasické granulomy nacházíme např. u tuberkulózy nebo sarkoidózy. U velké skupiny onemocnění však kompaktnost a ohraničenost granulomům chybí a jedná se spíše o nekohezivní shluky vícejaderných buněk, které se rozptylují na zánětlivém pozadí. K terminologickým zvláštnostem dodejme, že existují onemocnění, která si z granulomu ponechávají kompaktnost a ohraničenost, ale postrádají jeho patogenetický základ.
Stanovení grade lymfomatoidní granulomatózy
Obraz LyG obsahuje různý počet velkých B buněk s průkazem EBV, které často vykazují četné atypie [1]. Výše grade se posuzuje v závislosti na počtu přítomných EBV pozitivních buněk a nekróz s odlišením 3 prognosticky významných skupin [1,5,7,46]. U grade 1 se nachází polymorfní buněčný infiltrát v podstatě bez atypií, s malým podílem imunoblastů a nápadným zánětlivým pozadím, obvykle bez nekróz. Nález EBV pozitivních buněk může chybět [14]. Pro grade 2 je obvyklá větší atypie lymfocytů a imunoblastů a mírná mitotická aktivita a pro grade 3 výrazná buněčná atypie, monomorfní velké buňky i buňky silně atypické, vícejaderné až podobné RS (Reed-Sternberg) buňkám; obraz již odpovídá velkobuněčnému lymfomu, je přítomna malá a obvykle pouze periferní zánětlivá infiltrace, nápadné nekrózy a velmi četné EBV pozitivní buňky (tab. 4). Je velmi důležité rozlišit grade 3 od grade 1 a 2 [4,5]. Grade onemocnění se může měnit v čase a biopsie z odlišných lokalizací nemusí jevit ve stejný čas shodný grade [14].

Průkaz viru Epsteina-Barrové
Pro diagnostiku se používá metoda přímého průkazu viru v postižené tkáni. Pouhé vyšetření virové DNA pomocí polymerázové řetězové reakce PCR (polymerase chain reaction) v bioptickém materiálu není schopno odlišit přirozeně infiltrovanou tkáň EBV – pozitivními latentně infikovanými B-lymfocyty – od patologické proliferace těchto buněk. Proto je pro tyto účely vhodný průkaz EBV infekce přímo ve tkáňových řezech (při mikroskopickém vyšetření je možné identifikovat, zda jsou infikované právě atypické buňky patologického infiltrátu). Pro imunohistochemický průkaz EBV je v histologii k dispozici protilátka proti latentnímu membránovému proteinu 1 (LMP1). Za nejspolehlivější je však považovaná detekce EBV ve tkáních metodou in situ hybridizace se značenou fluorescenční sondou EBER1/2 (Epstein-Barr virus-encoded RNA) [49]. Stanovení samotné klonality B-buněk v diagnostice LyG příliš nepomůže. LyG grade 1 a 2 bývají často polyklonální, LyG grade 3 a některé typy grade 2 monoklonální – s přestavbou IgH genu pro těžký řetězec B-lymfocytu. T buněčný receptor (TCR-T cell receptor) je nezměněn [50].
Poznámka: Sérologické vyšetření EBV (průkaz heterofilních a specifických protilátek proti některým proteinovým antigenům EBV) se uplatňuje při zjišťování primoinfekce, ale při diagnostice reaktivace EBV je již daleko méně spolehlivé. Při reaktivaci infekce dosahují tyto protilátky jen nízkých hladin nebo se vůbec netvoří. Pro diagnostiku onemocnění souvisejících s virózou EB (např. tedy LyG) je sérologické vyšetření nevhodné: toto onemocnění vždy souvisí s poruchou funkce imunitního systému, tudíž i s poruchou tvorby protilátek). PCR lze užít při průkazu deoxyribonukleové kyseliny (DNA) EBV z periferní krve, ale s variabilním výsledkem [51].
Pokud jsou splněna všechna diagnostická imunohistologická kritéria, je stanovení diagnózy LyG poměrně jednoduché. V případě nejasností a nesplnění všech diagnostických podmínek, stejně tak při nepřítomnosti EBV pozitivity B-lymfocytů u grade 1, pomáhají v diagnostice další histologické rysy, přídatná klinická data, radiografický nález a typ diseminace choroby. Pro diagnózu je nezbytná přítomnost nodulárních polymorfních lymfoidních infiltrátů tvořených z malých lymfocytů, histiocytů, plazmatických buněk a různého počtu CD20 pozitivních velkých atypických mononukleárních buněk s četnými CD3 pozitivními T-lymfocyty na pozadí, současně s infiltráty v cévní stěně. Podpůrným nálezem jsou nekrózy celulárních infiltrátů, pozitivita EBER, mnohočetné postižení plic s vedlejším postižením kůže a nervového systému. Stanovení diagnózy bývá obvykle protrahované, neboť biologické chování, klinická manifestace a radiografické nálezy nejsou specifické a diagnostika je většinou komplexního rázu. Lymfomatoidní granulomatóza může imitovat řadu jiných chorob včetně pneumonie. Je diagnostikována v průměru mezi 3–6 měsíci od počátku symptomů, např. po neúspěšné antibiotické terapii pro suspektní pneumonii [4,52]. K pomocným vyšetřením při stagingu onemocnění patří vyšetření krve (krevní obraz, sedimentace, vyšetření subpopulací B-lymfocytů, stanovení laktátdehydrogenázy), vyšetření močového sedimentu, kvantita imunoglobulinů, diagnostická lumbální punkce, zobrazovací metody – RTG, výpočetní tomografie (CT, HRCT), CT/PET, zobrazení magnetickou rezonancí (MRI), vyšetření kostní dřeně a klinické údaje [4,14].
Diferenciální diagnóza
Diferenciální diagnostika plicních procesů je celkem složitá. Z histologického pohledu je nutné odlišit především jiné B - i T-lymfoproliferace, Hodgkinův lymfom a granulomatózní procesy, z radiografického pohledu a z pohledu klinických rysů ještě další nehematologické choroby.
Histologie
Největší podobnost s LyG vykazuje extranodální NK/T-lymfom, nazální typ pro predominanci T-lymfocytů, angiodestruktivní chování a rovněž asociaci s EBV infekcí [1]. Obě jednotky jsou společně nazývány angiocentrickými imunoproliferativními lézemi (angiocentric imunoproliferative lesion – AIL). K rozdělení na LyG a NK/T-lymfom došlo až s nástupem molekulárních metod a potvrzením klonality B-, resp. T-lymfocytů. Dosud je některými autory LyG považována za B-lymfom bohatý na T-lymfocyty (T-cell/histiocyte-rich large B-cell lymphoma – THRBCL) [9]. S tím má ale LyG společný jen výskyt velkých B-lymfocytů mezi převažujícími T-lymfocyty. Jinak je to naprosto odlišná nozologická jednotka postihující jiné orgány, často s infiltrací dřeně při prezentaci. Asociace s EBV infekcí či vaskulitida nejsou přítomny. Na rozdíl od difuzního velkobuněčného B lymfomu (DLBCL) je u LyG typická vaskulitida a přítomnost zánětlivého pozadí [1], je však především nutné odlišit EBV pozitivní DLBCL vyššího věku. Společné histomorfologické rysy může mít LyG s Hodgkinovým lymfomem (HL) pro přítomnost vícejaderných buněk. LyG ale imunohistochemicky vykazuje variabilní pozitivitu antigenu CD30 a negativitu antigenu CD15, na rozdíl od Reed-Sternebergových/Hodgkinových (RS/H) buněk u klasického HL, které jsou vždy CD30 pozitivní a často současně vykazují pozitivitu antigenu CD15 [1]. Z nehematologických diagnóz může LyG napodobit klasickou vaskulitidu, u které se objevuje polymorfonukleární infiltrace a intramurální fibrinoidní nekrotizace, což by u typické LyG být přítomno nemělo.
Radiografie
Obraz na prostém RTG snímku plic je nespecifický s širokou diferenciální diagnostikou. V diferenciální diagnostice dle CT obrazu přichází v úvahu zejména Wegenerova granulomatóza a vaskulitidy.
Klinické rysy
Diferenciálně diagnosticky je možné pomýšlet na infekci, absces, primární plicní tumor nebo metastázu, vaskulitidu, granulomatózní zánět (sarkoidóza, tuberkulóza – TBC) či lymfoproliferaci. Diferenciálně diagnostická rozvaha je zobrazena v tab. 5.
![Diferenciální diagnostika lymfomatoidní granulomatózy z klinického pohledu [1,4–6,35]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/9be4bb67cbf7b2d2bc31c44141b9a780.png)
Prognóza
I když je hranice mezi „polyklonalitou a monoklonalitou“ a současně mezi „benigním a maligním průběhem“ LyG někdy velmi obtížně definovatelná, prognóza LyG je špatná [37]. Mortalita onemocnění je obecně velmi vysoká (63,5 %) s mediánem přežití 14 měsíců [4] a více než 60 % nemocných umírá do 5 let. Většina úmrtí (94 %) nastává do 3 let od stanovení diagnózy. Mezi nejčastější příčiny úmrtí patří plicní selhání a infekční komplikace [4]. Věk < 25 let v době diagnózy, multiorgánové postižení, leukocytóza, postižení CNS, oboustranný plicní proces, hepatomegalie a přítomnost velkých atypických buněk v histologickém obraze se řadí k prognosticky nepříznivým rizikovým faktorům [4]. Za nejvýznamnější prognostický faktor je považováno dosažení kompletní remise po indukční terapii [7].
O prognóze rozhoduje především grade onemocnění. U pacientů s LyG grade 1 (low grade), u nichž je proces imunitně dependentní a většinou je spojen s polyklonalitou B-lymfocytů, bylo v některých případech pozorováno dosažení dlouhodobě trvající spontánní remise [4]. Nicméně i zde je riziko progrese a transformace do EBV pozitivního difuzního velkobuněčného B lymfomu. Diseminace onemocnění je spojena s nepříznivou prognózou [47]. LyG grade 2 častěji progreduje a transformuje se do agresivního difuzního velkobuněčného B lymfomu a LyG grade 3 primárně odpovídá obrazem i chováním difuznímu velkobuněčnému B lymfomu – jedná se o imunitně independentní monoklonální proces [20,44,46,53]. Onemocnění relativně dobře odpovídá na indukční terapii, pokud se terapie včas zahájí a adekvátně zvolí. Objevuje se ale častá rekurence onemocnění či progrese a transformace do lymfomu [54]. Transformace se objevuje u 1/3 případů grade 1 a 2/3 případů grade 2 [45]. Riziko jejího vzniku a progrese onemocnění jsou přímo úměrné délce trvání onemocnění. Prognóza je pak velmi nepříznivá [46]. Zajímavostí je, že u některých pacientů po úspěšné léčbě transformace s dosažením kompletní remise se může objevit následně low grade LyG v podobě relapsu [55]. V případě relapsu je tak vždy nutné proces histologicky verifikovat.
Léčba
Prospektivními studiemi nebyla dosud stanovena optimální léčebná strategie. Rozhodnutí o typu terapie je často svízelné, neboť prospektivních studií je početní minimum a v publikovaných kazuistických sděleních jsou prezentovány rozmanité léčebné modality pro různý grade onemocnění s nejednotnými výsledky. Z léčebných postupů je možné v individuálních případech volit vyčkávací strategii watch and wait (W+W), léčebně se podávají imunosupresiva (glukokortikoidy, cyklofosfamid – CF), cytostatika buď v monoterapii (chlorambucil) či v kombinaci (CHOP – cyklofosfamid, doxorubicin, vinkristin, prednison, COP – cyklofosfamid, vinkristin, prednison, CHOEP – cyklofosfamid, doxorubicin, vinkristin, etoposid, prednison, EPOCH – etopozid, doxorubicin, vinkristin, cyklofosfamid, prednison, C-MOPP – cyklofosfamid, mustargen, vinkristin, prokarbazin, prednison), dále antivirové (valganciklovir), antiproliferativní a imunomodulační látky (interferon α – INFα, rituximab – R). Literárně jsou popisovány i jednotlivé zkušenosti s autologní či alogenní transplantací u progredující či relabující choroby.
Katzensteinová et al prezentovali dosud největší soubor nemocných s LyG (152 nemocných), ve kterém byli nemocní rozděleni do 4 skupin podle strategie terapie (tab. 6). Mortalita (63,5 %) a přežití bez nemoci (disease free survival – DFS) byly podobné ve všech skupinách, nezávisle na typu indukční léčby. Pro malý počet pacientů ve skupinách nebyly analyzovány jednotlivé léčebné přístupy [4].
![Léčebná strategie LyG dle Katzensteinové et al [4]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/62d6af9c99c67c361ba9032e3f3977af.png)
Popis a zkušenost s jednotlivými léčebnými přístupy
Vyčkávací strategie „Watch and Wait“
V individuálních případech byla pozorována regrese onemocnění bez terapie, především u onemocnění s nižším grade (low grade 1; 14–27 % nemocných) [4,56]. Následně však může dojít k relapsu onemocnění [57].
Glukokortikoidy
Glukokortikoidy (GK) byly v minulosti podávány i samostatně, v současnosti většinou v kombinaci s imunochemoterapií. Glukokortikoidy v monoterapii vedou k rychlému klinickému zlepšení (např. zlepšení dušnosti, regrese neurologického deficitu), ale během krátké doby (měsíce) se pravidelně objeví relaps či progrese onemocnění. Tato léčba není vhodná pro dlouhodobou kontrolu onemocnění [4,35]. Léčba GK dále prohlubuje možný imunodeficitní stav, a tak zvyšuje riziko infekcí.
Glukokortikoidy se zkoušely podávat v kombinaci s CF, čímž se zvýšil počet kompletních remisí (46 % s mediánem trvání 5,2 let) a prodloužilo se celkové přežití, ale mortalita byla stále vysoká (53 % ve 3,5 letech) vzhledem k velké četnosti progrese a transformace do velkobuněčného B lymfomu [54]. Kombinované režimy s kortikoidy jako COP, MOPP, GK + azathioprim nebyly příliš efektivní a podskupiny jednotlivých kombinací nebylo možné hodnotit pro malý počet pacientů [4]. Kombinace CHOP byla v některých případech již úspěšnější [58], což svědčí o tom, že terapie musí být v některých případech shodná s léčbou pro agresivní lymfomy.
Interferon α
Účinnost interferonu α (INFα) je založena na jeho antiproliferativním (zásah do buněčného cyklu indukcí exprese inhibitorů cyklin dependentních kináz), imunomodulačním (stimulace cytotoxických lymfocytů, zvýšení produkce toxických cytokinů) a protivirovém (potlačení replikace virů) působení [59]. Je možno ho použít u LyG grade 1 a většiny případů grade 2 [14,20,60,61]. Léčba INFα bývá však často provázena rozmanitými vedlejšími účinky. Některé se objevují u všech nemocných, některé se objevují méně často a některé výjimečně. Mezi nejčastější nežádoucí účinky INFα patří tzv. interferonový syndrom s průvodními chřipkovými příznaky (horečka, zimnice, třesavka, bolesti hlavy, kloubů a svalů). Tyto příznaky se po zahájení terapie objevují prakticky u všech nemocných a s maximem v prvních dnech aplikace. Ke zmírnění vedlejších účinků je preventivně vhodné před každou aplikací podat paracetamol a aplikovat INFα ve večerních hodinách před ulehnutím ke spánku. Během 1. týdne aplikace často dochází ke zmírnění vedlejších účinků, ale u některých pacientů se vedlejší příznaky opakují po každé aplikaci. Vzhledem k intenzivním vedlejším účinkům někteří pacienti nemusejí terapii INFα tolerovat. Po odeznění akutních příznaků se často objevuje únava, slabost, pokles fyzické výkonnosti, zažívací obtíže, nechutenství, změny nálad, neurologické změny (parestezie, neuropatie), což často snižuje kvalitu života nemocných. INFα má též supresní účinek na kostní dřeň (leukopenie, neutropenie) a dalším vedlejším účinkem může být lehká nebo středně těžká reverzibilní alopecie [59].
Wilson et al prokázali efekt INFα u low grade 1 a 2 u 4 pacientů (1krát léčený relaps po režimu CHOP, 3 nepředléčení pacienti). Medián sledování byl 39 měsíců. V době publikace trvala kompletní remise 36, 43 a 60 měsíců, 1 pacient dosáhl parciální remise s trváním 16 měsíců – po přerušení terapie INFα zemřel na transformaci do velkobuněčného B lymfomu [14]. Dunleavy et al léčili INFα 31 nemocných s grade 1 a 2 po dobu 1 roku. Z 28 hodnocených pacientů 60 % dosáhlo kompletní remise, 21 % nemocných progredovalo do onemocnění s vyšším grade 3. Kompletní remise dosáhlo 9 z 10 pacientů s postiženim CNS. 5letého přežití bez progrese (progression free survival – PFS) dosáhlo 56 % pacientů [61].
Pro nedostatečný počet studií s nízkým počtem zařazených pacientů nelze doporučit dávkování ani trvání terapie INFα, nicméně v praxi se osvědčily dávky 10–40 milionů mezinárodních jednotek (MIU) 3krát týdně podkožně, iniciálně nejčastěji 7,5–10 MIU 3krát týdně s navýšením dávky dle tolerance a efektu [14,61]. Léčba trvala 1–3 roky, kratší doba nebyla zkoušena [14,30,61]. INFα může navodit u LyG grade 1 až 2 trvající remisi, pro indolentní chování je ale nemalé riziko relapsu [61]. Podává se v monoterapii, v kombinaci s chemoterapií nebo jako udržovací terapie [14,61–64].
Rituximab
Použití cílené terapie pomocí chimérické myší/lidské monoklonální protilátky anti CD20 připravené metodami genetického inženýrství používané v terapii B lymfoproliferativních onemocněních a autoimunitních chorob, je prezentováno u LyG pouze v podobě kazuistických sdělení.
Exprese CD20 pozitivity na EB pozitivních B-lymfocytech, imunomodulační efekt a nízká toxicita mají své opodstatnění v jeho užití. V účinku se předpokládá různá míra kombinace následujících mechanizmů: indukce apoptózy B-lymfocytů nesoucích znak CD20 (pre-B-lymfocyty a zralé lymfocyty), aktivace cytotoxických buněk závislých na protilátkách a cytotoxický účinek zprostředkovaný komplementem [65]. Léčba rituximabem je dobře tolerována. Vedlejší účinky provázejí většinou pouze první aplikaci infuze (akutní infuzní reakce v souvislosti s vyplavením cytokinů následující po lýze B-lymfocytů) a jsou převážně lehkého stupně. Mezi nejčastější projevy patří febrilie, třesavka, bronchospazmus, urtika, svědění, angioedém či mírná hypotenze nebo hypertenze. Profylaktické podání metylprednisolonu a paracetamolu snižuje incidenci a závažnost těchto reakcí. Případné reakce jsou terapeuticky dobře ovlivnitelné intravenózní aplikací kortikoidů, antihistaminik a současným přechodným pozastavením infuze. Závažné infuzní reakce se vyskytují ojediněle [59]. Rituximab je podáván u LyG v monoterapii, v kombinaci s chemoterapií nebo jako udržovací terapie. Vzhledem k odlišnému mechanizmu působení rituximabu ve srovnání s běžnými cytostatiky je kombinovaná léčba s cytostatiky vhodná a může být synergická.
Léčba rituximabem v monoterapii není u LyG standardizována co do velikosti celkové dávky. Ve většině kazuistických sdělení je rituximab podáván v běžné dávce 375 mg/m2 i.v. jako u jiných B lymfoproliferací, u LyG nejčastěji 1krát týdně, po dobu 4–5 po sobě jdoucích týdnů, v infuzi fyziologického roztoku nebo 5% glukózy, rychlostí 50–400 mg/hod. Počet podání se určuje dle efektu terapie. Jestliže se po 4 aplikacích za týden nedostaví léčebná odezva, léčebná strategie se většinou mění (imunochemoterapie).
V současné době nejsou k dispozici studie, které by hodnotily léčebnou odezvu a trvání remise po terapii rituximabem [28]. Literárně je dostupných 28 případů léčených rituximabem v monoterapii nebo v kombinaci s chemoterapií v letech 2003–2013 (2 pediatričtí pacienti, 26 dospělých) [28,34,38,43,66].
Rituximab podávaný u LyG grade 1–3 v monoterapii ve 4–5 dávkách (375 mg/m2 i.v.) u 9 pacientů vedl k dosažení 6 kompletních remisí, ke zlepšení radiologického nálezu u 2 pacientů a 2krát k progresi onemocnění [28,34,38]. V 1 případě rituximab vedl ke kompletní remisi i u LyG grade 3 [67]. V případech, v nichž došlo k progresi, není v dostupné literatuře uveden grade onemocnění [38,68].
V kombinaci s kortikoidy nebo s chemoterapií u 19 nemocných (nejčastěji rituximab + COP, rituximab + CHOP, rituximab + CHOEP, rituximab + cyklofosfamid, rituximab + vysokodávkovaná chemoterapie + autologní transplantace + rituximab) došlo v 6 případech k progresi onemocnění a v 7 případech ke kompletní remisi (u 5 pacientů nebyla léčebná odezva hodnocena) [28,38,43,66].
Otázkou je role rituximabu u CNS postižení. Z 28 pacientů mělo 8 CNS postižení. Přesto, že pouze 1 % rituximabu přechází přes hematoencefalickou bariéru, 3 nemocní měli benefit z podávání rituximabu. Ishiura et al popisují zkušenost s dosažením kompletní remise po monoterapii rituximabem u nemocného s izolovaným postižením CNS [69]. Zkušenost s intratekálním podáváním rituximabu u LyG nebyla prozatím publikována. Pro zjištění účinnosti rituximabu je třeba dalších klinických hodnocení.
Radioterapie
Radioterapie může být léčebnou volbou u lokalizovaného onemocnění (kožní léze) nebo jako konsolidační terapie po indukční léčbě [37,66]. Jsou publikovány kazuistiky s postižením CNS a dosažením kompletní remise po radioterapii [70,71], ale vzhledem ke špatné prognóze u CNS postižení je vhodné volit agresivnější terapii (imunochemoterapie, transplantace).
Transplantace
Literárně je prezentováno několik případů s úspěšným provedením autologní [62,72,73] či alogenní transplantace (tab. 7) [74].
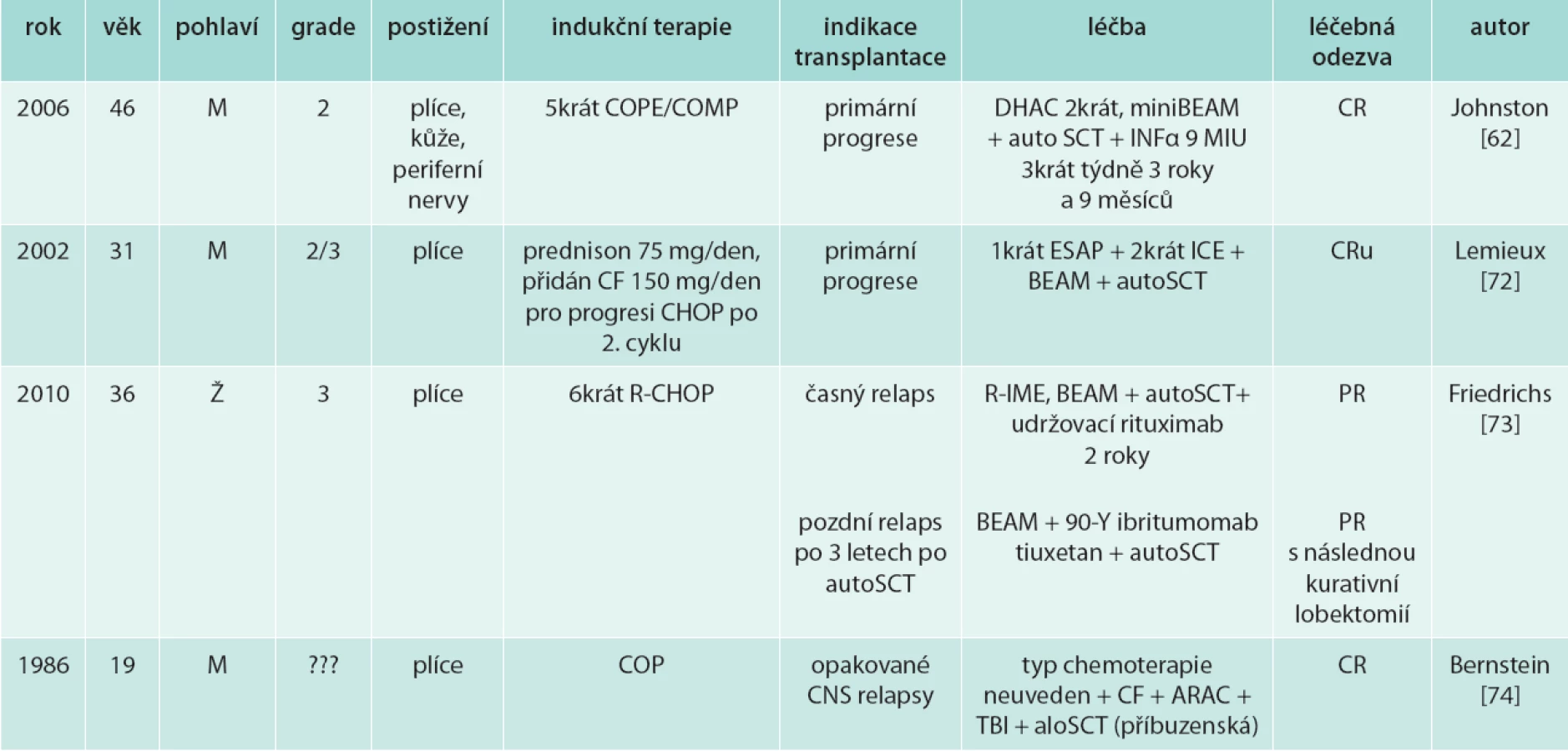
Pro velmi špatnou prognózu relabujícího onemocnění je nutné hledat další nové léčebné přístupy.
Kontroverzním přístupem je antivirová terapie ganciklovirem. Léčebnou remisi po 2 týdnech terapie ganciklovirem prezentoval Fassas et al u nemocného s LyG objevivší se po autologní transplantaci pro mnohočetný myelom [75]. Jordan et al popisují neúčinné podávání antivirové terapie u asymptomatické nemocné s LyG grade 2 [36].
Shrnutí léčby
Low grade LyG (grade 1 a 2)
Tam, kde je rozsah onemocnění malý, je možno přistoupit na strategii W+W. Všeobecně je doporučována imunomodulační terapie INFα nebo rituximabem pro polyklonalitu či oligoklonalitu procesu. Léčebně se v indukční terapii užívá INFα či rituximab s nebo bez kortikoidů. Touto léčbou bylo dosaženo dlouhodobých remisí onemocnění [14,28]. Léčba rituximabem je oproti INFα lépe tolerována, a proto by měla být použita jako lék 1. volby. U relapsu, progrese nebo transformace je indikována terapie jako u LyG 3, individuálně lze zvážit autologní transplantaci, neboť v současné době není léčba pro tyto nemocné standardizována. Pro prodloužení remise je vhodné podávat udržovací terapii – interferon nebo rituximab.
V National Cancer Institut (USA, Wilson WH) pokračuje od roku 1994 stále nábor pacientů do studie s léčbou INFα u grade 1 a 2, který je podáván 1 rok od dosažení kompletní remise. Cílem je zhodnotit léčebnou odezvu a dlouhodobý efekt terapie INFα [76].
High grade LyG (grade 3 a některé případy grade 2)
Přítomností klonality CD20 pozitivních buněk, imunitní nezávislosti a obrazu již velkobuněčného B lymfomu je indikována imunochemoterapie ve shodě s agresivním velkobuněčným B lymfomem [55]: podání rituximabu (R) s antracyklinovým režimem R-CHOP, event. (dose adjusted – DA) R-EPOCH [77]. U relapsu či progrese je indikována vysokodávkovaná léčba s autologní transplantací. Alogenní transplantace je experimentálním přístupem. V National Cancer Institut (USA, Wilson WH) pokračuje od roku 1994 nábor pacientů s LyG grade 3 nebo progredující LyG grade 1 a 2 po INFα do studie s léčbou režimem (DA) R-EPOCH (rituximab 375 mg/m2/den i.v. D1, etopozid 50 mg/m2/den kontinuálně i.v. D1–4, doxorubicin 10 mg/m2/den kontinuálně i.v. D1–4, vinkristin 0,4 mg/m2/den kontinuálně i.v. D1–4, cyklofosfamid 750 mg/m2/den i.v. na 1 hod i.v. D5, prednison 60 mg/m2/den 2krát denně p.o. D1–5). Dávky některých léků se mohou navyšovat dle tolerance v předchozím cyklu (DA). Po 2 cyklech se hodnotí léčebná odezva, minimálně se podává 6 cyklů [76]. Cílem je určit léčebnou odezvu a dlouhodobý efekt tohoto léčebného režimu u nemocných s LyG grade 3 a u nemocných se selháním léčby INFα.
Závěr
LyG patří k primárním plicním B lymfoproliferacím asociovaným s EBV infekcí. V posledních letech došlo k velkému posunu v chápání biologického chování této raritní nemoci, i když etiologie stále není plně objasněna. LyG může napodobit řadu jiných plicních onemocnění, nemá klinická ani radiologická specifika, a proto je diagnostika onemocnění komplexním a často i protrahovaným procesem. Lymfomatoidní granulomatóza představuje v závislosti na histologickém grade prognosticky velmi variabilní onemocnění s relativně krátkým celkovým přežitím. Současná léčba (monoklonální protilátky, polychemoterapie, INFα) jistě zlepšily prognózu onemocnění, ale výsledek léčby je stále pro velkou část pacientů nedostatečný a hledají se nové léčebné přístupy. Velmi slibně se jeví využití stimulace celulární imunity. Infuze kultivovaných EBV specifických cytotoxických T-lymfocytů získaných od HLA kompatibilních dárců je velmi nákladnou a pracnou terapií, ale pokud dojde ke změně podoby této léčby, mohla by být využitelná v rutinní praxi [77,78]. Další zdokonalení v porozumění biologického chování LyG, zvláště v určení přesné role EBV infekce v patogenezi, slibuje optimalizaci léčebné strategie u tohoto onemocnění.
V Čechách ani na Slovensku (dle databáze Bibliographia medica Čechoslovaca) nebyla zaznamenána žádná publikace popisující zkušenost s onemocněním typu lymfomatoidní granulomatózy. Dle registru Kooperativní lymfomové skupiny České republiky, do nějž se systematicky zadávají data dospělých pacientů s lymfoproliferacemi v České republice od roku 2000, jsou evidováni 2 pacienti s LyG. Dle konzultace s patology se však zdá, že případů bude v ČR jistě více. Protože LyG bývá často spjata s poruchou imunity, jsou zaznamenána též kazuistická sdělení z řad pediatrických pacientů, kteří mají např. prokázaný vrozený imunodeficitní stav.
Ráda bych poděkovala všem spoluautorům za podnětné připomínky, rady a formální opravy textu. Dále děkuji Radiologickému oddělení VFN Praha a FN Hradec Králové za zapůjčení a možnost prezentace radiografické obrazové dokumentace. Histologické obrázky byly pořízeny ve Fingerlandově ústavu LF UK a FN Hradec Králové. Za jejich vytvoření a možnost použití děkuji MUDr. P. Kašparové, Ph.D., MUDr. M. Nové a MUDr. K. Kamarádové.
Práce byla vypracována za podpory MZ ČR – RVO (FNHK, 00179906).
MUDr. Alice Sýkorová, Ph.D.
ali.sykorova@gmail.com
IV. interní hematologická klinika LF UK a FN Hradec Králové
www.fnhk.cz
Doručeno do redakce dne 6. 11. 2013
Přijato po recenzi dne 17. 12. 2013
Sources
1. Pittaluga S, Wilson WH, Jaffe ES. Lymphomatoid granulomatosis. In: Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon 2008 : 247–249. ISBN 139789283224310.
2. Hare SS, Souza CA, Bain G et al. The radiological spectrum of pulmonary lymphoproliferative disease. Br J Radiol 2012; 85 (1015): 848–864.
3. Liebow AA, Carrington CR, Friedman PJ. Lymphomatoid granulomatosis. Hum Pathol 1972; 3(4): 457–558.
4. Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer 1979; 43(1): 360–373.
5. Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol 2010; 34: e35-e48. Dostupné z DOI: <http://doi: 10.1097/PAS.0b013e3181fd8781>.
6. Colby TV. Current histological diagnosis of lymphomatoid granulomatosis. Mod Pathol 2012; 25 (Suppl 1): S39-S42.
7. Lipford EH, Margolick JB, Longo DL et al. Angiocentric immunoproliferative lesions: a clinicopathologic spectrum of post-thymic T-cell proliferations. Blood 1988; 72(5): 1674–1681.
8. Nichols PW, Koss M, Levine AM et al. Lymphomatoid granulomatosis: a T-cell disorder? Am J Med 1982; 72(3): 467–471.
9. Travis WD, Colby TV, Corrin B et al. Histological Typing of Lung and Pleural Tumors. 3rd ed. Springer: Berlin/New York 1999. ISBN-13 : 978–3540652199.
10. Guinee D, Jaffe ES, Kingma D et al. Pulmonary lymphamatoid granulomatosis: evidence for a proliferation of Epstein-Barr virus infected B lymphocytes with a prominent T-cell components and vasculitis. Am J Sur Pathol 1994; 18(8): 753–764.
11. Myers JL, Kurtin PL, Katzenstein AL et al. Lymphomatoid granulomatosis: Evidence of immunophenotypic diversity and relationship to Epstein-Barr virus. Am J Surg Pathol 1995; 19(11): 1300–1312.
12. Jaffe ES, Wilson WH. Lymphomatoid granulomatosis. In: Jaffe ES, Harris NL, Stein H et al. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon 2001 : 185–187.ISBN 92 83 22411 6..
13. Katzenstein AL, Peiper S. Detection of Epstein-Barr virus genomes in lymphomatoid granulomatosis: analysis of 29 cases using the polymerase chain reaction technique. Mod Pathol 1990; 3(4): 435–441.
14. Wilson WH, Kingma DW, Raffeld M et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood 1996; 87(11): 4531–4537.
15. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964; 1(7335):702–703.
16. Williams H, Crawford DH. Epstein-Barr virus: the impact of scientific advances on clinical practice. Blood 2006; 107(3): 862–869.
17. Young LS, Murray PG. Epstein-Barr virus and oncogenesis: from latent genes to tumours. Oncogene 2003; 22(33): 5108–5121.
18. Saha A, Robertson ES. Epstein-Barr virus-associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res 2011; 17(10): 3056–3063.
19. Heslop HE. How I treat EBV lymphoproliferation. Blood 2009; 114(19): 4002–4008.
20. Dunleavy K, Roschewski M, Wilson WH. Lymphomatoid granulomatosis and other Epstein-Barr virus associated lymphoproliferative processes. Curr Hematol Malig Rep 2012; 7(3): 208–215.
21. LeSueur BW, Ellsworth L, Bangert JL et al. Lymphomatoid granulomatosis in a 4-year-old boy. Pediatr Dermatol 2000; 17(5): 369–372.
22. Saxena A, Dyker KM, Angel S at al. Posttransplant diffuse large B cell lymphoma of “lymphomatoid granulomatosis” type. Virchows Arch 2002; 441(6): 622–628.
23. Beaty MW, Toro J, Sorbara L et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol 2001; 25(9): 1111–1120.
24. Hořejší V, Bartůňková J. Základy imunologie. Triton: Praha 2009. 4th ed: 247–262. ISBN 978–80–7387–280–9.
25. Tran H, Nourse J, Hall S et al. Immunodeficiency-associated lymphomas. Blood Rev 2008; 22(5): 261–281.
26. Dunleavy K, Chattopadhyah P, Kawada J et al. Immune characteristics associated with lymphomatoid granulomatosis and outcome following treatment with interferon-alpha. Blood (ASH Annual Meeting Abstracts) 2010; 116 : 963.
27. Karnak I, Ciftci AO, Talim B et al. Pulmonary lymphomatoid granulomatosis in a 4 year old. J Pediatr Surg 1999; 34(6): 1033–1035.
28. Hernández-Marqués C, Lassaletta A, Torrelo A et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol 2014; 36(2). Dostupné z DOI: <http://doi: 10.1097/MPH.0b013e31827e63a6>.
29. Araki F, Mimura T, Fukuoka S et al. Primary orbital lymphomatoid granulomatosis. Br J Ophthalmol 2009; 93(4): 554–556.
30. Bailie J, McNaughten B, Gray S et al. Unique presentation of testicular involvement in lymphomatoid granulomatosis. Onkologie 2012; 35(6): 372–375.
31. Cameron JR, Cackett P. Lymphomatoid granulomatosis associated with bilateral exudative retinal detachments. Arch Ophthalmol 2007; 125(5): 712–713.
32. Dister F, Ghaye B. Lymphomatoid granulomatosis. JBR-BTR 2012; 95(3): 140–141.
33. James WD, Odom RB, Katzenstein AL. Cutaneous manifestations of lymphomatoid granulomatosis. Report of 44 cases and a review of the literature. Arch Dermatol 1981; 117(4): 196–202.
34. Zaidi A, Kampalath B, Peltier WL et al. Successful treatment of systemic and central nervous system lymphomatoid granulomatosis with rituximab. Leuk Lymphoma 2004; 45(4): 777–780.
35. Roschewski M, Wilson WH. Lymphomatoid granulomatosis. Cancer J 2012; 18(5): 469–474.
36. Jordan K, Grothey A, Grothe W et al. Successful treatment of mediastinal lymphomatoid granulomatosis with rituximab monotherapy. Eur J Haematol 2005; 74(3): 263–266.
37. Cadranel J, Wislez M, Antoine M. Primary pulmonary lymphoma. Eur Respir J 2002; 20(3): 750–762.
38. Chung JH, Wu CC, Gilman MD et al. Lymphomatoid Granulomatosis: CT and FDG-PET findings. Korean J Radiol 2011; 12(6): 671–678.
39. Webb WR, Muller NL, Naidich DP. High-resolution CT of the lung. Lippincott Williams and Wilkins: Philadelphia 2009. 4th ed. ISBN 978078176909.
40. Roarke MC, Nguyen BD. PET/CT characterization and monitoring of disease activity in lymphomatoid granulomatosis. Clin Nucl Med 2007; 32(3): 258–259.
41. Arai H, Oshiro H, Yamanaka S et al. Grade I lymphomatoid granulomatosis with increased uptake of [18F] fluorodeoxyglucose in positron emission tomography: a case report. J Clin Exp Hematop 2009; 49(1): 39–44.
42. Suzuki H, Takeda H, Kishi H et al. Reactivation of Epstein – Barr virus is involved in the pathogenesis of lymphomatoid granulomatosis. Nihon Kokyuki Gakkai Zasshi 2006; 44(7): 492–498.
43. Jung KH, Sung HJ, Lee JH et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy 2009; 55(5): 386–390.
44. Kawai N, Miyake N, Nishiyama Y et al. FDG-findings of the brain in lymphomatoid granulomatosis. Ann Nucl Med 2006; 20(10): 683–687.
45. Pisani RJ, DeRemee RA. Clinical implications of the histopathologic diagnosis of pulmonary lymphomatoid granulomatosis. Mayo Clin Proc 1990; 65(2): 151–163.
46. Jaffe ES, Wilson WH. Lymphomatoid granulomatosis: pathogenesis, pathology and clinical implications. Cancer Surv 1997; 30 : 233–248.
47. Jaffe ES. Pulmonary lymphocytic angiitis: a nosologic quandary. Mayo Clin Proc 1988; 63(4): 411–413.
48. Tichý T, Musilová K, Škarda J. Morfologie granulomů a základní histologie vybraných plicních granulomatóz. Program konference. Dostupné z WWW: <http://www.geum.org/sarko/3_Tichy.html>.
49. Hamilton-Dutoit SJ, Raphael M, Audouin J et al. In situ demonstration of Epstein Barr virus small RNAs (EBER1) in acquired immunodeficiency syndrome-related lymphomas: correlation with tumor morphology and primary site. Blood 1993; 82(2): 619–624.
50. McNiff JM, Cooper D, Howe G et al. Lymphomatoid granulomatosis of the skin and lung. An angiocentric T-cell-rich B cell lymphoproliferative disorder. Arch Dermatol 1996; 132(12): 1464–1470.
51. Roubalová K. Laboratorní diagnostika herpetických virů. Med Pro Praxi 2010; 7(5): 241–244.
52. Jaffre S, Jardin F, Dominique S et al. Fatal haemoptysis in a case of lymphomatoid granulomatosis treated with rituximab. Eur Respir J 2006; 27(3): 644–646.
53. Guinee DG Jr, Perkins SL, Travis WD et al. Proliferation and cellular phenotype in lymphomatoid granulomatosis: implications of a higher proliferation index in B cells. Am J Surg Pathol 1998; 22(9): 1093–1100.
54. Fauci AS, Haynes BF, Costa J et al. Lymphomatoid granulomatosis. Prospective clinical and therapeutic experience over 10 years. N Engl J Med 1982; 306(2): 68–74.
55. Armitage JO. My treatment approach to patients with diffuse large B-cell lymphoma. Mayo Clin Proc 2012; 87(2): 161–171.
56. Medeiros LJ, Peiper SC, Elwood L et al. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Human Pathology 1991; 22(11): 1150–1157.
57. Takeishi G, Moroki K, Kawasoe T et al. Spontaneous regression and regrowth of central nervous system lymphomatoid granulomatosis: case report. Neurol Med Chir (Tokyo) 2011; 51(11): 801–804.
58. Drasga RE, Williams SD, Wills ER et al. Lymphomatoid granulomatosis. Successful treatment with CHOP combination chemotherapy. Am J Clin Oncol 1984; 7(1): 75–80.
59. Klener P (ed). Biomodulační léčba (imunoterapie). In: Klener P, Abrahámová J, Fait V et al. Klinická onkologie. Galén: Praha 2002 : 223–230. ISBN 8072621513.
60. Shapiro RS, Chauvenet A, McGuire W et al. Treatment of B-cell lymphoproliferative disorders with interferon alfa and intravenous gamma globulin. N Engl J Med 1988; 318(20): 1334.
61. Dunleavy K, Chattopadhyay P, Kawada J et al. Immune characteristics associated with lymphomatoid granulomatosis and outcome following treatment with interferon-alpha. Blood 2010; 116(424): Abstrakt 963.
62. Johnston A, Coyle L, Nevell D. Prolonged remission of refractory lymphomatoid granulomatosis after autologous hemopoietic stem cell transplantation with post-transplantation maintenance interferon. Leuk Lymphoma 2006; 47 : 323–328.
63. Shanti RM, Torres-Cabala CA, Jaffe ES et al. Lymphomatoid granulomatosis with involvement of the hard palate: a case report. J Oral Maxillofac Surg 2008; 66(10): 2161–2163.
64. Oosting-Lenstra SF, van Marwijk KM. Failure of CHOP with rituximab for lymphomatoid granulomatosis. Neth J Med 2007; 65(11): 442–447.
65. Boye J, Elter T, Engert A. An overview of the current clinical use of the anti CD-20 monoclonal antibody rituximab. Ann Oncol 2003; 14(4): 520–535.
66. Mohyuddin GR, Sultan F, Khaleeq G. A rare presentation of a rare disease: pulmonary lymphomatoid granulomatosis. Case Rep Pulmonol 2012; 2012 : 371490. Dostupné z DOI: <http://doi: 10.1155/2012/371490>.
67. Castrale C, El Haggan W, Chapon F et al. Lymphomatoid granulomatosis treated successfully with rituximab in a renal transplant patient. J Transplant 2011; 2011 : 865957. Dostupné z DOI: <http://doi: 10.1155/2011/865957>.
68. Polizzotto MN, Dawson MA, Opat SS. Failure of rituximab monotherapy in lymphomatoid granulomatosis. Eur J Haematol 2005; 75(2): 172–173.
69. Ishiura H, Morikawa M, Hamada M et al. Lymphomatoid Granulomatosis Involving Central Nervous System Successfully treated With Rituximab alone. Arch Neurol 2008; 65(5): 662–665.
70. Nair BD, Joseph MG, Catton GE et al. Radiation therapy in lymphomatoid granulomatosis. Cancer 1989; 64(4): 821–824.
71. Petrella TM, Walker IR, Jones GW et al. Radiotherapy to control CNS lymphomatoid granulomatosis: a case report and review of literature. Am J Hematol 1999; 62(4): 239–241.
72. Lemieux J, Bernier V, Martel N et al. Autologous hematopoietic stem cell transplantation for refractory lymphomatoid granulomatosis. Hematology 2002; 7(6): 355–358.
73. Friedrichs B, Thiel E. Relapsed pulmonary lymphomatoid granulomatosis grade III: curative treatment with radioimmune therapy and stem cell transplantation. Dtsch Med Wochenschr 2010; 35(38): 1857–1860.
74. Bernstein ML, Reece ER, de Chadarévian JP et al. Bone marrow transplantation in lymphomatoid granulomatosis. Report of a case. Cancer 1986; 58(4): 969–972.
75. Fassas A, Jagannath S, Desikan KR et al. Lymphomatoid granulomatosis following autologous stem cell transplantation. Bone Marrow Transplant 1999; 23(1): 79–81.
76. Wilson WH, Grossbard ML, Pittaluga S et al. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood 2002; 99(8): 2685–2693.
77. Macsween KF, Crawford DH. Epstein-Barr virus-recent advances. The Lancet Infectious Diseases 2003; 3(3): 131–140.
78. Bollard CM, Gottschalk S, Leen AM et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood 2007; 110(8): 2838–2845.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 3
Most read in this issue
- Crush syndróm súčasnosti – rabdomyolýza intoxikovaných
- Bolest u chronické pankreatitidy a karcinomu pankreatu – možnosti léčby
- Lymfomatoidní granulomatóza – minulost a současnost
- Patogenéza zmien vnútorného prostredia pri akútnom svalovom kompartment syndróme