
Sekundární humorální imunodeficience u nemocných se systémovým lupus erythematodes
Secondary humoral immunodeficiency in patiens with systemic lupus erythematosus
Introduction:
Systemic lupus erythematosus (SLE) is a chronic autoimmune multisystem disease. The aim of our study was to clarify the frequency of decreased serum immunoglobulin levels in SLE patients. There were evaluated 799 results of serum immunoglobulin levels gained from 157 patients fulfilling revised ACR criteria in the retrospective study.
Results:
The immunoglobulin levels under the normal range were found in 29/157 (18.5 %) patients. The most frequent was isolated reduction of IgG 12/157 (7.6 %), two persons fulfilled criteria for selective IgA deficiency, and one case possible diagnosis of common variable immunodeficiency (CVID). Additionally we report two cases of SLE patients complicated by severe hypogammaglobulinaemia and infectious complications with necessity of long-term immunoglobulin substitution therapy. The diagnosis of CVID is highly probable in the first case. The second case presents sever drug-induced hypogammaglobulinaemia. This female with lymphoma history and multiorgan impairment due to acute SLE was treated with rituximab after convention therapy failure.
Conclusion:
Humoral immunodeficiency may occur in SLE patients. The monitoring of serum immunoglobulin levels could be a routine in these patients. The CVID diagnosis is possible in patients suffering from recurrent sinopulmonary infections, especially in combination with absence of lupus activity. Rituximab therapy could cause long-term suppression of B lymphocytes with secondary humoral deficiency requiring immunoglobulin substitution therapy.
Key words:
CVID – humoral immunodeficiency – lupus – rituximab – substitution therapy
Authors:
Pavlína Králíčková 1; Eva Malá 1; Doris Vokurková 1; Ondřej Souček 1; Irena Krčmová 1; Zbyněk Hrnčíř 2
Authors‘ workplace:
Ústav klinické imunologie a alergologie LF UK a FN Hradec Králové, přednosta prof. RNDr. Jan Krejsek, CSc.
1; II. interní gastroenterologická klinika LF UK a FN Hradec Králové, přednostka prof. MUDr. Marcela Kopáčová, Ph. D.
2
Published in:
Vnitř Lék 2015; 61(9): 778-794
Category:
Original Contributions
Overview
Úvod:
Systémový lupus erythematodes (SLE) je chronické autoimunitní onemocnění charakterizované multisystémovým postižením. Cílem naší studie bylo ozřejmit výskyt snížených hodnot koncentrací imunoglobulinů (s-Ig) v séru v populaci nemocných se SLE. V retrospektivní studii bylo zhodnoceno 799 výsledků vyšetření s-Ig získaných od 157 nemocných splňujících revidovaná klasifikační kritéria.
Výsledky:
Sérová koncentrace imunoglobulinů nižší než dolní hranice normy byla zjištěna celkem u 29 ze 157 nemocných (18,5 %). Nejčetnější byl výskyt izolovaného snížení IgG 12 ze 157 (7,6 %), 2 nemocné naplňovaly kritéria selektivního deficitu IgA a v jednom případě se jednalo o vzácnou koincidenci s běžnou variabilní imunodeficiencí (common variable immunodeficiency – CVID). Článek podrobněji prezentuje 2 případy nemocných se SLE komplikované těžkou hypogamaglobulinemií a infekcemi vyžadujícími dlouhodobou substituční léčbu imunoglobuliny. V prvním případě se jedná o vysoce pravděpodobný případ běžné variabilní imunodeficience a v případě druhém pak o léky indukovanou hypogamaglobulinemii. Tato žena s anamnézou maligního lymfomu a současným závažným multiorgánovým postižením při SLE byla léčena rituximabem po selhání konvenční léčby.
Závěr:
S protilátkovými deficiencemi se můžeme setkat i u nemocných se SLE. Opakované měření sérové koncentrace imunoglobulinů by mělo být součástí rutinního sledování těchto nemocných. Na možnost CVID bychom měli myslet v případě opakujících se sinopulmonárních infekcí, zejména v kombinaci s absencí aktivity SLE. Léčba rituximabem může vést i u nemocných se SLE k dlouhodobé supresi B-lymfocytů s rozvojem sekundárního protilátkového deficitu, který v některých případech vyžaduje substituční léčbu imunoglobuliny.
Klíčová slova:
CVID – lupus – protilátkový imunodeficit – rituximab – substituční léčba
Úvod
Systémový lupus erythematodes (SLE ) je chronické autoimunitní onemocnění charakterizované multisystémovým postižením, proměnlivým průběhem i prognózou. Udávaná prevalence SLE v populaci se pohybuje mezi 20–150 případy na 100 000 obyvatel [1–4], s vyšší prevalencí u afroameričanů až 406 na 100 000 obyvatel [2]. Odhadované rozmezí incidence je udáváno mezi 1–25 na 100 000 v Severní a Jižní Americe, Evropě i Asii [3,5,6].
Studium asociace mezi SLE a primárními imunodeficity přispívá k dalšímu porozumění patofyziologii této závažné choroby. Zatímco vztah SLE anebo lupus-like manifestace byl poměrně dobře popsán v případě vrozených poruch komplementu a u žen přenašeček chronické granulomatózní choroby, o vztahu k primárním protilátkovým deficitům, zejména běžné variabilní imunodeficienci, chybí v literatuře dostatek údajů [7].
Cílem naší studie bylo tedy ozřejmit výskyt snížených koncentrací hlavních tříd imunoglobulinů v séru (s-Ig) v populaci nemocných se SLE.
Soubor a metodika
V retrospektivní studii, schválené etickou komisí FN v Hradci Králové, bylo zhodnoceno celkem 799 výsledků vyšetření s-Ig od 157 nemocných splňujících revidovaná klasifikační kritéria ACR z roku 1997 a SLICC/2012 s přihlédnutím k doporučení České revmatologické společnosti [8]: rekrutovaná kohorta zahrnovala i překryvné syndromy SLE – revmatoidní artritida a SLE – systémová sklerodermie.
Z laboratorní databáze Ústavu klinické imunologie a alergologie byly vybrány výsledky vyšetření s-Ig zaslaných pod diagnózou SLE z II. interní kliniky, oddělení revmatologie, FN v Hradci Králové za období 1. 1. 2007–31. 12. 2013. Správnost diagnózy pak byla ověřena z dostupné dokumentace. Jako abnormální byly považovány hodnoty více než či méně než 2 směrodatné odchylky od průměrné hodnoty běžné populace, tzn. mimo rozmezí IgG 7,3–19,5 g/l, IgA 0,8–4,8 g/l, IgM 0,4–3 g/l. Jako primární humorální imunodeficience byly označeny ty případy, které naplňovaly diagnostická kritéria pro selektivní deficit IgA (SDIgA) či běžnou variabilní imunodeficienci (common variable immunodeficiency – CVID) dle PAGID/ESID [9]. Data byla následně zpracována pomocí statistického programu Excel 2010.
Výsledky
Celkem bylo zhodnoceno 799 výsledků vyšetření sérových koncentrací hlavních tříd imunoglobulinů získaných od 157 nemocných (142 žen, 15 mužů), věkové rozložení ke dni zhodnocení 47,5 ± 15,6 (16–85), aritmetický průměr ± směrodatná odchylka (rozmezí).
Jak ukazuje graf, snížené hodnoty s-Ig byly zjištěny celkem u 29 ze 157 nemocných (18,5 %). Nejčetněji se jednalo o izolované snížení v třídě IgG 12 z 29, v třídě IgA 8 z 29 a IgM 4 z 29. Snížení ve 2 třídách současně (IgG a IgA) bylo zaznamenáno jedenkrát. Snížení ve všech 3 třídách imunoglobulinů pak u 4 z 22 nemocných. Dva pacientské případy jsou prezentovány níže. Ve 3. případě se jednalo o symptomy provázející bílkovinné ztráty při lupusové nefropatii těžkého stupně. Po nasazení imunosupresivní léčby pak u této nemocné došlo k postupné normalizaci hodnot s-Ig. Sekundární příčiny hypogamaglobulinemie převažovaly nad primárními. Pouze 2 pacienti ze 157 (1,27 %) naplnili diagnostická kritéria pro selektivní deficit IgA a 1 ze 157 (0,6 %) pro běžnou variabilní imunodeficienci. Sekundární příčiny hypogamaglobulinemie byly tvořeny především ztrátami při nefropatii či toxicky, polékově. K řadě případů se nebylo možno k etiologii jednoznačně vyjádřit z důvodů nedostupnosti laboratorního vyšetření před vzplanutím základního onemocnění.

Pacient 1 – těžká protilátková imunodeficience charakteru běžné variabilní imunodeficience
Žena (*1971) byla doporučena revmatologem na imunologické pracoviště pro těžkou hypogamaglobulinemii provázenou recidivujícími sinusitidami a bronchitidami.
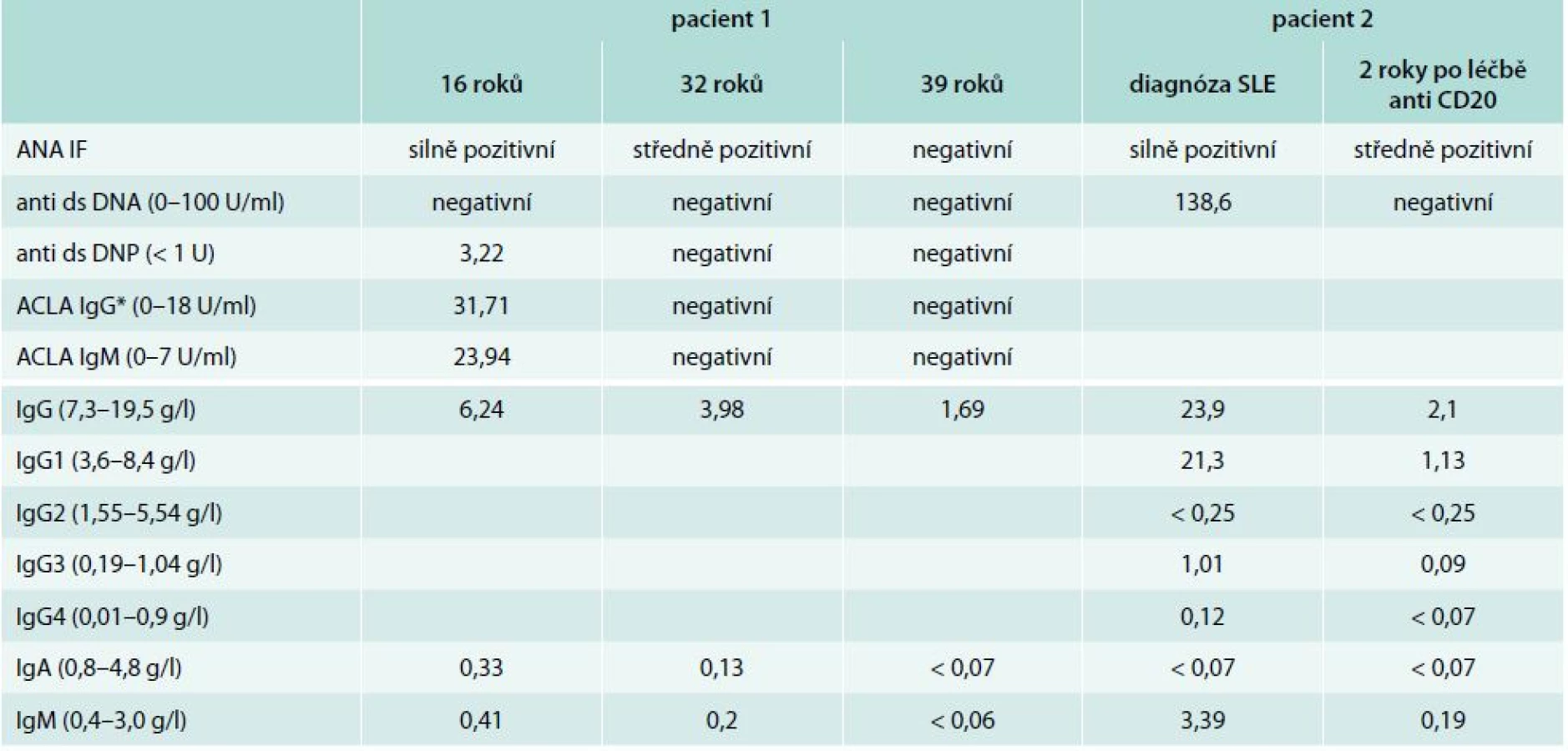
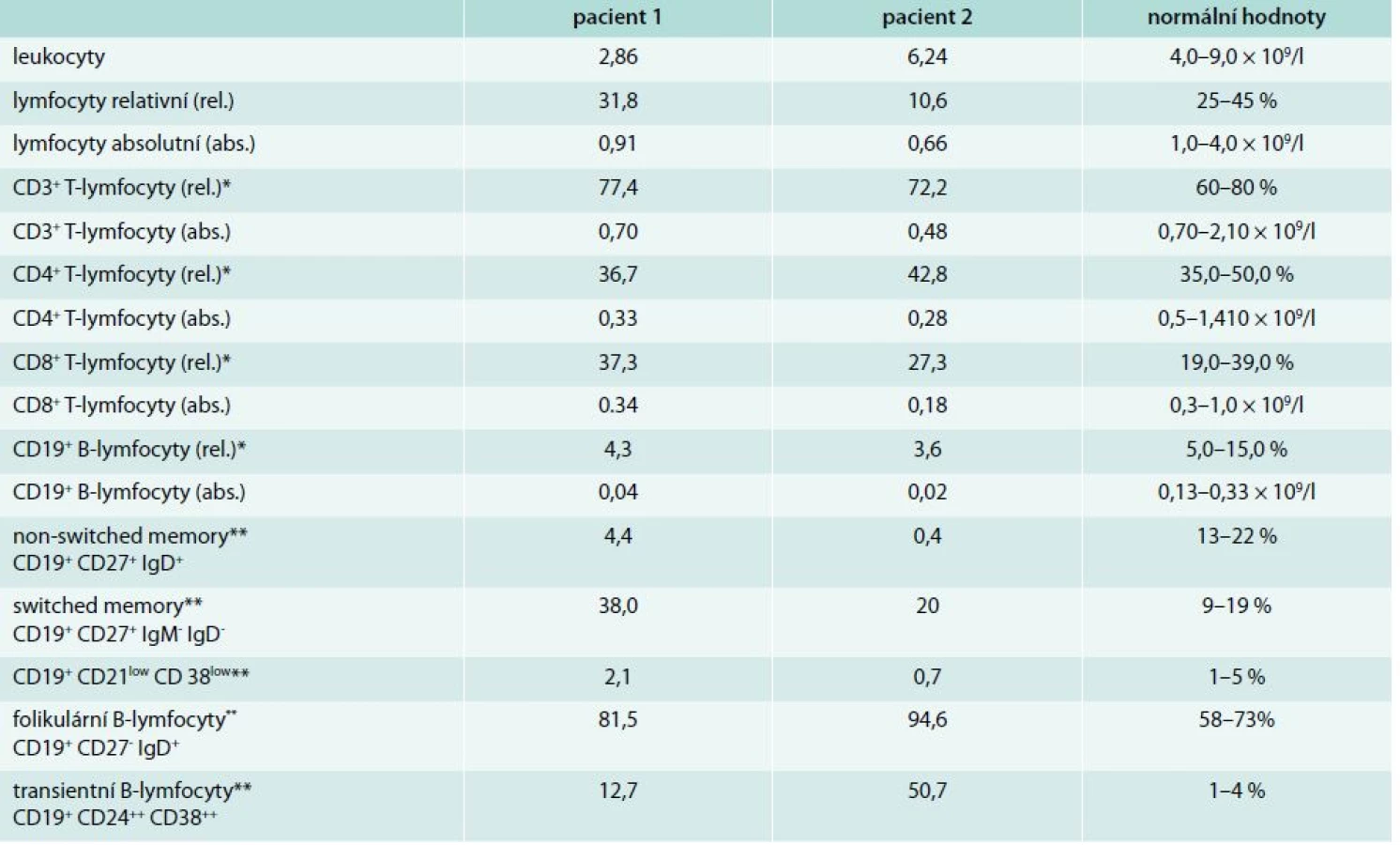
Do 8 let věku byla zcela zdráva, v návaznosti na prodělanou angínu u ní propukla artritida drobných kloubů ruky, exantém na trupu a neurologické obtíže charakteru chorea minor. Stav byl v té době uzavřen jako revmatická horečka. V 16 letech náhle prodělala cévní mozkovou příhodu s tranzitorní pravostrannou hemiparézou. Magnetická rezonance prokázala ischemické postižení v oblasti temporálního laloku. Laboratorně byla zjištěna pozitivita antinukleárních protilátek ANA-IF +++ a zvýšení protilátek proti deoxyribonukleoproteinu anti ds-DNP 3,22 U (norma: < 1) a antikardiolipinových protilátek IgG 23,94 U/ml (0–10), IgM 31,71 U/ml (0–7). Na základě anamnézy a laboratorních a zobrazovacích vyšetření byla stanovena diagnóza SLE a neprodleně zahájena imunosupresivní léčba systémovými kortikoidy s dobrým klinickým efektem. V 26 letech byla poprvé odebrána sérová koncentrace imunoglobulinů: IgG 6,24 g/l (7,5–10,5), IgA 0,33 g/l (0,8–4,8), IgM 0,41 g/l (0,4–3,0). V 29 letech došlo k exacerbaci aktivity SLE ve formě centrální parézy n. facialis. MRI prokázalo významný nárůst ischemických ložisek v bílé mozkové hmotě a krátkou stenózu na a. cerebri anterior a a. cerebri media. Neprodleně zahájená léčba pulzy cyklofosfamidu (celkem 12 pulzů à 800 mg měsíčně) vedla k remisi onemocnění s plnou úpravou neurologického nálezu. Dlouhodobá imunosupresivní léčba byla rozšířena o hydroxychloroquin 200 mg/den. V 32 letech pacientka otěhotněla a předčasně porodila v 27. týdnu těhotenství pro eklampsii a hypotrofii plodu. Od té doby se aktivita SLE snižovala a bylo možné postupně snižovat dávku metylprednisolonu na 4 mg denně. Antikardiolipinové protilátky, anti dsDNP a ANA se znormalizovaly. Paradoxně však byl zaznamenán další pokles s-Ig (IgG 3,98 g/l, IgA 0,13 g/l, IgM 0,20 g/l). Od 37 let si začala stěžovat na zvýšenou nemocnost. Jednalo se o recidivující sinusitidy a bronchitidy, převážně bakteriálního původu. Vyšetření imunologem prokázalo těžkou hypogamaglobulinemii ve všech hlavních třídách – IgG 1,68 g/l, IgA < 0,07 g/l, IgM < 0,06 g/l spojenou s poruchou tvorby specifických protilátek – nízká hodnota specifických protilátek proti polysacharidovému pneumokokovému antigenu 9,14 mg/ml (20–200) i antitetanovému toxoidu 0,07 IU/ml (> 0,5 IU/ml), bez odezvy na očkování vakcínami TETAVAX a PNEUMO 23, obě vyrobeny Sanofi Pasteur, Lyon, Francie. Tab. 1 zobrazuje vývoj hladiny sérových imunoglobulinů a autoprotilátek v čase. Vyšetřením buněčné imunity bylo zjištěno snížení absolutního počtu B-lymfocytů v periferní krvi 0,03 × 109/l (0,13–0,33), snížení absolutního počtu CD4+ T-lymfocytů 0,29 × 109/l (0,5–1,4) a současně velmi nízký počet paměťových B-lymfocytů (tab. 2). Byly vyloučeny ztráty imunoglobulinů ledvinami. Zobrazovací vyšetření neprokázala žádnou další možnou příčinu hypogamaglobulinemie. Poléková etiologie se jevila s ohledem na klinický a laboratorní průběh onemocnění jako málo pravděpodobná, a proto jsme se přiklonili k názoru, že došlo k rozvoji běžné variabilní imunodeficience. Po zahájení pravidelné substituční léčby imunoglobuliny v dávce 250 mg/kg/měsíc podkožně došlo k normalizaci sérové koncentrace IgG (7,4 g/l) a současně klinicky k významnému poklesu nemocnosti se spotřebou antibiotik 1–2krát ročně pro infekci horních cest dýchacích. Aktivita SLE je i nadále zcela uspokojivá.
Pacient 2 – těžká protilátková imunodeficience ze sekundárních příčin
26letá žena s negativní rodinnou anamnézou nádorových a autoimunitních onemocnění byla vyšetřena ultrasonograficky před plánovanou miniinterrupcí. Byla zjištěna masivní břišní lymfadenopatie. Další vyšetření diagnostikovala folikulární B lymfom stadia IIIA, IPI skóre 1 (International Prognostic Index). Podaná chemoterapie R-CHOP (rituximab, cyklofosfamid, adriamycin, vinkristin, prednison) v kombinaci s radioterapií retroperitonea (LD 30,6 Gy) vedla ke kompletní remisi. Po zhodnocení všech rizik bylo pokračováno udržovací léčbou interferonem α v dávce 3 MU 3krát týdně. Po 8 letech léčby začalo docházet k postupnému nárůstu počtu infekcí dýchacích cest s nutností opakované antibiotické léčby. Následně se přidaly i subfebrilie. Po několika měsících se stav akutně zhoršil a nemocná byla přijata na interní oddělení pod obrazem akutní pleuritidy. I přes nitrožilně podávanou antibiotickou léčbu stav rychle progredoval. Došlo k rozvoji oboustranného srdečního selhávání v důsledku perimyokarditidy. Stav byl komplikován akutní renální insuficiencí s nutností dialyzační léčby a pseudomembranózní kolitidou, pravděpodobně v souvislosti s antibiotickou léčbou. Laboratorní vyšetření prokázala pozitivitu ANA-IF +++ a protilátek proti dvouvláknové DNA (anti ds DNA) 138,6 U/ml (0–100), proti extrahovatelným nukleárním antigenům ENA imunoblotem – protilátky anti Sjoegrenův syndrom A (SS/A) Ro +, proti směsi jaderných ribonukleoproteinů anti Sm +, a U1-RNP +, pozitivita Coombsova testu. Biopsie ledvin prokázala akutní tubulární nekrózu v kombinaci s mezangiální proliferativní lupusovou nefritidou, třídy II. Endomyokardiální biopsie nebyla výtěžná. Byla stanovena diagnóza systémového lupus erythematodes a neprodleně zahájena pulzní léčba kortikoidy (metylprednisolon 3krát 1 g) komplikovaná vznikem akutní pankreatitidy. Od dalších pulzů kortikoidů bylo upuštěno a byl nasazen rituximab (375 mg/kg, 4 cykly). Klinický stav nemocné se rychle upravoval. Bylo dosaženo remise onemocnění. Po čase byla propuštěna do domácího ošetřování kardiopulmonálně kompenzovaná bez nutnosti dialyzační léčby. Jako dlouhodobá imunosupresivní léčba byl zvolen metylprednisolon v dávce 16 mg denně.
Po následující 2 roky byla pacientka v dobrém klinickém stavu, bez zvýšené frekvence infekčních onemocnění, bez aktivity SLE. Náhle však byla akutně přijata na infekční kliniku pro těžkou gastroenteritidu způsobenou Campylobacter jejuni komplikovanou oběhově významnou dehydratací. Rehydratace a antibiotická léčba vedly k úzdravě.
Imunologem byla nemocná vyšetřena v té době podruhé. Poprvé v době diagnózy SLE (ještě před zahájením imunosupresivní léčby) a poté těsně před vznikem gastroenteritidy (tab. 1). Zatímco první vyšetření prokázalo deficit IgA kombinovaný s deficitem podtřídy IgG2, druhé vyšetření pak těžkou hypogamaglobulinemii ve všech 3 hlavních třídách imunoglobulinů a sníženou přítomnost anamnestických specifických protilátek proti polysacharidovému pneumokokovému antigenu 8,34 mg/ml (20–200) i antitetanovému toxoidu 0,18 IU/ml (nad 0,5 IU/ml). Při vyšetření buněčné imunity dominovala významně nízká hodnota B-lymfocytů v periferní krvi (tab. 2). Doplněná vakcinace tetanickým anatoxinem potvrdila poruchu tvorby specifických protilátek. Očkování proti pneumokokům nebylo realizováno pro potenciální riziko exacerbace aktivity SLE. Po zhodnocení všech faktorů vč. anamnézy polyvalentní lékové alergie (penicilin, piperacilin/tazobactam, meronemum, ciprofloxacin, sulfometoxatol-trimetoprim) bylo rozhodnuto o zahájení substituční léčby imunoglobuliny. Nemocná je nitrožilní cestou substituována dávkou 150 mg/kg/měsíc. Klinicky je v dobrém stavu, k dalším epizodám závažnějších bakteriálních infekcí nedošlo. V současné době, 4 roky po ukončení léčby rituximabem, stále nedošlo k vzestupu absolutního počtu B-lymfocytů v periferní krvi (0,03 × 109/ml, norma 0,13–0,33), s odpovídající protilátkovou odezvou.
Diskuse
Výskyt systémového lupus etyhematodes u nemocných s primárními imunodeficity bývá nejčetněji spojován s homozygotními mutacemi v oblasti komplementu, zejména deficitu proteinů, které se podílejí na aktivaci komplementu klasickou cestou. Nejtěsnější asociace je udávána s deficitem C1q složky, při němž se až u 93 % nositelů homozygotní mutace rozvine SLE. Dále pak již četnost klesá (75 % u deficitu C4, 57 % u C1r/s, do 24 % u deficitu C2). Na druhou stranu deficit komplementu prokážeme pouze u 1 % nemocných se SLE [7]. U žen přenašeček X vázané chronické granulomatózní choroby byly popsány případy fotosenzitivity, diskoidního lupusu a ev. aftózní stomatitidy [10].
U protilátkových imunodeficiencí byla sledována frekvence výskytu u nemocných se selektivním deficitem IgA, nejčetnějším primárním humorálním imunodeficitem v populaci. Prevalence v různých studiích kolísá mezi 1–4,6 %, což odpovídá frekvenci 50krát vyšší, než tomu je v běžné populaci [11,12]. Zdá se, že vyšší frekvenci najdeme zejména u nemocných s juvenilní formou onemocnění [13]. Do našeho souboru byla zahrnuta pouze jedna mladá žena, jejíž příznaky začaly v dětství, ta však netrpěla žádnou primární humorální imunodeficiencí. Frekvence SDIgA v našem souboru dospělých nemocných činila 1,27 %. Nebyl shledán významný rozdíl v klinické manifestaci ani tíži onemocnění v populaci nemocných se SLE a současně s přítomným SDIgA či bez něj [12,13].
Běžná variabilní imunodeficience je primární protilátková imunodeficience charakterizovaná významným snížením sérové koncentrace imunoglobulinů IgG, IgA nebo IgM, které je spojeno s poruchou tvorby specifických protilátek při vyloučení možných sekundárních příčin [9]. Ačkoliv 32 % nemocných trpí infekčními komplikacemi, převážně bakteriálního původu, u zbývajících 68 % lze diagnostikovat neinfekční, zánětlivé či autoimunitní komplikace. Neinfekční zánětlivé komplikace nejčastěji postihují plicní parenchym ve formě lymfocytární intersticiální pneumonitidy s/bez tvorby granulomů, dále zažívací trakt (malabsorpce v důsledku celiakia-like postižení, nespecifické střevní záněty, nodulární lymfoidní hyperplazie), či jaterní parenchym (nodulární regenerativní hyperplazie, granulomy, chronická hepatitida, biliární cirhóza). Autoimunitní komplikace nejčastěji postihují krvetvorbu ve formě idiopatické trombocytopenické purpury, autoimunitní hemolytické anémie, ev. Evansova syndromu. Tyto komplikace mohou i o řadu let předcházet manifestaci CVID a po zahájení adekvátní substituční léčby obvykle nerecidivují. S vyšší frekvencí se vyskytuje i přítomnost protilátek proti IgA, revmatoidní artritida, alopecie, tyreopatie. Diagnostika je vždy výrazně ztížena sníženou schopností tvořit protilátky, takže diagnóza bývá stanovena pouze na základě klinického obrazu, zobrazovacích vyšetření a histologie. Tyto komplikace nejsou zásadním způsobem ovlivnitelné substituční léčbou imunoglobuliny, nezbytné bývá nasazení adekvátní imunosupresivní léčby [14,15].
SLE jako autoimunitní komplikace CVID byl popsán pouze v nečetných kazuistických sděleních, s frekvencí výskytu < 1 %. Fernandez-Castro et al porovnávají ve své práci 2 případy nemocných se SLE a současnou diagnózou CVID s dalšími 16 případy publikovanými mezi lety 1982–2005 [16]. V databázi PubMed pak od té doby byly publikovány další 4 případy [17–19]. Diagnóza SLE může předcházet diagnóze CVID, jak tomu bylo i v našem případě, či méně často druhotně, na pozadí již známé poruchy tvorby specifických protilátek [20]. Nejčastějšími symptomy předcházejícími diagnózu SLE byly sinopulmonární infekce. Aktivita SLE se se vznikem protilátkového deficitu u 2/3 nemocných snižovala. I u naší nemocné lze z tab. 1 vysledovat na jedné straně pokles jednotlivých autoprotilátek v čase s možností snižování imunosupresivní léčby, při současném postupném poklesu sérové koncentrace imunoglobulinů na straně druhé. U části nemocných však byly autoprotilátky zachyceny opakovaně i v období, kdy již byla významně snížená s-Ig. Při vyšetření buněčné imunity byl u 1/5 nemocných zjištěn snížený relativní počet CD4+ T-lymfocytů a u 40 % nemocných snížená lymfocytární aktivace po mitogenech. Většina případů byla léčena substituční léčbou imunoglobuliny [16]. Přesná patogeneze vzniku koincidence těchto dvou onemocnění, stojících zdánlivě na opačných pólech, nebyla doposud objasněna.
Zatímco zvýšená hodnota s-Ig diagnózu SLE podporuje, jejich deficit může mít řadu příčin. V některých případech je diferenciální diagnostika mezi primárními a sekundárními příčinami obtížná. Zásadním problémem bývá fakt, že nejsou anamnesticky dostupná data z období před propuknutím systémového onemocnění pojiva. Mezi nejčastější sekundární příčiny patří léky indukovaná hypogamablobulinemie a hypogamaglobulinemie v důsledku renálních ztrát při lupusové nefritidě.
Lupusová nefritida se nejčastěji manifestuje jako nefritický syndrom. Až 40 % nemocných může mít i syndrom nefrotický, který bývá spíše spojen se zvýšenými ztrátami imunoglobulinů. Ztrátami je nejvíce ovlivněna sérová koncentrace IgG. Koncentrace IgA a IgM bývají sníženy až při závažném poškození ledvin. V případě nefritidy třídy III/IV a V bylo zaznamenáno v době diagnózy snížení sérové koncentrace IgG u 26 % nemocných [21]. Na rozdíl od léky indukované hypogamaglobulinemie, imunosupresivní léčba naopak vede po čase ke snížení ztrát. Z klinické zkušenosti vyplývá, že i značné ztráty nemusí být provázeny zvýšenou nemocností [21,22]. Toto lze vysvětlit neporušenou schopností produkce protilátek při akutní infekci [23].
V léčbě SLE je používána řada léků, které mohou vést ke snížené tvorbě imunoglobulinů či negativně zasahovat do T buněčné odpovědi. Jedná se především o glukokortikoidy, azathioprin, antimalarika, cyklofosfamid, méně často metotrexát či cyklosporin A. K novějším léčivům patří v léčbě lupusové nefritidy mykofenolát mofetil, či v některých případech takrolimus. V léčbě SLE je uplatňována i biologická léčba, zejména u případů s multiorgánovým postižením při selhání konvenční léčby [24], a to např. monoklonální protilátka anti CD20 – rituximab.
Rituximab je genetickým inženýrstvím získaná chimérická monoklonální protilátka produkovaná suspenzí uměle kultivovaných savčích buněk. Deplece CD20+ buněk (B-lymfocytů) se využívá zejména při léčbě maligních lymfomů a chronické lymfatické leukemie. K dalším indikacím, v řadě zemí off-lable, patří i léčba refrakterní idiopatické trombocytopenické purpury, autoimunitní hemolytické anémie či neinfekčních komplikací u nemocných s běžnou variabilní imunodeficiencí. Použití rituximabu se postupně rozšiřuje i do oblasti autoimunitních onemocnění, především revmatoidní artritidy, Wegenerovy granulomatózy, mikroskopické polyangiitidy a v neposlední řadě i SLE.
Deplece B-lymfocytů po léčbě rituximabem obvykle trvá 6–9 měsíců, po této době obvykle dochází k jejich obnově s následnou indukcí tvorby paměťových B-lymfocytů. Pouze u části nemocných je snížena i sérová koncentrace imunoglobulinů. Doposud nebylo objasněno, proč u některých nemocných dochází k významně prodloužené imunorestauraci, či dokonce doživotní poruše tvorby protilátek vyžadujících ať již přechodnou, či dlouhodobou substituční léčbu imunoglobuliny. Tíže suprese pravděpodobně závisí na typu základního onemocnění, preexistujícím protilátkovém imunodeficitu, kombinaci s dalšími léky s potenciálním imunosupresivním efektem i délce jeho podávání [25–27]. Za více rizikovou lze považovat skupinu nemocných s hematologickými malignitami, zejména pokud byli léčeni vysokodávkovanou chemoterapií, event. transplantací kostní dřeně [28–32]. Doposud zůstává otevřená i otázka míry rizika infekcí u nemocných s prokázanou poruchou tvorby protilátek [33,34]. Roberts et al pozorovali, že ve skupině nemocných léčených rituximabem pro multisystémové autoimunitní onemocnění mělo 26 % před zahájením léčby již patrné snížení sérové koncentrace IgG. V průběhu dalšího sledování byla hypogamablobulinemie přítomna u 56 % (u 4 % se jednalo hodnoty IgG < 4 g/l). Nižších hodnot dosahovali nemocní s předcházející léčbou cyklofosfamidem. Asociace s dávkou rituximabu zde nebyla prokázána. Substituční léčba imunoglobuliny byla zahájena pro infekční komplikace u 12 ze 288 nemocných (4,2 %) [35,36]. V případě námi prezentované kazuistiky 2 došlo pravděpodobně ke kumulaci rizikových faktorů: anamnéza maligního lymfomu, kdy byl již poprvé podáván v rámci systémové terapie i rituximab, a možný preexistující protilátkový imunodeficit.
Substituční léčba imunoglobuliny by měla být zahájena u nemocných s nízkou sérovou koncentrací IgG a současně přítomnými závažnými či rekurujícími infekcemi. Při léčbě primárních imunodeficitů bývá doporučována dávka 400–800 mg/kg/měsíc s úpravou dle aktuálního klinického stavu [37]. U nemocných se sekundárními imunodeficity stačí obvykle ke kontrole infekcí dávka nižší [38]. Kromě intravenózní substituční cesty je možno se stejnou efektivitou použít i léčbu podkožními preparáty [39]. Obecně lze tedy doporučit, aby sérové koncentrace imunoglobulinů byly zjištěny minimálně v období diagnózy základního onemocnění, dále pak před zahájením léčby rituximabem a za 6 měsíců od jeho ukončení.
Závěr
Závěrem bychom rádi shrnuli, že i mezi nemocnými se SLE lze identifikovat nemocné s primárním humorálním imunodeficitem. Na možnost výskytu běžné variabilní imunodeficience je vhodné pomýšlet zejména u nemocných s recidivujícími sinopulmonálními infekcemi při současné nízké aktivitě základního onemocnění. Monitorování sérových koncentrací imunoglobulinů je vhodné z důvodů identifikace deficitu a v indikovaných případech i zahájení substituční léčby imunoglobuliny.
Doručeno do redakce 29. 3. 2015
Přijato po recenzi 16. 5. 2015
MUDr. Pavlína Králíčková, Ph.D.
pavlina.kralickova@fnhk.cz
Ústav klinické imunologie a alergologie LF UK a FN Hradec Králové
www.fnhk.cz
Sources
1. Lawrence RC, Helmick CG, Arnett FC et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum 1998; 41(5): 778–799.
2. Chakravarty EF, Bush TM, Manzi S et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum 2007; 56(6): 2092–2094.
3. Pons-Estel GJ, Alarcon GS, Scofield L et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum 2010; 39(4): 257–268.
4. Furst DE, Clarke AE, Fernandes AW et al. Incidence and prevalence of adult systemic lupus erythematosus in a large US managed-care population. Lupus 2013; 22(1): 99–105.
5. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus 2006; 15(5): 308–318.
6. Peschken CA, Esdaile JM. Rheumatic diseases in North America‘s indigenous peoples. Semin Arthritis Rheum 1999; 28(6): 368–391.
7. Carneiro-Sampaio M, Liphaus BL, Jesus AA et al. Understanding systemic lupus erythematosus physiopathology in the light of primary immunodeficiencies. J Clin Immunol 2008; 28(Suppl 1): S34-S41.
8. Horák P, Tegzová D, Závada J et al. Doporučení České revmatologické společnosti pro diagnostiku a sledování nemocných se systémovým lupus erythematotes. Čes Revmatol 2013; 21(2): 59–70.
9. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999; 93(3): 190–197.
10. Battersby AC, Cale AM, Goldblatt D et al. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease. J Clin Immunol 2013; 33(8): 1276–1284.
11. Liblau RS, Bach JF. Selective IgA deficiency and autoimmunity. Int Arch Allergy Immunol 1992; 99(1): 16–27.
12. Rankin EC, Isenberg DA. IgA deficiency and SLE: prevalence in a clinic population and a review of the literature. Lupus 1997; 6(4): 390–394.
13. Cassidy JT, Kitson RK, Selby CL. Selective IgA deficiency in children and adults with systemic lupus erythematosus. Lupus 2007; 16(8): 647–650.
14. Resnick ES, Moshier EL, Godbold JH et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119(7): 1650–1657.
15. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999; 92(1): 34–48.
16. Fernandez-Castro M, Mellor-Pita S, Citores MJ et al. Common variable immunodeficiency in systemic lupus erythematosus. Semin Arthritis Rheum 2007; 36(4): 238–245.
17. Geneviève M, Bonnet F, Michaux C et al. Lupus nephritis associated with common variable immunodeficiency: favourable outcome with intravenous immunoglobulin treatment. Rev Med Interne 2012; 33(6): e31-e33. Dostupné z DOI: <http://dx.doi.org/10.1016/j.revmed.2011.05.002>.
18. Suyama K, Kawasaki Y, Abe Y et al. Development of common variable immunodeficiency in IgA - and IgG2-deficient patients with systemic lupus erythematosus. Pediatr Nephrol 2012; 27(3): 489–492.
19. Torres-Salido M, Cortes-Hernandez J, Balada E et al. Systemic lupus erythematosus as a first presentation of common variable immunodeficiency associated with infrequent mannose-binding lectin gene polymorphisms. Rheumatol Int 2011; 31(4): 537–541.
20. Al Hamzi H, Al Shaikh A, Arnaout RK. Poor specific antibody response immunodeficiency (dysgammaglobulinemia) predates systemic lupus erythematosus. Lupus 2013; 22(9): 961–966.
21. Yap D, Yung S, Ma M et al. Serum immunoglobulin G level in patients with lupus nephritis and the effect of treatment with corticosteroids and mycophenolate mofetil. Lupus 2014; 23(7):678–683.
22. Yong PF, Aslam L, Karim MY et al. Management of hypogammaglobulinaemia occurring in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2008; 47(9): 1400–1405.
23. McIntyre P, Craig JC. Prevention of serious bacterial infection in children with nephrotic syndrome. J Paediatr Child Health 1998; 34(4): 314–317.
24. Lan L, Han F, Chen JH. Efficacy and safety of rituximab therapy for systemic lupus erythematosus: a systematic review and meta-analysis. J Zhejiang Univ Sci B 2012; 13(9): 731–744.
25. Delbe-Bertin L, Aoun B, Tudorache E et al. Does rituximab induce hypogammaglobulinemia in patients with pediatric idiopathic nephrotic syndrome? Pediatr Nephrol 2013; 28(3): 447–451.
26. Marco H, Smith RM, Jones RB et al. The effect of rituximab therapy on immunoglobulin levels in patients with multisystem autoimmune disease. BMC Musculoskelet Disord 2014; 15 : 178. Dostupné z DOI: <http://dx.doi.org/10.1186/1471–2474–15–178>.
27. De La Totte, I, Leandro MJ, Valor L et al. Total serum immunoglobulin levels in patients with RA after multiple B-cell depletion cycles based on rituximab: relationship with B-cell kinetics. Rheumatology (Oxford) 2012; 51(5): 833–840.
28. Worch J, Makarova O, Burkhardt B. Immunreconstitution and infectious complications after rituximab treatment in children and adolescents: what do we know and what can we learn from adults? Cancers (Basel) 2015; 7(1): 305–328.
29. Irie E, Shirota Y, Suzuki C et al. Severe hypogammaglobulinemia persisting for 6 years after treatment with rituximab combined chemotherapy due to arrest of B lymphocyte differentiation together with alteration of T lymphocyte homeostasis. Int J Hematol 2010; 91(3): 501–508.
30. Walker AR, Kleiner A, Rich L et al. Profound hypogammaglobulinemia 7 years after treatment for indolent lymphoma. Cancer Invest 2008; 26(4): 431–433.
31. Shortt J, Spencer A. Adjuvant rituximab causes prolonged hypogammaglobulinaemia following autologous stem cell transplant for non-Hodgkin‘s lymphoma. Bone Marrow Transplant 2006; 38(6): 433–436.
32. Kelesidis T, Daikos G, Boumpas D et al. Does rituximab increase the incidence of infectious complications? A narrative review. Int J Infect Dis 2011; 15(1): e2-e16. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijid.2010.03.025>.
33. Casulo C, Maragulia J, Zelenetz AD. Incidence of hypogammaglobulinemia in patients receiving rituximab and the use of intravenous immunoglobulin for recurrent infections. Clin Lymphoma Myeloma Leuk 2013; 13(2): 106–111.
34. Furst DE. Serum immunoglobulins and risk of infection: how low can you go? Semin Arthritis Rheum 2009; 39(1): 18–29.
35. Roberts DM, Jones RB, Smith RM et al. Immunoglobulin G replacement for the treatment of infective complications of rituximab-associated hypogammaglobulinemia in autoimmune disease: A case series. J Autoimmun 2015; 57 : 24–29.
36. Roberts DM, Jones RB, Smith RM et al. Rituximab-associated hypogammaglobulinemia: Incidence, predictors and outcomes in patients with multi-system autoimmune disease. J Autoimmun 2015; 57 : 60–65.
37. Eijkhout HW, van der Meer JW, Kallenberg CG et al. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann Intern Med 2001; 135(3): 165–174.
38. Compagno N, Malipiero G, Cinetto F et al. Immunoglobulin replacement therapy in secondary hypogammaglobulinemia. Front Immunol 2014; 5 : 626. Dostupné z DOI: <http://dx.doi.org/10.3389/fimmu.2014.00626>.
39. Compagno N, Cinetto F, Semenzato G et al. Subcutaneous immunoglobulin in lymphoproliferative disorders and rituximab-related secondary hypogammaglobulinemia: a single-center experience in 61 patients. Haematologica 2014; 99(6): 1101–1106.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2015 Issue 9
Most read in this issue
- Terapeutický potenciál mikronizované purifikované flavonoidní frakce (MPFF) diosminu a hesperidinu v rámci léčby chronického žilního onemocnění
- Účelnost zavádění kaválních filtrů z pohledu internisty
- Návrh optimálního léčebného postupu v léčbě nízkorizikového karcinomu štítné žlázy
- Sekundární humorální imunodeficience u nemocných se systémovým lupus erythematodes