
Genetické změny u Waldenströmovy makroglobulinemie
Gene mutations connected to Waldenstöm macroglobulinemia
Waldenstöm macroglobulinemia (WM) is a rare lymphoproliferative disorder, currently classified as a monoclonal gammopathy, with incidence rate of 3 per million. The disease is characterized by presence of clonal B lymphocytes in the bone marrow and by presence of monoclonal immunoglobulin IgM in serum. It is mostly an indolent disorder, with median overall survival 6 years. Molecular pathogenesis of WM remains unclear, but deletion of 6q and 13q, trisomy of chromosomes 4 and 8 seem to be typical. Mutations of MYD88L265P and CXCR4WHIM are very common for WM and affect growth and survival of malignant cells. This work is aimed at the current knowledge of chromosomal aberrations and gene mutations connected to the pathophysiology of WM.
Key words:
chromosomal aberrations – somatic mutations – Waldenström macroglobulinemia
Authors:
Kateřina Kutálková 1; Lenka Sedlaříková 1,2; Zdeněk Adam 3; Sabina Ševčíková 1,2
Authors‘ workplace:
Babákova myelomová skupina, Ústav patologické fyziologie LF MU Brno
1; Oddělení klinické hematologie FN Brno
2; Interní hematologická a onkologická klinika LF MU a FN Brno, pracoviště Bohunice
3
Published in:
Vnitř Lék 2016; 62(1): 40-43
Category:
Reviews
Overview
Waldenströmova makroglobulinemie (WM) je vzácné lymfoproliferativní onemocnění ze skupiny monoklonálních gamapatií, s incidencí 3 případy na 1 milion obyvatel. Toto onemocnění je charakterizováno infiltrací kostní dřeně klonálními B-lymfocyty a přítomností monoklonálního imunoglobulinu třídy IgM v séru. Nejčastěji se jedná o indolentní onemocnění s mediánem přežití 6 let. Molekulární podstata není zcela objasněná, ale delece, trizomie chromozomů 4 a 8 a delece 13q se zdají být typické pro WM. Mutace MYD88L265P a CXCR4WHIM se u WM vyskytují velice často a mají vliv na růst a přežití nádorových buněk. Tato práce se zaměřuje na současné poznatky o chromozomových aberacích a genových mutacích spojených s patofyziologií WM.
Klíčová slova:
chromozomové aberace – somatické mutace – Waldenströmova makroglobulinemie
Úvod
Waldenströmova makroglobulinemie (WM) je vzácné lymfoproliferativní onemocnění charakterizované akumulací maligních lymfoplazmatických buněk v kostní dřeni, lymfatických uzlinách, slezině a nadbytečnou produkcí sérového IgM [1–3]. Patogeneze WM není dosud dostatečně objasněna. Nicméně v současnosti se pozornost upírá především na mutace genů MYD88 a CXCR4. Mutace MYD88 byla pozorována až u 90 % pacientů s WM a má potenciál stát se diagnostickým nástrojem pro odlišení WM od ostatních IgM monoklonálních gamapatií, případně predikovat vývoj WM. Z klinického hlediska může být přítomnost těchto mutací potenciálně velmi důležitá i pro léčbu WM.
V tomto čísle časopisu vychází přehledový článek Adama et al, který se zabývá klinickou podstatou WM. Tento doprovodný článek se proto zaměří pouze na genetické změny u této choroby.
Chromozomové aberace u WM
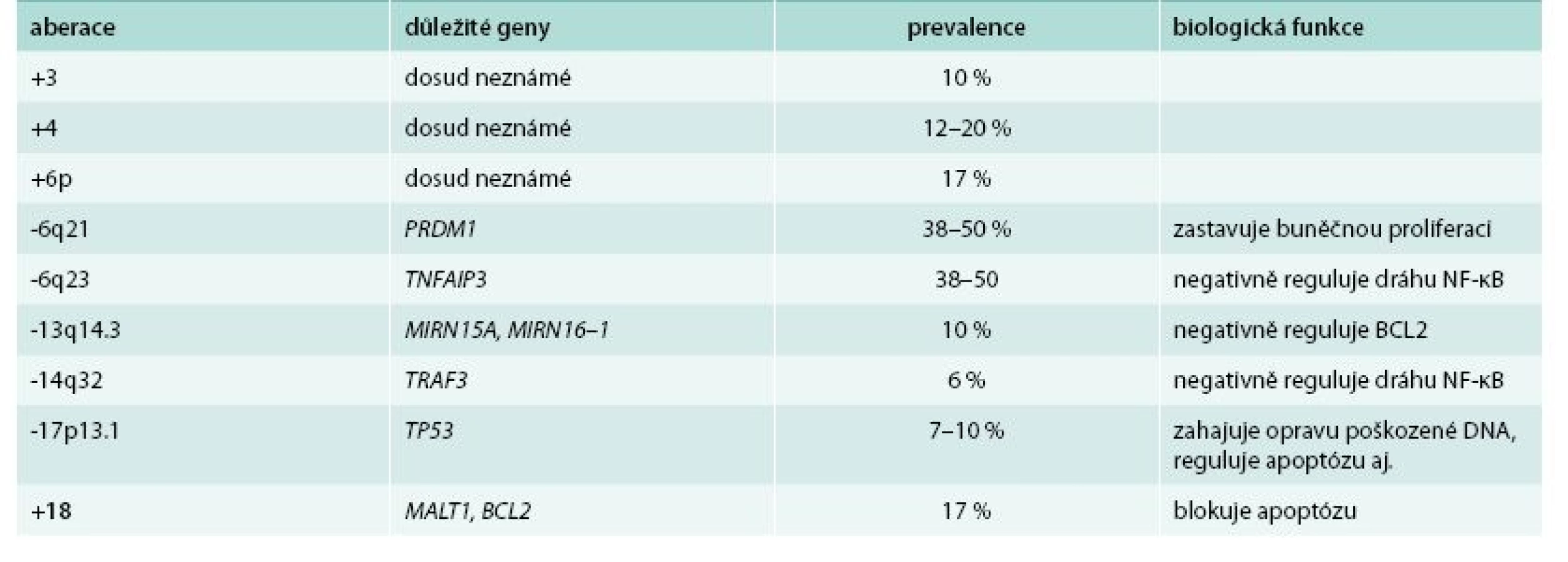
Karyotypování maligních buněk WM odhalilo pestrou paletu cytogenetických aberací, ovšem žádná z nich není jedinečná a typická pro tuto chorobu. Nejčastěji (až v 50 % případů) je nalezena delece chromozomu 6q, která je spojená s negativními klinickými a laboratorními parametry. Ovšem efekt na přežití zůstává nejasný. Typicky dochází k delecím v oblasti 6q21–23, a to přibližně u 40–60 % pacientů [3], přičemž 41 % z nich mělo současně inzerce v oblasti 6p [5].
V lokusu 6q se nachází několik genů potenciálně účinkujících v patogenezi WM. Mezi významné patří PRDM1 a TNFAIP3. PRDM1 slouží jako represor buněčné proliferace a přes represi PAX5 a následně XBP1 hraje roli v diferenciaci buněk B lymfocytární linie a sekreci imunoglobulinů. U WM byla zvýšená hladina XBP1 prokázána u 61 % pacientů. TNFAIP3 je nádorovým supresorem, při jehož inaktivaci dochází k trvalé aktivaci signální dráhy NF-κB, která ovlivňuje celou řadu biologických procesů včetně inhibice apoptózy a buněčné proliferace [6].
V pokročilejších stadiích nemoci se objevují i další delece – oblasti 13q14, obsahující geny pro mikroRNA miR15a a miR-16–1 (obě negativně regulují expresi genu BCL2 kódující antiapoptotický protein) a delece oblasti 17p13 s genem TP53 [7,8].
Pro odlišení WM od jiných nemocí, zvláště pak od mnohočetného myelomu (MM), je významná nepřítomnost translokací zahrnujících gen pro těžký imunoglobulinový řetězec na chromozomu 14q32 [9]. V této oblasti jsou nicméně detekovány delece zasahující gen TRAF3, který kóduje důležitý negativní regulátor nekanonické dráhy NF-κB [4]. Pro WM je typickou chromozomovou aberací trisomie chromozomu 4, jelikož u jiných malignit se nachází jen zřídka [10]. Dále byly u WM zaznamenány zisky v chromozomech 3q, 18, 8q a Xq27–28 a ztráty u 7q22, 8p, 11q22–24, 13q14 a 17p11–13 [4]. Nejčastěji pozorované aberace a geny jimi zasažené jsou shrnuty v tab. 1.
Somatické mutace
MYD88
MYD88 (Myeloid differentiation primary response gene 88) slouží jako adaptorová molekula pro toll-like receptory (TLR) a receptor pro IL1 (interleukin 1). Poté, co se k receptoru naváže příslušný ligand, MYD88 dimerizuje a v tomto stavu spouští autofosforylaci IRAK4 a IRAK1 (kinázy asociované s receptorem pro IL1). Fosforylovaná IRAK1 uvolňuje TRAF6 (s receptorem pro TNF asociovaný faktor 6), který následně vede k aktivaci komplexu IKK, kinázy zodpovědné za spuštění dráhy NF-κB [11].
Při celogenomovém sekvenování patologických buněk byla u WM objevena jednonukleotidová záměna tyminu za cytosin na pozici 38182641 v oblasti chromozomu 3p22.2. Následnou záměnou aminokyselin leucinu za prolin vzniká mutantní protein MYD88L265P [10]. Tato substituce tvoří aktivnější protein, který napomáhá přežití maligních buněk tím, že spontánně sestavuje komplex s IRAK1 a IRAK4 [12]. Zároveň při alternativním sestřihu vznikají další 2 izoformy, jejichž význam nebyl dosud objasněn [13]. Tato mutace MYD88L265P se rovněž často vyskytuje současně s delecí 6q, která je typická pro WM, jak již bylo zmíněno dříve [14,15].
Pacienti s MYD88L265P oproti MYD88WT (wild type, nemutovaný) mají ve většině případů vysokou infiltraci lymfocytů do kostní dřeně, a to až 80 % všech buněk [14], a rovněž vyšší hodnoty sérového IgM [14]. Mutace MYD88L265P byla prokázána u více než 90 % případů WM. U některých pacientů dokonce v homozygotní podobě. Avšak tito pacienti se klinickým obrazem nijak nelišili od pacientů heterozygotních [11].
V buňkách zdravých osob se tato mutace nenachází. Nebyla nalezena ani u MM, a to včetně IgM sekretujícího typu. V malé míře (asi 7 %) se může nacházet u MZL (lymfomu marginální zóny), u případů, jejichž klinické projevy se částečně překrývají s WM [11]. Dále se nachází u 29 % difuzních velkobuněčných B lymfomů (subtypu aktivovaných B-buněk), 9 % MALT lymfomů (lymfomy odvozené od slizniční tkáně) [12] a 3 % chronických lymfatických leukemií [16]. Mutace MYD88L265P by se mohla stát potenciálním diagnostickým nástrojem pro odlišení WM od ostatních monoklonálních gamapatií, především IgM sekretujícího typu, což může být jinak obtížné. Na druhou stranu, u pacientů s lymfoplazmocytárním lymfomem bez sekrece IgM se mutace MYD88L265P nacházela stejně často jako u pacientů s WM [11].
MYD88L265P se nachází i u pacientů s IgM MGUS (monoklonální gamapatie nejasného významu), ale hodnoty pozorované v jednotlivých studiích se významně liší – od 10 % [17] až po 80 % [18], podle citlivosti použité metody. Při použití kvantitativní AS-PCR (alelově specifické PCR) je mutace odhalena i při nízkých koncentracích klonálních buněk. Xu et al v roce 2013 pozorovali, že pacienti IgM MGUS s vysokou expresí MYD88L265P progredovali do WM. K tomuto vývoji onemocnění dochází ve většině případů, nicméně ne ve všech. Je tedy třeba dalších rozsáhlejších studií, které by mohly pomoci objasnit, zda přítomnost mutace MYD88L265P dokáže predikovat vývoj WM. Hladina exprese mutantního MYD88 analyzovaná pomocí qPCR tak má potenciál stát se důležitým prognostickým markerem. MYD88L265P u IgM MGUS by mohla být zásadní mutací, která dává časné nádorové buňce růstovou výhodu oproti ostatním klonům, a potenciálně umožní další mutace vedoucí k progresi do symptomatické WM [15].
CXCR4
CXCR4 je chemokinový receptor spřažený s G proteinem. Po navázání ligandu SDF1 (také označovaného jako CXCL12) se spouští několik signálních drah, vedoucích k různým odezvám: aktivací G proteinu může inhibovat adenylátcyklázu, aktivovat Src tyrozinové kinázy, fosfolipázu C-β (PLC-β) a PI3K, což vede k regulaci pochodů jako je transkripce, migrace a adheze buněk. CXCR4 ovlivňuje i dráhy na G proteinu nezávislé, např. JAK/STAT [19].
Při pokusech na myších s chybějícími CXCR4 nebo SDF1 byla pozorována pozdní gestační letalita s poruchami lymfopoézy B buněk, kolonizace kostní dřeně a tvorby srdečního septa, což naznačuje, že kromě funkce klasického chemokinového receptoru u dospělých má i další funkce při vývoji [20,21].
WM je první onkologické onemocnění, u kterého byla prokázána mutace CXCR4 [13], ačkoli jeho dysregulace je pozorována poměrně často [22]. Tyto mutace jsou nalézány u vzácné vrozené poruchy imunity, tzv. WHIM syndromu (warts, hypogammaglobulinemia, infections, and myelokathexis syndrome – syndrom bradavic, hypogamaglobulinemie, infekcí a myelokatexie), a proto bývají nazývány WHIM-like. Jde o mutace ovlivňující C konec proteinu. Tato oblast se vyznačuje přítomností většího počtu serinů, jejichž fosforylace vede ke spuštění signalizační kaskády buňky.
Rozeznáváme 2 typy mutací: tzv. nonsense mutace (NS; ztráta 15–20 aminokyselin v důsledku předčasného ukončení translace) a mutace mající za následek posun čtecího rámce (frameshift, FS; ovlivňují řetězec o délce přibližně 40 aminokyselin). NS a FS mutace se objevují s takřka stejnou četností [13]. Nejčastější z WHIM-like mutací nalézaných u WM, tedy CXCR4S338X, brání internalizaci receptoru, která by měla nastat po stimulaci SDF1. Déle trvající stimulace signálních drah (za pomoci kináz AKT, ERK a BTK) vede k přežívání nádorových buněk a také k rezistenci k některým léčivům [23].
Ve studii Treona et al bylo pozorováno, že 30 % pacientů vykazovalo mutace v genu pro CXCR4. U 98 % z nich se současně vyskytovala forma MYD88L265P, zatímco 94 % pacientů s MYD88WT bylo také CXCR4WT. Porovnáním se subpopulacemi zdravých buněk bylo potvrzeno, že se jedná o somatické mutace [17].
Klinické projevy jednotlivých kombinací zmíněných mutací se výrazně liší. Pacienti s MYD88L265PCXCR4NS vykazovali výrazně vyšší hladiny IgM, časnější symptomatické onemocnění a velkou infiltraci kostní dřeně, zatímco s MYD88WTCXCR4WT se projevy objevovaly ve vyšším věku a infiltrace kostní dřeně byla malá. Kombinace MYD88L265PCXCR4WT a MYD88L265PCXCR4FS vykazovaly shodně střední míru infiltrace. Přestože jsou projevy nemoci u CXCR4NS agresivnější, nemá tato mutace vliv na délku přežití. Pacienti s mutovaným CXCR4 vykazovali méně často adenopatie, což je nejspíše způsobené zvýšeným tropizmem nádorových buněk ke stromatu kostní dřeně. Zvýšená adheze napomáhá přežití maligních buněk a větší tvorbě IgM, která je zvláště patrná u CXCR4NS [17].
Z klinického hlediska může být přítomnost těchto mutací potenciálně velmi důležitá i pro léčbu WM. Určení stavu mutace MYD88 a CXCR4 může pomoci identifikovat nové podskupiny WM, které se liší s ohledem na klinický vývoj onemocnění a prognózu. Přítomnost somatických mutací MYD88 a CXCR4 má vliv rovněž na účinek nových léků, které by mohly změnit další vývoj této choroby. Příkladem je ibrutinib, inhibitor BTK kinázy, tedy enzymu působícího ve spolupráci s proteinem MYD88, který je účinný pro léčbu WM pacientů s MYD88L265P. Na druhou stranu, bylo prokázáno, že buňky s CXCR4WHIM jsou vůči ibrutinibu výrazně odolnější než CXCR4WT [23].
Další mutace
Celogenomové sekvenování odhalilo i další mutované geny, ovšem jejich mutace nebyly rekurentní. Somatická varianta ARID1A (AT-rich interactive domain 1A) byla pozorována u 17 % pacientů. Ve všech případech se vyskytovala společně s MYD88L265P. Tito pacienti měli větší invazi do kostní dřeně (90 % vs 50 %), nižší hematokrit a méně krevních destiček [11].
Při pátrání po mutacích, které by byly exkluzivní pro pacienty bez mutovaného MYD88, objevili Treon et al u 2 ze 3 pacientů mutaci v genu MLL2 (leukemie smíšeného původu 2). Oba pacienti měli zvýšený počet cirkulujících klonálních B-lymfocytů se zvýšenou expresí CD23, což pro WM není příliš typické [10]. MLL2 kóduje histonovou metyltransferázu, která kontroluje genovou transkripci modifikací čtvrtého lyzinu na histonu 3 [24].
Copy number variations (CNV)
CNV jsou krátké segmenty DNA, jejich počet je mezi jednotlivými jedinci variabilní. Tyto variabilní regiony tvoří 12 % genomu a jsou tak důležitou složkou genetické diverzity. Je obtížné rozlišit, jestli je daný CNV pouze polymorfizmem, nebo už je patogenní a podílí se na onemocnění [25]. U WM byly nalezeny ve větší míře na chromozomu 6, ale většina jich byla roztroušena po ostatních chromozomech. Nejčastější z delecí se nacházely v genech HIVEP2, ARID1B a BCLAF1, pozorované u pacientů s i bez viditelných delecí 6q [13].
Závěr
I když WM zůstává indolentním onemocněním, nové poznatky o aberacích spojených s touto nemocí zlepšují naše poznání patofyziologie WM, která není doposud dostatečně objasněna. Významnou roli hrají chromozomové aberace, zvláště delece dlouhého raménka chromozomu 6. Geny z deletovaných oblastí (např. PRMD1, TNFAIP3, TRAF3) se podílejí na kontrole buněčného cyklu a proliferace a jejich ztráta podporuje přežívání maligních buněk. Průlomovým objevem ve výzkumu WM je nález vysoce rekurentní mutace genu MYD88 a CXCR4. Mutovaný MYD88 byl pozorován až u 90 % pacientů. Tato mutace může v budoucnu napomáhat v diagnostice, při níž by mohla odlišit WM od nemocí s překrývajícím se klinickým obrazem, ale je také potenciálním terčem pro cílenou terapii. Kontrola přítomnosti mutace v genu MYD88 by mohla sloužit jako jednoduchý nezávislý rizikový faktor pro progresi do WM především u pacientů s IgM MGUS. Mutace CXCR4 není tak častá, zato se jedná o zcela novou, u nádorových onemocnění doposud nepozorovanou mutaci, která se rovněž nabízí jako cíl pro léčbu.
Tato práce byla podpořena grantem MZ ČR IGA NT14575.
RNDr. Sabina Ševčíková, Ph.D.
sevcik@med.muni.cz
Babákova myelomová skupina, Ústav patologické fyziologie LF MU, Brno
babak.med.muni.cz
Doručeno do redakce 18. 5. 2015
Přijato po recenzi 2. 7. 2015
Sources
1. Adam Z, Šmardová J, Ščudla V. Waldenstrőmova makroglobulinémie – klinické projevy a diferenciální diagnostika a prognóza nemoci. Vnitř Lék 2007; 53(12): 1325–1337.
2. Owen RG, Pratt G, Auer RL et al. Guidelines on the diagnosis and management of Waldenstrom macroglobulinaemia. Br J Haematol 2014; 165(3): 316–333.
3. Adam Z, Pour L, Krejčí M et al. Léčba Waldenströmovy makroglobulinemie – zkušenosti jednoho pracoviště. Vnitř Lék 2009; 55(11): 9–1.
4. Ocio EM, Schop RF, Gonzalez B et al. 6q deletion in Waldenström macroglobulinemia is associated with features of adverse prognosis. Br J Haematol 2007; 136(1): 80–86.
5. Braggio E, Keats JJ, Leleu X et al. Identification of Copy Number Abnormalities and Inactivating Mutations in Two Negative Regulators of Nuclear Factor-κB Signaling Pathways in Waldenstrom’s Macroglobulinemia. Cancer Res 2009; 69(8): 3579–3588.
6. Braggio E, Philipsborn C, Novak A et al. Molecular pathogenesis of Waldenstrom’s macroglobulinemia. Haematologica 2012; 97(9): 1281–1290.
7. Cimmino A, Calin GA, Fabbri M et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102(39): 13944–13949.
8. Chang H, Samiee S, Li D et al. Analysis of IgH translocations, chromosome 13q14 and 17p13.1(p53) deletions by fluorescence in situ hybridization in Waldenstrom’s macroglobulinemia: a single center study of 22 cases. Leukemia 2004; 18(6): 1160–1162.
9. Schop RFJ, Kuehl WM, Van Wier SA et al. Waldenstrom macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood 2002; 100(8): 2996–3001.
10. Terre C, Nguyen-Khac F, Barin C et al. Trisomy 4, a new chromosomal abnormality in Waldenstrom’s macroglobulinemia: a study of 39 cases. Leukemia 2006; 20(9): 1634–1636.
11. Treon SP, Xu L, Yang G et al. MYD88 L265P Somatic Mutation in Waldenstrom’s Macroglobulinemia. N Engl J Med 2012; 367(9): 826–833.
12. Ngo VN, Young RM, Schmitz R et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470(7332): 115–119.
13. Hunter ZR, Xu L, Yang G et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014; 123(11): 1637–1646.
14. Kim JA, Im K, Park SN et al. MYD88 L265P Mutations Are Correlated with 6q Deletion in Korean Patients with Waldenstrom Macroglobulinemia. Biomed Res Int: 2014; 2014 : 363540. Dostupné z DOI: <http://dx.doi.org/10.1155/2014/363540>.
15. Xu L, Hunter ZR, Yang G et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013; 121(11): 2051–2058.
16. Puente XS, Pinyol M, Quesada V et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475(7354): 101–105.
17. Treon SP, Cao Y, Xu L et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014; 123(18): 2791–2796.
18. Jimenez C, Sebastian E, Chillon MC et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom’s macroglobulinemia. Leukemia 2013; 27(8): 1722–1728.
19. Kucia M, Jankowski K, Reca R et al. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J. Mol. Histol 2004; 35(3): 233–245.
20. Nagasawa T, Hirota S, Tachibana K et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996; 382(6592): 635–638.
21. Zou YR, Kottmann AH, Kuroda M et al. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998; 393(6685): 595–599.
22. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Acta-Biomembr 2007; 1768(4): 952–963.
23. Cao Y, Hunter ZR, Liu X et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia 2015; 29(1): 169–176.
24. Pasqualucci L, Trifonov V, Fabbri G et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet 2011; 43(9): 830–837.
25. Redon R, Ishikawa S, Fitch KR et al. Global variation in copy num
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 1
Most read in this issue
- Toxická epidermální nekrolýza
- Změny v prognóze a v léčbě Waldenströmovy makroglobulinemie: přehled literatury a vlastní zkušenosti
- Význam vyšetrovania alanínaminotransferázy u darcov krvi pre redukciu rizika prenosu hepatitíd B a C hemoterapiou
- Genetické změny u Waldenströmovy makroglobulinemie