
Familiární hypercholesterolemie v České republice v roce 2016
Familial hypercholesterolemia in the Czech Republic in 2016
Familial hypercholesterolemia (FH) is the most frequent autosomal dominant hereditary disease which is characterized by a decreased LDL-cholesterol catabolism and early clinical manifestation of atherosclerosis affecting blood vessels. The MedPed (Make early diagnosis to Prevent early deaths) project aims to diagnose patients with FH as early as possible, so that they can profit the most from a therapy started in a timely manner and avoid premature cardiovascular events. Currently, as of 31 October 2016, the Czech national database keeps records of 6 947 patients with FH from 5 223 families. Considering the prevalence of FH equalling 1 : 250, this represents 17.4 % of the overall expected number of patients with FH in the Czech Republic. Determining the mutation responsible for FH, now using a next generation sequencing technology in the Czech Republic, brings with it higher diagnostic accuracy, better cooperation of patients and in particular facilitation of cascade screening in families. Although we are among the most successful countries in the world with regard to FH detection, the majority of patients are still undiagnosed. Moreover, as it turns out, most FH patients do not reach the target values with the current therapeutic possibilities. In this regard the newly approved hypolipidemic drugs, PCSK9 inhibitors, to be hopefully available also in the Czech Republic in the near future for chosen patients with FH at high risk, hold great promise.
Key words:
cascade screening – familial hypercholesterolemia – LDL-cholesterol – MedPed
Authors:
Tomáš Freiberger 1; Martina Vaclová 2; Lukáš Tichý 3; Vladimír Soška 4,5,6; Vladimír Bláha 7; Lenka Fajkusová 3; Richard Češka 2; Michal Vrablík 2
Authors‘ workplace:
Genetická laboratoř Centra kardiovaskulární a transplantační chirurgie, Brno
1; III. interní klinika 1. LF UK a VFN v Praze
2; Centrum molekulární biologie a genové terapie FN Brno
3; Oddělení klinické biochemie a ICRC – oddělení kardiovaskulárních chorob, FN u sv. Anny v Brně
4; Katedra laboratorních metod, LF MU Brno
5; II. interní klinika LF MU a FN U sv. Anny v Brně
6; III. interní gerontometabolická klinika LF UK a FN Hradec Králové
7
Published in:
Vnitř Lék 2016; 62(11): 924-928
Category:
Reviews
Overview
Familiární hypercholesterolemie (FH) je nejčastějším autosomálně dominantně dědičným onemocněním, které je charakterizováno snížením katabolizmu LDL-cholesterolu a předčasnou klinickou manifestací aterosklerotického postižení cév. Projekt MedPed (Make early diagnosis to Prevent early deaths) má za cíl diagnostikovat pacienty s FH co nejdříve, aby co nejvíce profitovali ze včasně zahájené terapie a nedospěli k předčasné kardiovaskulární příhodě. V současné době (k 31. 10. 2016) evidujeme v celonárodní české databázi 6 947 pacientů s FH z 5 223 rodin. Při prevalenci FH 1 : 250 to představuje 17,4 % z celkového očekávaného počtu pacientů s FH v ČR. Určení mutace zodpovědné za FH, k němuž je nyní v ČR využívána moderní technologie sekvenování nové generace, vede ke zpřesnění diagnostiky, zvýšení spolupráce ze strany pacientů a především usnadnění kaskádovitého screeningu v rodinách. Přestože patříme z hlediska záchytu FH k nejúspěšnějším zemím na světě, většina pacientů zůstává stále nediagnostikována. Navíc se ukazuje, že při současných léčebných možnostech většina pacientů s FH nedosahuje cílových hodnot. V tomto směru jsou velkým příslibem nově schválené léky, inhibitory PCSK9, které budou snad v nejbližší době k dispozici i v České republice pro vybrané vysoce rizikové pacienty s FH.
Klíčová slova:
familiární hypercholesterolemie – kaskádovitý screening – LDL-cholesterol – MedPed
Úvod
Familiární hypercholesterolemie (FH) je nejčastějším autosomálně dominantně dědičným onemocněním, které je charakterizováno snížením katabolizmu LDL-cholesterolu (LDL-C) a předčasnou klinickou manifestací aterosklerotického postižení cév [1]. Dlouhá léta tradovaná prevalence heterozygotní FH (heFH) 1 : 500 ve většině rozvinutých zemí [2,3] byla ve světle dat z recentních studií poopravena na hodnotu přibližně dvojnásobnou [4,5], což představuje celosvětově více než 30 milionů jedinců postižených touto chorobou. V populacích ovlivněných genetickým efektem zakladatele a s vysokou mírou příbuzenských sňatků, jako je tomu např. v Libanonu, jižní Africe nebo v oblasti Quebecu v Kanadě, je prevalence FH ještě vyšší. S tím souvisí také prevalence homozygotní formy FH (hoFH) pozměněná z původní hodnoty 1 : 1 000 000 na nynější hodnotu 1 : 300 000 [4].
FH byla poprvé popsána v roce 1938 jako familiární xantomatóza: prof. C. Muller spojil rodinný výskyt xantomů, vysoké hladiny cholesterolu a ischemické choroby srdeční s vrozenou poruchou metabolizmu pramenící z defektu jednoho genu [6]. Dalším průlomovým momentem v historii charakterizace FH byl rok 1963, kdy Khachadurian popsal heterozygotní a homozygotní fenotyp FH u pacientů v Libanonu a vymezil kodominantní typ dědičnosti [7]. Na tyto práce navázali v roce 1973 Goldstein a Brown, kteří popsali LDL receptor a jeho úlohu v katabolizmu cholesterolu a defekt v genu pro LDL receptor označili za příčinu FH [8]. Za tento objev se také v roce 1985 oba pánové stali laureáty Nobelovy ceny.
Pokud není FH léčena, relativní riziko předčasné koronární příhody je u pacientů s heFH výrazně vyšší než u ostatních jedinců a pacienti s hoFH jsou typicky postižení akutní kardiovaskulární příhodou do 20 let věku a často se nedožijí 30 let [9,10]. Situace pacientů s FH se podstatně zlepšila po uvedení statinů na trh v roce 1987. Celá řada studií, včetně metaanalýz, prokázala, že statiny jsou bezpečné léky redukující významně koncentraci LDL-C v cirkulaci i riziko kardiovaskulárních ischemických příhod a kardiovaskulární mortalitu [11]. Vedle režimových opatření jsou potentní statiny (atorvastatin a rosuvastatin) u FH jednoznačně lékem první volby, často s nutností přidání dalších léků, jako ezetimibu nebo sekvestrantů žlučových kyselin. V poslední době se objevily nové možnosti léčebného ovlivnění hypercholesterolemie a rizika ischemické kardiovaskulární ataky, zejména v podobě inhibitorů molekuly PCSK9, které podstatně rozšířily naše možnosti dosáhnout u pacientů s FH cílové hodnoty LDL-C. Dosavadní studie potvrdily vysoký potenciál a přidanou hodnotu léčby inhibitory PCSK9 [12,13], která už je schválená a snad bude v blízké budoucnosti k dispozici i v České republice.
Přestože FH je klinicky i laboratorně velmi dobře definována, přes pokročilé možnosti diagnostiky i dostatek informací o vysokém riziku předčasné manifestace aterosklerózy související s celoživotně zvýšenými hladinami LDL-C i přes dostupnost efektivní hypolipidemické terapie je FH stále nedostatečně diagnostikována, neléčena nebo léčena neadekvátně [1,14]. Navíc i při využití stávající dostupné léčby většina pacientů s FH nedosahuje cílových hodnot LDL-C [15,16]. A to i v České republice, která patří v záchytu FH díky projektu MedPed (Make early diagnosis to Prevent early deaths) k nejúspěšnějším zemím na světě.
Diagnostika FH
Diagnostika je založena na opakovaném vyšetření sérových koncentrací LDL-C (nejlépe bez léčby) a pečlivě odebrané rodinné anamnéze. Hodnoty LDL-C nad 95. percentilem populačně, věkově a pohlavně specifických hodnot svědčí pro možnou FH. Výskyt předčasné klinické manifestace aterosklerózy nebo hypercholesterolemie u probanda, jednoho z jeho rodičů, prarodičů nebo sourozenců je významným faktorem podporujícím diagnózu FH, stejně jako přítomnost šlachových xantomů či v mladém věku (do 40 let) objevivších se xantelazmat nebo arcus lipoides na rohovce. Koncentrace triglyceridů je obvykle v normě, ale vyšší hodnoty FH nevylučují. Samozřejmostí je vyloučení sekundární příčiny hypercholesterolemie, např. hypotyreóza nebo nefrotický syndrom. Diagnóza je potvrzena určením kauzální mutace v některém z genů, jejichž defekt je zodpovědný za vznik FH (LDLR, APOB nebo PCSK9) [14].
Zřejmě nejšířeji akceptovanými a v našich podmínkách nejvhodnějšími diagnostickými kritérii FH jsou Dutch lipid network criteria, která využívají bodový systém k hodnocení, zda se jedná o jistou, pravděpodobnou nebo možnou FH (tab. 1) [17]. Zohledňují jak údaje z rodinné anamnézy (hypercholesterolemie nebo výskyt předčasné kardiovaskulární příhody), tak přítomnost fyzikálních známek FH (šlachové xantomy, arcus lipoides na rohovce před 40. rokem věku), hodnoty LDL-C nebo průkaz kauzální mutace v genu LDLR, APOB nebo PCSK9, která má obzvlášť velkou váhu a je téměř dostačující pro stanovení jisté diagnózy FH (8 bodů z 9 potřebných).
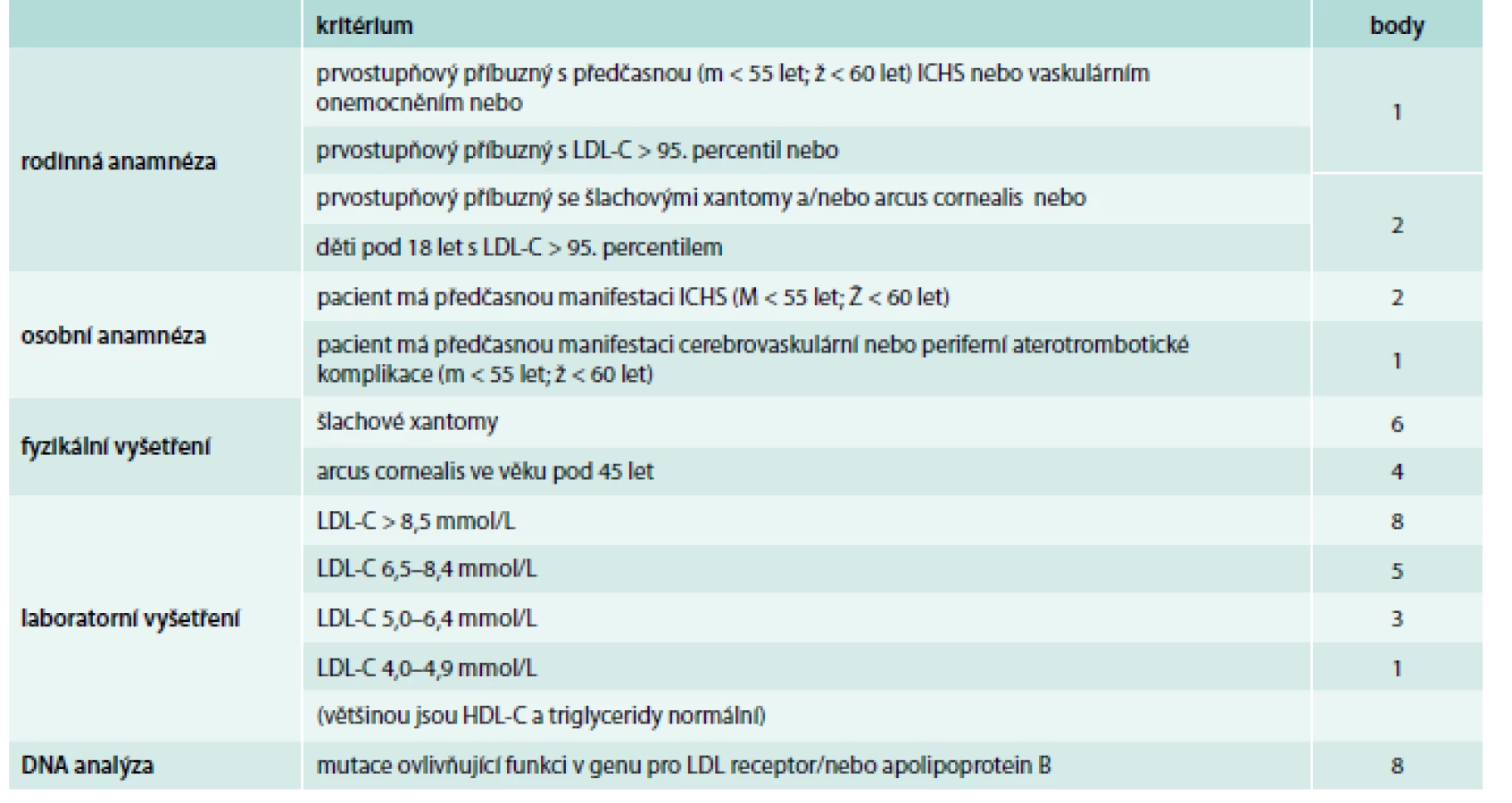
Diferenciálně diagnosticky je nutno odlišit především familiární kombinovanou hyperlipidemii – polygenní poruchu vyznačující se variabilním lipidovým fenotypem, často se pojící s inzulinovou rezistencí, dále polygenní hypercholesterolemii, a u těžkých forem heFH či hoFH je potřeba myslet hlavně na sitosterolemii, která má autosomálně recesivní typ dědičnosti a může být potvrzena vyšetřením koncentrace fytosterolů v séru a molekulárně genetickým vyšetřením (mutace v genu ABCG5 nebo ABCG8) [18].
Jakkoliv je riziko předčasné klinické manifestace aterosklerózy dáno především celoživotně zvýšenými koncentracemi LDL-cholesterolu, řada dalších faktorů může riziko pacienta s FH významně modifikovat a je nutné se jimi zabývat. Více ohroženi jsou především pacienti se symptomatickou nebo i subklinickou aterosklerózou, kuřáci, muži, pacienti starší 40 let, kteří nebyli dosud léčeni, hypertonici, diabetici nebo pacienti se zvýšenými hladinami lipoproteinu(a) [19].
Molekulární aspekty FH
Většina případů familiární hypercholesterolemie je podmíněna defektním genem pro LDL receptor (LDLR) nebo apolipoprotein B-100 (APOB), vzácně jsou příčinou mutace v genu PCSK9 vedoucí ke zvýšení funkce tohoto proteinu. Ojediněle však byly popsány i mutace v jiných genech způsobující fenotyp FH, např. v genu STAP1 nebo APOE [19]. Fenotyp hoFH může být determinován také mutacemi v genu LDLRAP1, které jsou spojeny s autosomálně recesivní formou onemocnění, což znamená, že rodiče pacienta jsou zdraví nositelé defektu na jedné alele, zatímco k manifestaci onemocnění je potřeba zdědit defektní alelu od obou rodičů. Existuje částečná korelace genotypu a fenotypu, pokud vyšší hladiny LDL-C a těžší fenotyp jsou spojeny s tzv. nulovými mutacemi v genu LDLR, které vedou k poklesu aktivity LDL receptoru na úroveň < 2 %, než je tomu u tzv. defektních mutací, které mají za následek snížení aktivity LDL receptoru na 2–25 % jeho normální funkce.
Určení mutace v některém z genů, jejichž defekt je zodpovědný za vznik FH, znamená potvrzení diagnózy FH, a tím také potvrzení působení zvýšeného LDL-C již od narození. Zejména je ale znalost mutace důležitá pro účinný kaskádovitý screening, protože umožní jednoznačné potvrzení či vyloučení FH u ostatních členů rodiny. Znalost mutace v rodině také zvyšuje ochotu příbuzných přijít k vyšetření, což dokládají i údaje z české celonárodní databáze, podle níž je v rodinách se známou kauzální mutací průměrný počet pacientů s FH v jedné rodině 1,77, zatímco v rodinách bez detekované mutace je to jen 1,18. Přínos molekulárně genetického vyšetření podtrhují i výsledky recentní komunitní studie z USA, které ukázaly, že u pacientů s hypercholesterolemií byla přítomnost mutace nezávislým prediktorem manifestního kardiovaskulárního postižení [20]. Je ale potřeba zdůraznit, že negativní výsledek molekulárně genetického vyšetření neznamená vyloučení FH. Může být dán omezenou senzitivitou použitých metod, polohou mutace mimo analyzované oblasti genu nebo tím, že defekt leží v jiném genu. Část pacientů s klinickou diagnózou FH ovšem může mít ve skutečnosti polygenní hypercholesterolemii, která je způsobena určitými variantami (polymorfizmy) ve více různých genech, které každá jednotlivě vedou k malému zvýšení LDL-C, ovšem kolektivně mohou vést k podstatnému nárůstu LDL-C a mohou imitovat FH. Talmud et al určili 12 takových genů malého účinku, jejichž varianty nejvíce přispívaly ke zvýšení LDL-C. Podle kombinace různých variant těchto 12 genů je kalkulováno tzv. LDL skóre, jehož výše určuje polygenní vliv na koncentraci LDL-C. Bylo zjištěno, že výše LDL skóre může pomoci rozlišit pacienty s FH (mají nízké skóre) od pacientů s polygenní hypercholesterolemií (vysoké skóre), ale tento postup ještě vyžaduje ověření více studiemi na vyšším počtu pacientů [21]. Polygenní hypercholesterolemii má až 30 % pacientů s klinickou diagnózou FH [22].
Diagnostické možnosti se v poslední době významně zvýšily nástupem moderní technologie sekvenování nové generace (NGS). Při použití celoexomového sekvenování byla v nedávné studii kauzální mutace odhalena u 20 % pacientů s klinicky jistou diagnózou FH, u nichž předchozí analýza nevedla k nálezu mutace [22]. Ovšem aplikace cíleného NGS u pacientů s hypercholesterolemií v primární péči vedla k detekci mutace způsobující FH jen u 2 % jedinců [23].
Iniciativy ke zvýšení záchytu FH a počtu léčených pacientů
V České republice byl před 18 lety spuštěn program aktivního vyhledávání pacientů s FH, MedPed, kterým jsme se připojili k mezinárodnímu úsilí, jehož hlavním cílem je významně snížit riziko předčasného úmrtí u pacientů s FH [24]. Hlavními prostředky jsou včasné stanovení diagnózy a včasná a dlouhodobá léčba postižených osob, přičemž důraz je kladen zejména na vyhledávání ohrožených jedinců mezi příbuznými již diagnostikovaných pacientů, a také na molekulárně genetickou diagnostiku, která je v ČR v současnosti založena na cíleném sekvenování nové generace a kopíruje tak trend, který se prosazuje ve vyspělých zemích.
Síť projektu MedPed v ČR je nyní tvořena 69 aktivními centry a spolupracovníky. Sestává ze 2 párů národních center v Praze a v Brně, 15 regionálních center pro dospělé a 10 pro děti, 21 specializovaných pracovišť pro dospělé a 5 pro děti a dalších 14 spolupracujících lékařů.
V současné době, k 31. 10. 2016, máme v databázi evidováno 6 947 pacientů z 5 223 rodin. Počet pacientů s FH/FDB zařazených do databáze tak činí 17,4 % z očekávaného počtu asi 40 000 pacientů v ČR (při uvažované prevalenci 1 : 250). U 712 pacientů byla diagnóza stanovena do 19 let věku. Vzorek DNA je k dispozici od 4 399 nepříbuzných pacientů a mutace v genu pro LDL receptor nebo ApoB byla prokázána u 1 332 z nich. Kauzální mutace v genu PCSK9 nebyla v české populaci doposud detekována. V ČR evidujeme také 14 pacientů s homozygotní formou FH, resp. FDB.
I nástup nových léků na trh oživil zájem o FH a ve světě se objevila řada iniciativ zaměřená na zvýšení záchytu a počtu léčených pacientů s FH, a tím významnému omezení počtu předčasných úmrtí a kardiovaskulárních příhod v důsledku předčasně rozvinutého aterosklerotického postižení cév. V Austrálii, Pacifiku a jižní Americe proběhla 10 countries study vedená G. Wattem [25], v USA se již několik let úspěšně rozvíjí společná platforma pacientů a lékařů FH foundation [26]. V Evropě probíhá projekt FH studies collaboration vedený K. Rayem pod hlavičkou EAS (European Atherosclerosis Society) [1] a také projekt ScreenPro FH zaměřený na zlepšení diagnostiky a léčby FH v regionu jižní a východní Evropy a střední Asie, zejména v méně rozvinutých zemích, vedený R. Češkou [27]. V obou těchto iniciativách je Česká republika aktivně zapojena.
Závěr
Familiární hypercholesterolemie je závažná porucha lipidového metabolizmu, která je nejčastějším dědičným metabolickým onemocněním vůbec a vede k předčasným úmrtím v důsledku klinické manifestace aterosklerózy. Přesto, že ČR patří v záchytu FH k nejúspěšnějším zemím na světě, i u nás je stále většina pacientů nediagnostikována. Navíc část diagnostikovaných pacientů není adekvátně léčena. Ale i u pacientů, kteří adekvátně léčeni jsou, nedosahujeme ve většině případů cílových hodnot LDL-cholesterolu. V tomto směru jsou příslibem nové léky, které snad budou v blízké budoucnosti k dispozici pro nejvíce ohrožené pacienty s FH i v ČR a které pomohou zvýšit procento pacientů s dosaženými cílovými hodnotami LDL-cholesterolu.
Práce byla podpořena granty AZV ČR 15–28277A a 16–29084A. Poděkování patří všem spolupracujícím lékařům (jejich seznam je uveden na http://www.athero.cz/projekt-medped/pro-odborníky/pracoviště-medped), koordinátorkám Editě Firoňové, Elišce Mitvalské a Marii Plotěné, pracovníkům zajišťujícím molekulární diagnostiku, Martině Slezáčkové a Mgr. Petře Zapletalové, společnosti Galén Symposion a Mgr. Haně Středové za pomoc s logistikou projektu a sponzorujícím společnostem Amgen, AOP Orphan, Krka, MSD, Pfizer a Sanofi.
MUDr. Tomáš Freiberger, Ph.D.
tomfre@cktech.cz
Centrum kardiovaskulární a transplantační chirurgie,
Brno
www.cktch.cz
Doručeno do redakce 4. 11. 2016
Přijato po recenzi 24. 11. 2016
Sources
1. Vallejo-Vaz AJ, Kondapally Seshasai SR, Cole D et al. Familial hypercholesterolaemia: A global call to arms. Atherosclerosis 2015; 243(1): 257–259. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2015.09.021 >.
2. Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutarylcoenzyme A reduktase activity associated with over production of cholesterol. Proc Natl Acad Sci USA 1973; 70(10): 2804–2808.
3. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL (eds) et al. The metabolic and molecular bases of inherited disease. 8th ed. McGraw-Hill: New York 2001. 2. Vol: 2863–2914. ISBN 978–0071363211.
4. Benn M, Watts GF, Tybjaerg-Hansen A et al. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012; 97(11): 3956–364. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2012–1563>. Erratum in J Clin Endocrinol Metab 2014; 99 : 4758–4759.
5. Sjouke B, Kusters DM, Kindt I et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J 2015; 36(9): 560–565. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehu058>
6. Müller C. Angina pectoris in hereditary xanthomatosis. Arch Intern Med 1939; 64 : 675–700.
7. Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med 1964; 37 : 402–407.
8. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232(4746): 34–47.
9. Huijgen R, Kindt I, Defesche JC et al. Cardiovascular risk in relation to functionality of sequence variants in the gene coding for the low-density lipoprotein receptor: a study among 29,365 individual stested for 64 specific low-density lipoprotein-receptor sequence variants. Eur Heart J 2012; 33(18): 2325–2330. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehs038>.
10. Cuchel M, Bruckert E, Ginsberg HN et al. [European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia]. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014; 35(32): 2146–2157. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehu274>.
11. Baigent C, Blackwell L, Emberson J, Holland LE et al. Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376(9753): 1670–1681.
12. Raal FJ, Stein EA, Dufour R et al. [RUTHERFORD-2 Investigators]. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 385(9965): 331–340. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(14)61399–4>.
13. Kastelein JJ, Ginsberg HN, Langslet G et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36(43): 2996–3003. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehv370>.
14. Nordestgaard BG, Chapman MJ, Humphries SE et al. [European Atherosclerosis Society Consensus Panel]. Familial hypercholesterolaemia is under diagnosed and under treated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J 2013; 34(45): 3478–3490a. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/eht273>.
15. Pijlman AH, Huijgen R, Verhagen SN et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in the Netherlands. Atherosclerosis 2010; 209(1): 189–194. Dostupné z DOI: http://dx.doi.org/10.1016/j.atherosclerosis.2009.09.014>.
16. Perez de Isla L, Alonso R, Watts GF et al. [SAFEHEART Investigators]. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART Registry follow-up. J Am Coll Cardiol 2016; 67(11): 1278–1285. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2016.01.008>.
17. van Aalst-Cohen ES, Jansen AC, Tanck MW et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. Eur Heart J 2006; 27(18): 2240–2246.
18. Watts GF, Gidding S, Wierzbicki AS et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol 2014; 171(3): 309–325. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2013.11.025>.
19. Santos RD, Gidding SS, Hegele RA et al. [International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel]. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol 2016; 4(9): 850–861. Dostupné z DOI: <http://dx.doi.org/10.1016/S2213–8587(16)30041–9>.
20. Khera AV, Won HH, Peloso GM et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol 2016; 67(22): 2578–2589. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2016.03.520>.
21. Talmud PJ, Shah S, Whittall R et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 2013; 381(9874): 1293–1301. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(12)62127–8>
22. Futema M, Plagnol V, Li K et al. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet 2014; 51(8): 537–544. Dostupné z DOI: <http://dx.doi.org/10.1136/jmedgenet-2014–102405>.
23. Norsworthy PJ, Vandrovcova J, Thomas ER et al. Targeted genetic testing for familial hypercholesterolaemia using next generation sequencing: a population-based study. BMC Med Genet 2014; 15 : 70. Dostupné z DOI: <http://dx.doi.org/10.1186/1471–2350–15–70>.
24. Freiberger T, Vrablík M. 15 let projektu MedPed v České republice. Hypertenze a kardiovaskulární prevence 2013; 2(5): 58–60.
25. Watts GF, Ding PY, George P et al. Translational Research for Improving the Care of Familial Hypercholesterolemia: The “Ten Countries Study” and Beyond. J Atheroscler Thromb 2016; 23(8): 891–900. Dostupné z DOI: <http://dx.doi.org/10.5551/jat.35949>.
26. O‘Brien EC, Roe MT, Fraulo ES et al. Rationale and design of the familial hypercholesterolemia foundation Cascade Screening for Awareness and DEtection of Familial Hypercholesterolemia registry. Am Heart J 2014; 167(3): 342–349. e17. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ahj.2013.12.008>.
27. Ceska R et al. ScreenPro FH – Screening Project for Familial Hypercholesterolemia in Central, Southern and Eastern Europe: Rationale and Design. Submitted. Dostupné z WWW: <http://screenprofh.com/>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 11
Most read in this issue
- Dieta při dyslipidemii a metabolickém syndromu
- Hyperlipoproteinemie u dětí
- Hyperlipoproteinemie a dyslipidemie jako vzácná onemocnění: diagnostika a léčba
- Kyselina močová jako rizikový faktor kardiovaskulárních onemocnění