
Genetická determinace dyslipidemií – co přinesly výsledky celogenomových screeningů a další směry výzkumu
Genetic determination of dyslipidemia – What tell us the results of genome-wide association studies?
Dyslipidemia (high levels of plasma triglycerides and total cholesterol/LDL-cholesterol and low HDL-cholesterol) is considered as one of the major factors in the development of atherosclerosis and subsequent myocardial infarction. The final value of lipid parameters results from joint action of genetic predispositions and lifestyle factors (primarily smoking status, physical activity and in lower extent also diet). It is estimated that genetic factors are responsible for 40–80 % of the variability of plasma lipid values. Currently are as predictors DL analyzed mainly single nucleotide polymorphisms (SNPs). A fundamental shift in knowledge of genetic determination DL bring genome-wide association studies (GWAs). These revealed several dozen major polymorphisms in a DNA sequence related to lipid levels. Rather surprisingly, these variants are usually not substitutions of the amino acids, or causing a premature stop codon, but substitutions outside the genes. GWAS also found a number of variants within the genes whose function in lipid metabolism was completely unknown (e.g. gene for sortilin). Polymorphisms in genes for APOE, SORT1, LDLR (affect levels of total cholesterol and LDL-cholesterol), CETP, APOA1, ABCA-1, GALNT-2 (influence HDL-cholesterol) and finally in genes for APOA5, LPL or TRIB1 (affect the levels of triglycerides) but explains max. 30 % of the variability of plasma lipids. It is supposed, that rare polymorphisms/mutations and genetic factors unrelated directly to alterations in the DNA sequence (DNA methylation, histone modifications, regulatory RNA molecules) are responsible for the remaining proportion of DL determination.
Key words:
gene – cholesterol – interaction – mutation – polymorphism – triglycerides
Authors:
Jaroslav Alois Hubáček
Authors‘ workplace:
Centrum experimentální medicíny IKEM, Praha
Published in:
Vnitř Lék 2016; 62(11): 868-876
Category:
Reviews
Overview
Dyslipidemie (vysoké hladiny plazmatických triglyceridů a celkového cholesterolu/LDL-cholesterolu a nízké hladiny HDL-cholesterolu) jsou považovány za jeden z hlavních faktorů rozvoje aterosklerózy a následného infarktu myokardu. Konečná hodnota těchto parametrů je výsledkem společného působení genetických predispozic a životního stylu (primárně kuřáckým statusem, fyzickou aktivitou a mírně i složením diety) každého jedince. Genetické faktory determinují odhadem 40–80 % z variability plazmatických lipidů. V současné době jsou jako prediktory dyslipidemie analyzovány především jednonukleotidové polymorfizmy – SNPs. Zásadní posun ve znalostech genetické determinace DL přinesly celogenomové screeningy (GWAs). Ty odhalily několik desítek klíčových variant v sekvenci DNA spjatých s hladinami lipidů. Zajímavé je, že se většinou nejedná o záměny měnící aminokyseliny, nebo způsobující předčasné stop kodony, ale o záměny ležící mimo geny. GWAs rovněž nalezly řadu variant v genech, jejichž funkce v metabolizmu lipidů byla zcela neznámá (např. v genu pro sortilin). Polymorfizmy např. v genech APOE, SORT1, LDLR (jsou spojeny s hladinami celkového cholesterolu a LDL-cholesterolu), CETP, APOA1, ABCA-1, GALNT-2 (ovlivní hladiny HDL-cholesterolu) a konečně v genech pro APOA5, LPL nebo TRIB1 (determinují hladiny triglyceridů) však vysvětlí maximálně 30 % z variability plazmatických lipidů. Další proporce variability je ovlivněna vzácnými polymorfizmy/mutacemi a genetickými faktory, nesouvisejícími přímo se změnami sekvence DNA (DNA metylací, modifikací histonů, regulačními RNA molekulami).
Klíčová slova:
gen – cholesterol – interakce – mutace – polymorfizmus – triglyceridy
Úvod
Za zvýšenou hladinu plazmatických lipidů (dyslipidemii – DL) považujeme hodnoty plazmatického cholesterolu > 5 mmol/l, plazmatických triglyceridů > 1,8 mmol/l a hodnoty HDL-cholesterolu < 1,0 mmol/l u mužů a 1,2 mmol/l u žen [1–3]. Tyto hodnoty jsou spíše obecným doporučením, přesná čísla nejsou překvapivě uvedena ani v dokumentech odborných společností [4], které se omezují na konstatování, že hodnoty plazmatických lipidů musí být co nejnižší a hodnoceny dle klinického stavu jedince a řeší primárně otázku, jak hluboko může cholesterol klesnout [5].
DL je považována, vedle kouření, diabetu, obezity a hypertenze, za jeden ze závažných rizikových faktorů rozvoje aterosklerózy [6]. Nicméně je třeba zdůraznit, že ačkoli vztah vysokých triglyceridů k celkové úmrtnosti byl jednoznačně prokázán [7,8], v případě hladin celkového cholesterolu je tomu tak pouze u jedinců do 5. dekády věku [9].
Analýzy studie MONICA a post-MONICA prokázaly, že v České populaci hodnoty plazmatických lipidů v posledních dekádách postupně klesají [10]. Příčin může být celá řada, ať už se jedná o změnu stravovacích zvyklostí (má vliv především na hladiny triglyceridů), zvýšenou fyzickou aktivitu, či vysokou preskripci různých dyslipidemik, převážně statinů.
Co se ale nijak nemění, je genetické „pozadí“ populace. Odhaduje se, že genetické faktory jsou odpovědné za finální hladiny plazmatických lipidů ze 40–80 % [11]. Nejvyšší odhady byly získány ze sourozeneckých studií, nejnižší pak z populačních studií.
V minulém tisíciletí byly „zlatým standardem“ pro analýzu genetického pozadí dyslipidemií/hodnot plazmatických lipidů asociační studie [12]. Geny byly vybírány primárně dle znalostí o jejich funkci – analyzovaly se geny pro lipoproteinové receptory, strukturální proteiny lipoproteinů nebo pro enzymy metabolizující triglyceridy nebo cholesterol. Genetické varianty byly rovněž selektovány dle předpokládané funkční důležitosti, např. zda měnily aminokyselinové složení proteinu, nebo zda způsobily předčasný stop kodon a podobně. Tyto studie obvykle trpěly velkým rizikem falešně pozitivních a falešně negativních výsledků (tj. replikovatelnost nálezů byla nízká), protože do nich bylo zahrnováno relativně málo analyzovaných jedinců, obvykle maximálně několik set, ale často i méně než jedno sto.
Přes tyto nedostatky byly i v této éře výzkumu dyslipidemií detekovány varianty významně a replikovatelně ovlivňující hladiny plazmatických lipidů. Jako příklad lze zmínit především gen pro apolipoprotein E (APOE), jehož 3alelický polymorfizmus je dosud jedním z nejsilnějších známých genetických determinantů hodnot plazmatických lipidů. Nositelé alely APOE2 (záměna arginin158 → cystein, rs429358, oproti nejčastěji se vyskytující alele APOE3) mají nižší a nositelé alely APOE4 (záměna cystein112 → arginin, rs7412) pak vyšší hodnoty plazmatického cholesterolu [13].
Na přelomu tisíciletí bylo několik genů objeveno pomocí komparativního sekvenování. Byly porovnány vybrané oblasti genomů několika savčích druhů a v místech s vysokou homologií byly detekovány nové, dosud nerozpoznané geny. Tímto způsobem byl objeven a popsán gen pro apolipoprotein A5 (APOA5), jehož 3 varianty (T-1131 → C, rs662799; serin19 → tryptofan, rs3135506; a glycin185 → cystein, rs2075291; detekovaná prakticky výhradně u asiatů) jsou významně spojeny s hladinami plazmatických triglyceridů [14–16].
Monogenní poruchy metabolizmu lipidů
Monogenních poruch metabolizmu lipidů je celá řada (Tangierovo onemocnění, ARH, sitosterolemie, deficience lipoproteinové nebo jaterní lipázy a další) a jejich základní charakteristikou je, že jsou způsobeny jednou mutací v jednom genu (tab. 1) [17] a až na výjimky (familiární hypercholesterolemie – FH, familiární defekt ApoB-100 – FDB) je jejich výskyt v populaci velice vzácný – pro některá onemocnění bylo popsáno pouze několik desítek jedinců na celém světě.
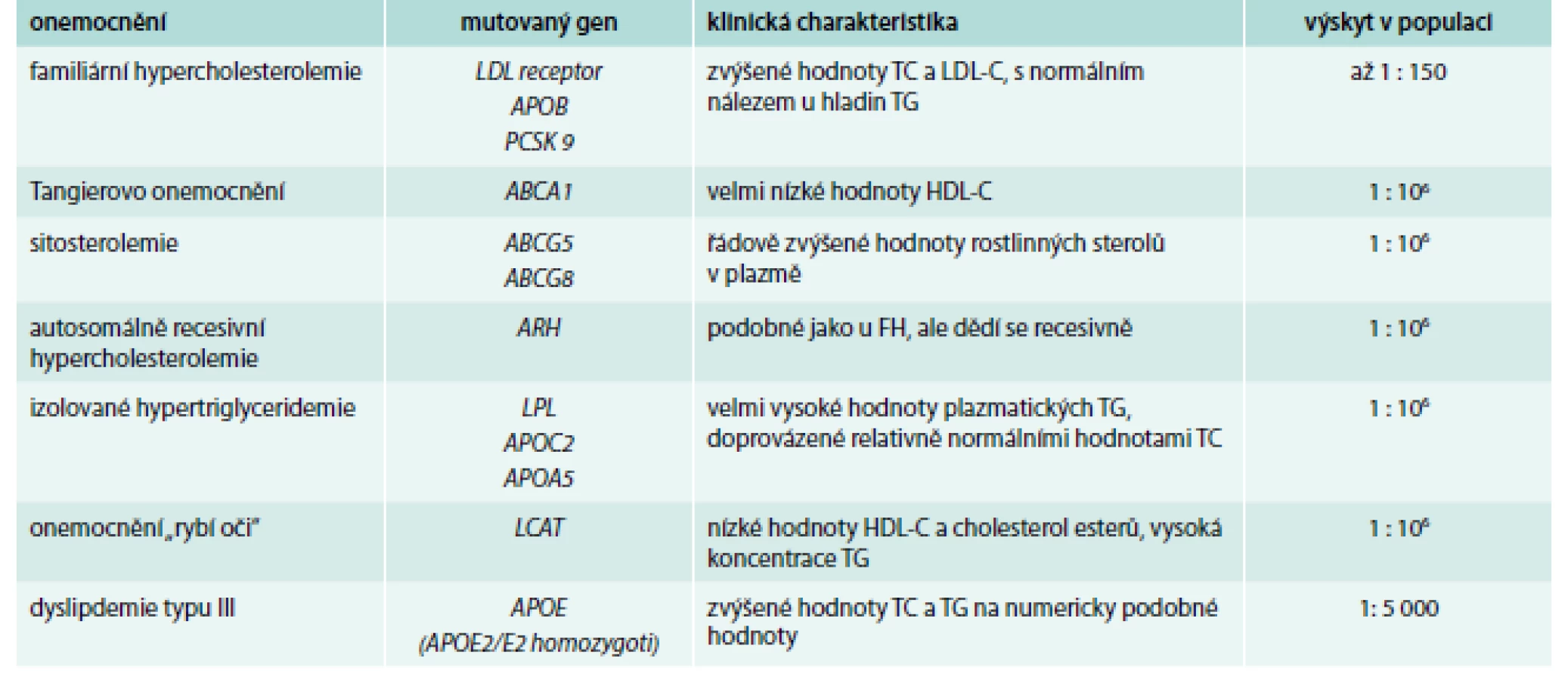
Mezi nejčastější monogenní poruchu metabolizmu lipidů patří familiární hypercholesterolemie (FH) [18]. Poslední analýzy ukazují, že výskyt tohoto onemocnění v heterozygotní formě může být i více než 1 : 150 (oproti původním odhadům 1 : 500). Heterozygoti FH mají hladiny celkového cholesterolu obvykle mezi 8–12 mmol/l, homozygoti i více než 20 mmol/l [19]. Ke snížení vysokého rizika kardiovaskulárních onemocnění v raném věku podstupují tito jedinci intenzivní dyslipidemickou léčbu, ať již farmakologickou, nebo invazivní, a dodržují dietní doporučení [20–23].
V případě FH je mutován jeden ze 3 genů, apolipoprotein B, LDL receptor, nebo, vzácně, PCSK9 (proprotein konvertáza subtilisin/kexin typ 9). Není vyloučeno, že u jedinců, u nichž nebyla detekována kauzální mutace v některém z těchto genů, bude mutován gen jiný (bezúspěšně byl screenován např. gen pro sortilin [24]). Mutací především v genu pro LDL receptor jsou popsány stovky (aktualizovaný seznam lze nalézt na https://grenada.lumc.nl/LOVD2/UCL-Heart/home.php?select_db=LDLR) a řada pacientů si nese svoji jedinečnou, výhradní mutaci, nicméně extenzivní analýzy ukázaly, že některé mutace jsou mnohem častější v určitých populacích. V české populaci tak mezi pacienty s FH (kromě mutace dříve známé jako APOB3500 v genu pro apolipoprotein B způsobující záměnu arginin3527 za glutamovou kyselinu) najdeme „často“ mutace glycin592 → kyselina glutamová a asparagin266 → kyselina glutamová v genu pro LDL receptor [25].
FH byla dlouho označována jako příkladné autosomálně dominantní onemocnění. Poznatky z posledních let však toto zařazení značně zpochybňují. Primárně se ukázalo, že řada nositelů mutací v genu pro LDL receptor má normální, zcela nepatologické hodnoty jak celkového, tak LDL-cholesterolu (Hubáček a Vrablík, připravováno k publikaci). Dále byl londýnskými kardiovaskulárními genetiky popsán fenomén „polygenní FH“ [26]. Prokázali, že kumulace dostatečného množství negativních, běžně se vyskytujících alel 12 (nebo 6, dle různých definicí) genů má za následek takové spektrum plazmatických lipidů, které je klinicky nerozlišitelné od monogenní FH (viz níže kapitola Genetické skóre).
Polygenní poruchy – celogenomové screeningy
Naprostá většina dyslipidemií však není způsobena mutací v jediném genu, ale jedná se o polygenní záležitost způsobenou řadou variant s relativně nízkým vlivem v různých genech. Jejich vliv se navíc může do určité míry lišit mezi muži a ženami a záviset na věku jedince, a varianty mohou interagovat jak mezi sebou, tak s prostředím [27]. GWAs (genome wide association study) prokázaly, že těchto variant bude méně, než se dosud předpokládalo.
Naše znalosti o genetice dyslipidemií [28] se v posledním desetiletí výrazně prohloubily díky celogenomovým scrreeningům (GWAs).
GWAs [29] jsou designovány tak, aby bylo možné, pomocí tzv. čipů, relativně snadno a relativně rychle analyzovat stovky tisíc jednonukleotidových polymorfizmů (single nucleotide polymorphisms – SNPs) na rozsáhlých skupinách jedinců. Tyto SNPs byly pro nejnovější čipy vybírány tak, aby jejich populační frekvence byla alespoň 1% (původní GWAs však používaly čipy se SNPs s populační frekvencí > 5 %) a aby pokrývaly lidský genom co nejrovnoměrněji, bez ohledu na jejich předpokládanou funkční významnost. To může vést k chybám, při nichž nejsou detekovány některé důležité polymorfizmy jako příčinné, protože nebyly k analýzám vybrány a nejsou součástí nějakého provázaného uskupení polymorfizmů (haplobloku). Vzhledem k obrovskému množství analyzovaných polymorfizmů je také stanoven extrémní požadavek na významnost výsledků na < 5 × 10–8. Aby bylo dosaženo tak významných výsledků, je nezbytně nutné zahrnout do analýz tisíce jedinců a významné výsledky dále replikovat na několika nezávislých, ale podobně rozsáhlých studiích.
Přes nepopiratelné klady GWAs je nutné zmínit i jejich nevýhody. Protože tako technologie poněkud „předběhla“ dobu, pro GWAs byly (a jsou) primárně používané velice heterogenní a rozličně analyzované studie. Pro první GWAs studie byly navíc využity prakticky výhradně studie západoevropských populací, a chybí tak informace např. o slovanských populacích, či jiných etnikách. Vysoce významný výsledek ze západoevropských populací pak nemusí být potvrzen v jiných národnostních skupinách/etnikách, jak ukázaly např. analýzy genu pro transkripční faktor MLXIPL [30–32]. Obecně lze v literatuře pozorovat nedostatek replikačních studií výsledků získaných prostřednictvím GWAs.
GWAs studie, oproti původním nereálným očekáváním, odhalily primárně varianty s relativně malým efektem na výsledné hodnoty plazmatických lipidů. Nicméně je na tomto místě třeba zdůraznit, že vliv dietních intervencí nebo fyzické aktivity na hodnoty plazmatických lipidů je nižší než v řadě jednotlivých polymorfizmů. Obvykle se jednou variantou vysvětlí okolo 5–15 % z konečné hodnoty sledovaného parametru s tím, že v případě rozsáhlých metaanalýz statisticky významně vychází i vliv variant s efektem mezi 1 a 5 %. Každá individuální varianta tak vysvětluje relativně nízkou proporci finální hladiny a je jisté, že se neobejdeme bez analýz řady variant najednou [33].
Obrovské počty zahrnutých jedinců s sebou nesou další, často opomíjené riziko. Použití klasické před-GWAs statistické analýzy ukáže na statisticky extrémně významný výsledek, který však může být z klinického a biologického hlediska zcela bezvýznamný a pozorované rozdíly jsou občas i nižší, než je obvyklá přesnost měření analyzovaného parametru.
Rovněž výběr anonymních variant, u něhož bylo primárním požadavkem rovnoměrné pokrytí genomu, má svá úskalí. Kromě již zmíněné nevýhody, že na původní čipy byly zahrnuty jen varianty s frekvencí nad 5 % v běžné populaci, je nutné podtrhnout, že ne všechny varianty vybrané pomocí GWAs reprezentují skutečně ty klíčové varianty v kauzálním genu/kauzální oblasti. Detailní analýza oblasti genu pro PCSK9 [34] kupříkladu ukázala, že ve skutečnosti není podstatnou a funkční variantou ovlivňující hladiny cholesterolu varianta detekovaná pomocí GWAs, ale s ní úzce provázaná, ale méně frekvenční varianta s téměř 4násobně silnějším efektem, která ale na čipu zahrnuta nebyla.
Nedá se očekávat, že by se seznam těchto variant v příštích letech výrazně rozšířil. Nicméně zcela jistě se objeví další vzácné varianty zvyšující riziko KVO, či varianty, které nejsou součástí rozsáhlejších haplotypů, a které tedy nebylo možné detekovat pomocí GWAs ale bude možné je objevit pomocí celogenomového nebo exomového sekvenování. Řada významných variant je uložena v genech či oblastech, jejichž přesnou funkci je třeba objasnit.
Příklady variant detekovaných GWAs s vlivem na lipidové spektrum
První celogenomové screeningy změřené na detekci variant spojených s plazmatickými lipidy byly publikovány téměř současně [35–37]. Velice záhy, s narůstajícím počtem zahrnutých subjektů, narůstaly i počty detekovaných variant s tím, že tyto varianty vykazovaly nižší a nižší vliv na plazmatické lipidy [38–40].
Většina pomocí GWAs detekovaných variant neovlivňuje pouze některý z lipidových parametrů, ale pochopitelně je spojena, i když často s rozdílným efektem, s celým lipidovým spektrem. Nejsilnější varianty ovlivňují hladiny triglyceridů až o 30 %, hladiny celkového cholesterolu nebo LDL-cholesterolu (LDL-C) o 10–15 %.
Příklady genů, jejichž varianty byly primárně nejsilněji asociované s hladinami celkového cholesterolu a LDL-C, jsou shrnuty v tab. 2.
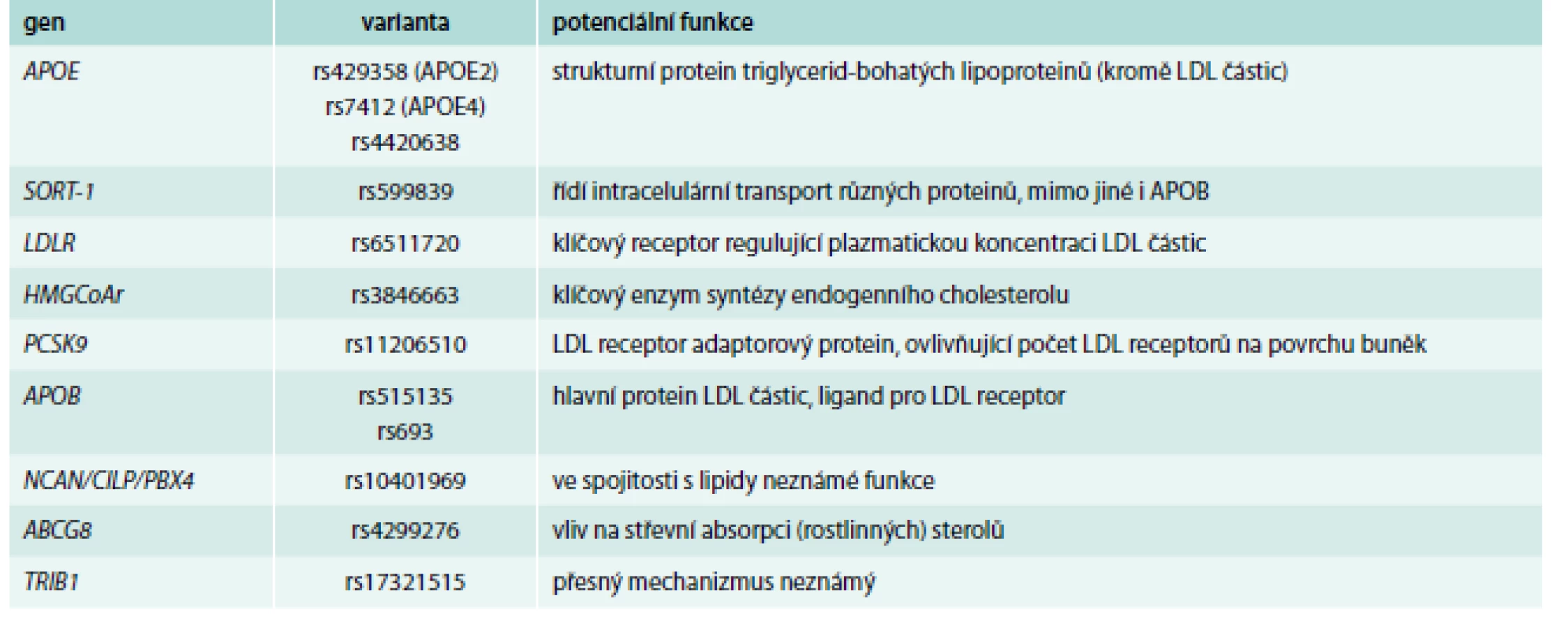
Patrně nejsilnějším genetickým determinantem zde je stále gen pro APOE, i když výsledky GWAs ukázaly i na další polymorfizmy, kromě klasického 3alelického systému. GWAs dále nalezly, dle očekávání, varianty v genech pro klíčové proteiny metabolizmu a transportu cholesterolu (triglyceridů), tedy v genech pro APOB, LDL receptor a HMGCoA reduktázu. Pomocí GWAs byly ale detekovány i další varianty, a to v genech, o jejichž úloze v metabolizmu lipidů se v době prvních výsledků prakticky nic nevědělo. Mezi ně patří např. gen pro sortilin, což je protein pomáhající transportovat APOB skrz Golgiho aparát a ovlivňující tak tvorbu LDL částic. Další varianty ukázaly na důležitost některého z genů clusteru NCAN/CILP/PBX4, ale mechanizmus ovlivnění lipidových parametrů zde jasný není.
Přestože se pomocí mendeliánských randomizačních studií [41] neprokázalo, že by HDL-C byl kauzálně spjat s kardiovaskulárním onemocněním a je jeho pouhým markerem [42], jeho hodnoty jsou stále intenzivně sledovány a diskutovány. Proto je vhodné se blíže zmínit i o genech, jejichž varianty hladiny HDL-cholesterol ovlivní (příklady viz tab. 3, s. 871).
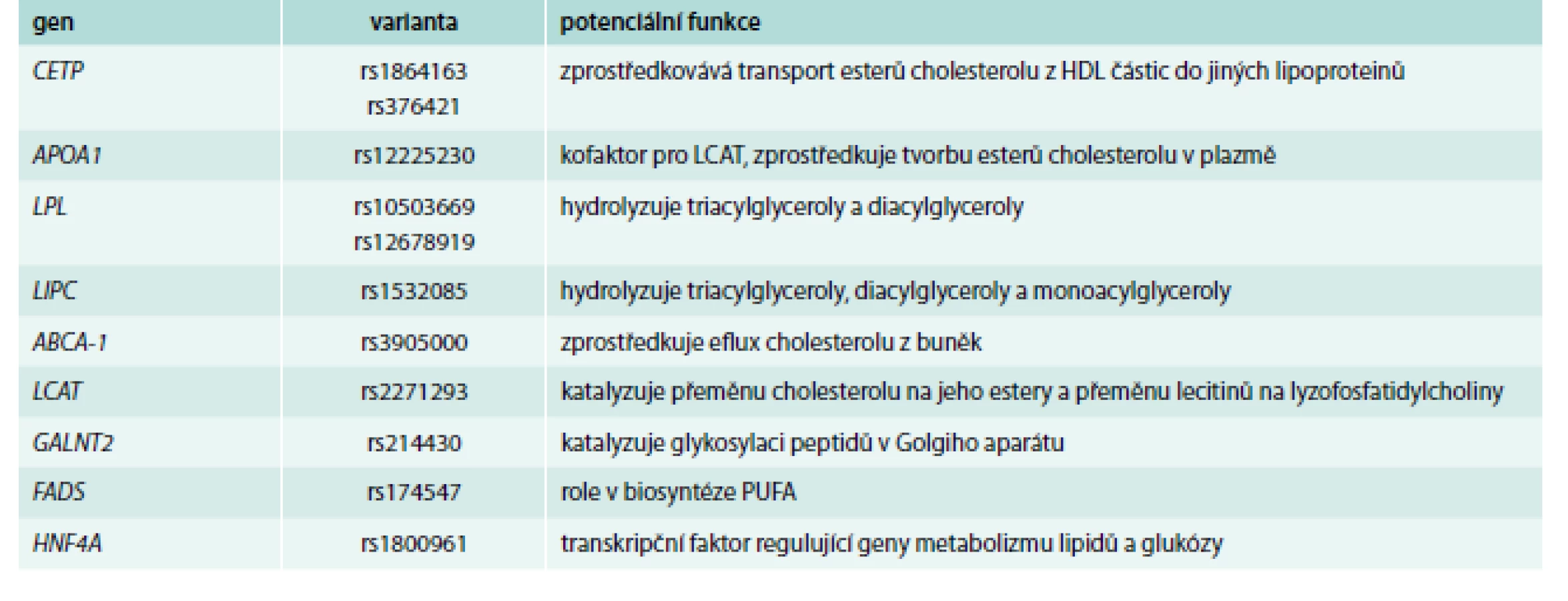
Patří mezi ně jak geny, jejichž varianty byly spojované s HDL-C již z dob asociačních studií s tím, že GWAs často popsaly nové dříve neanalyzované varianty těchto genů; tak o geny zcela nové, jako např. gen pro ANGPTL4, HNF4A nebo GALNT2 [43].
Naopak, poněkud stranou zájmu kliniků (nikoli však stranou zájmu genetiků) jsou hladiny triglyceridů (TG), které ani nebývají zahrnuty do výpočtu rizika rozvoje KVO. A možná právě zde byly objeveny varianty, které jsou silnějšími determinanty, než v případě hladin cholesterolu (tab. 4) [44–46] a jednotlivé alely ovlivní TG i z více než 15 %.
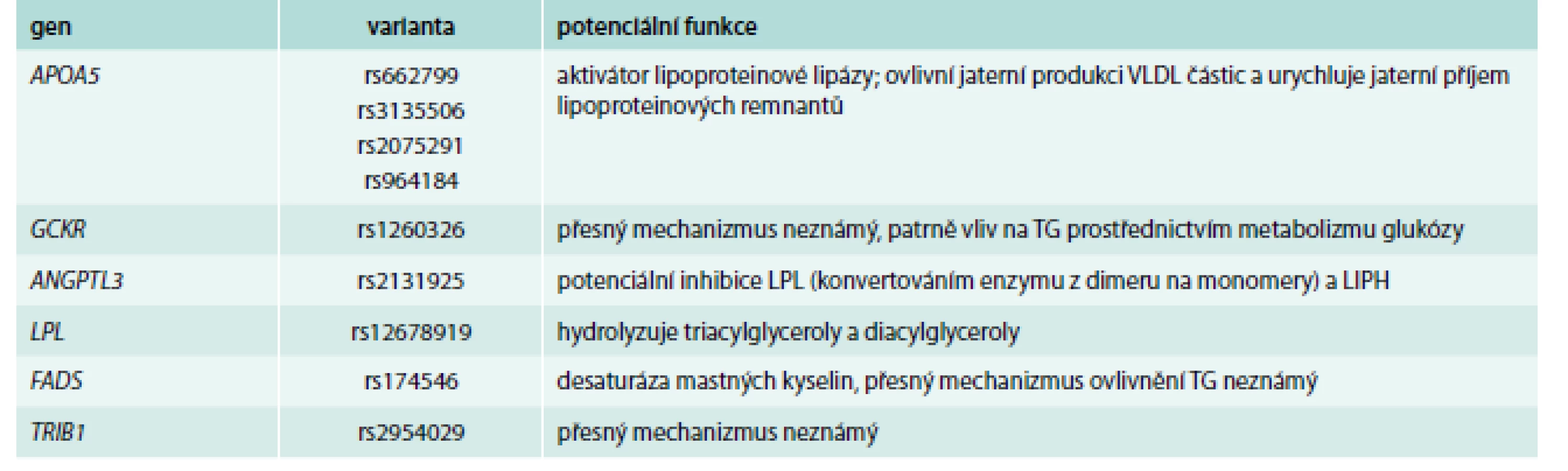
Jednoznačně nejsilnějším prediktorem plazmatických triglyceridů jsou varianty v již zmíněném genu pro apolipoprotein A5, jehož popsání však spadá do éry před GWAs [47]. Nejdůležitější variantou je zde patrně záměna T-1131 → C, každá C alela je spojena se zvýšením hladin plazmatických triglyceridů o 16 % [48]. V této rozsáhlé metaanalýze byl detekován i mírný vliv této varianty na ostatní lipidové parametry, s výjimkou LDL-C. Mezi dalšími geny s vysokou potencí ovlivnit plazmatické TG najdeme velmi dobře známou lipoproteinovou lipázu, ale také zcela nové „hráče“ jako GCKR, FRMD5, CAPN3 nebo TRIB1, u kterých dosud není znám přesný mechanizmus ovlivnění TG.
Genetické skóre
Jak již bylo řečeno, dyslipidemie jsou primárně polygenní záležitostí, což znamená, že se na jejich rozvoji podílí určitým způsobem až několik desítek polymorfizmů v řadě na sobě nezávislých genů.
Prvním příkladem práce analyzující polygenní efekt v predikci dyslipidemie může být pokus o definici tzv. polygenní familiární hypercholesterolemie (FH) [26]. Klasické FH onemocnění bývalo prezentováno jako onemocnění autosomálně dominantního charakteru, současné znalosti však hovoří proti tomuto zařazení. FH je způsobeno primárně (ne však výhradně, jak jsme se dlouhou dobou domnívali) mutacemi v LDL receptoru, nebo v jeho ligandu, apolipoproteinu B.
Současné poznatky ukazují, že existuje mnoho jedinců, u nichž je klinicky definována FH, avšak bez přítomnosti kauzální mutace. Právě analýza několika běžných, ale pro hladinu LDL-C zásadních variant v genech pro SORT1, PCSK9, LDL receptor, APOB, APOE, HMGCoA reduktázu, ABCG5/ABCG8 transportéry, HFE a ST3GAL4, nalezla výrazné rozdíly ve frekvencích rizikových alel těchto genů mezi pacienty a kontrolami. O tom, kolik přesně a jaké konkrétně SNPs v kterých genech je vhodné v tomto případě analyzovat, se dosud vedou spory, počty se pohybují od 6 do 33 variant [26,49].
V případě analýzy polygenního skóre v běžné populaci [50], využívajícího 20 SNPs, se ukázalo, že jedinci nesoucí maximálně 22 „negativních“ alel, mají v průměru o 1,5 mmol/l nižší hodnoty LDL-cholesterolu než jedinci s alespoň 33 „negativními“ alelami.
Vyšší počty SNPs pro analýzu prediktivního genetického skóre byly použity v případě hypertriglyceridemie. Zde je v návrhu panelu více než 30 SNPs, mezi nimi např. varianty v genech pro APOA5, LPL, ANGPTL3, MLXIPL, GCKR, FRMD5, CILP2, nebo TRIB1. I zde se dá očekávat snížení „potřebných“ variant, protože některé z nich ovlivní variabilitu plazmatických triglyceridů i z méně než z 1,5 % [44,45].
Vzhledem k počtu zahrnutých genů a potenciálnímu počtu negativních a pozitivních alel je pochopitelné, že prakticky nelze nalézt jedince nesoucí pouze negativní, nebo naopak pouze pozitivní alely.
Protokoly dosud publikovaných studií využívajících genetické skóre používají v zásadě 2 přístupy. Buď se riziko počítá z prosté přítomnosti negativních nebo pozitivních alel anebo se používá tzv. váženého skóre, při jehož užití je nutné nejdříve určit významnost jednotlivých variant, a tyto pak nemají při výpočtu finálního rizika identickou váhu. V obou případech je problém, že tento výpočet rizika nebere v úvahu potenciální interakce mezi jednotlivými variantami (a již vůbec ne interakce s prostředím), v nichž může dojít k multiplikaci rizika, nebo naopak k jeho nelineárnímu snížení.
Obecně lze uzavřít, že použití genetických skóre v predikci rizika rozvoje dyslipidemií je určitě krokem vpřed a mohlo/mělo by být používáno v odhadu rizika těchto onemocnění. Jak bylo zmíněno výše, bude třeba vytvořit etnicky a populačně specifické panely polymorfizmů, protože ne všechny varianty mají stejný nebo alespoň podobný vliv bez ohledu na geografickou lokalizaci a vnější vlivy okolního prostředí, což bude vyžadovat provedení celé řady replikačních studií.
Další směry výzkumu genetiky dyslipidemií v éře po GWAs studiích
Primárním úkolem současných genetických studií by mělo být replikovat nálezy původních GWAs studií na všech populacích a etnikách, protože ne všechny varianty mají univerzální celosvětový vliv. Příčiny rozdílů mezi populacemi mohou být způsobeny rozličnými faktory, ať už se jedná o vnější prostředí, či celkové genetické charakteristiky populací.
Do GWAs studií byly zahrnuty SNPs charakterizující určité oblasti genomu. Některé studie se soustředily na podrobnou sekvenci nově objevených oblastí, což vedlo k objevu variant se silnějším vlivem, než měly varianty původně anotované [34,51,52].
Se stále snižujícími náklady na genetické analýzy je stále častěji využívána i možnost jak exomového, tak celogenomového sekvenování. Přes nesporné klady těchto studií (objev zcela nových variant ovlivňujících zásadně lipidový metabolizmus) je právě extrémní množství informací získané z DNA analyzovaných jedinců komplikací. U každého jedince mohou být detekovány i tisíce nových, jedinečných mutací a bývá velice obtížné zjistit, které z nich jsou skutečně kauzální [53] a které zcela bezvýznamné.
Podobně mohou k objevu dalších „nových“ genů stále přispět i studie rodinné, od nichž ale nelze očekávat explozi nových nálezů použitelných v populační klinické praxi.
Překvapivě je stále velice málo studií za měřených na interakce mezi geny a prostředím, především dietou a fyzickou aktivitou, přestože je jasné, že právě tyto interakce budou pro výsledné hladiny plazmatických lipidů kruciální [28,54].
Klinické využití poznatků z GWAs studií
Přes rozsáhlý prediktivní potenciál jsou bohužel poznatky z GWAs studií využívány v klinické lipidologii (a kardiologii obecně) pouze minimálně.
Velký potenciál přitom nabízí genetické analýzy především v oblasti „monogenních“ onemocnění, jako je FH, u nichž je možné jednoduše a jednou pro vždy i s pomocí neinvazivního (a korespondenčně zaslaného) vzorku detekovat přítomnost kauzálních mutací v genech pro LDL receptor, APOB nebo PCSK9 a vhodnost/nutnost dalšího klinického vyšetření a případné léčby. Podobný potenciál má analýza genetických skóre jak v případě polygenní pseudo-FH a nebo polygenní dyslipidemie.
Epigenetické regulace a hladiny plazmatických lipidů
Pod pojem epigenetika zahrnujeme tu část dědičnosti, která nevychází pouze a výhradně z pořadí a záměn nukleotidů v sekvenci DNA a kterou můžeme do určité míry ovlivnit naším životním stylem (kouřením, cvičením, výživou apod.), anebo kterou nějak ovlivní životní prostředí okolo nás (průmyslové znečištění životního prostředí, podnební pás, v němž dotyčný jedinec žije apod) [55]. Zjednodušeně můžeme za první vizionářské zmínky o epigenetice považovat dosud striktně odmítané učení P. B. Lamarcka, který předpokládal existenci biologických podkladů pro nenáhodné, na prostředí závisející a z generace na generaci přenosné změny fenotypu, či nedávno zmíněnou teorii tzv. „memů“ [56].
Mezi epigenetické mechanizmy zahrnujeme primárně metylace DNA a modifikace nukleosomálních histonů (acytelyce, metylace a další) [57].
DNA metylace je významný epigenetický modifikátor. Hraje významnou roli v genomovém imprintingu, regulaci embryogeneze a ochraně genomu před nebezpečnou aktivitou transponovatelných elementů. Ze 4 nukleotidů vyskytujících se v DNA mohou být 2 metylovány, ale ve skutečnosti je metylován téměř výhradně pouze cytozin, a to často v oblastech s vysokým výskytem dinukleotidových CG opakování (o celkové délce alespoň 200 bp a s obsahem G a C vyšším než 50 %) obecně jde především o oblasti centromer, repetitivních sekvencí a transpozonů.
S narůstajícím věkem (a v závislosti na řadě faktorů vnějšího prostředí) se metylace DNA spíše snižuje, ale neplatí to obecně pro všechny oblasti/geny. Jsou rovněž pozorovány pohlavní rozdíly a DNA metylace je vyšší u mužů než u žen.
Metylace modifikuje dostupnost DNA sekvence pro přepis do RNA, a v případě, že regulační části DNA jsou odlišně metylované, mají tyto úpravy za následek rozdíly v transkripci genů. Je známo, že DNA metylace není stabilní, ale může být ovlivněna faktory životního prostředí, např. kouřením nebo stravovacími návyky. Studie dvojčat ukázaly, že jejich DNA metylace jsou téměř totožné v raném věku a v případě, že sourozenci v dospělosti žijí odděleně v jiných vnějších podmínkách, podobnost jejich DNA metylace se snižuje [58]. Řada studií, jak experimentálních, tak epidemiologických prokázala, že nutriční deficit během těhotenství, kojení a časného vývoje jedinců má negativní konsekvence (zprostředkované právě DNA metylací) na vývoj onemocnění i o řadu desítek let později.
Byly opakovaně popsány rozdíly v celkové metylaci DNA mezi pacienty s KVO a zdravými kontrolami [59,60]. I v případě již zmíněného onemocnění FH byly pozorovány v porovnání se zdravými jedinci odlišnosti jak v hypometylaci, tak v hypermetylaci určitých genomových oblastí [61], tak v expresi různých genů [62], ale je třeba tyto výsledky replikovat na dalších nezávislých populacích. V případě polygenních dyslipidemií jsou studie zaměřené na metylaci DNA ojedinělé a byly do nich zahrnuty pouze desítky jedinců, je tedy předčasné dělat jakékoli závěry.
Pod pojmem modifikace histonů rozumíme jejich acetylaci, fosforylaci, metylaci, sumoylaci a ubikvitinizaci. Modifikace primárně mění náboj histonů a „těsnost“ jejich vazby s molekulou DNA. Modifikace histonů má podobné důsledky jako metylace DNA. Změna konformace histonů pak ovlivní expresi jednotlivých genů podobně jako metylace DNA [61].
V poslední dekádě je intenzivně studována regulační úloha malých RNA molekul a dlouhých nekódujících molekul RNA [63,64]. Mezi nekódující RNA (ncRNA) patří krátké RNA (obecně < 50 bp); např. mikroRNA (miRNA), malé interferující RNA (siRNA) a dlouhé nekódující RNA (lncRNA) s délkou přes 200 bp.
MiRNA [65] jsou vysoce konzervované a extrémně stabilní krátké molekuly RNA (obsahují obvykle 20–22 nukleotidů), které post-transkripčně modulují genovou expresi. Tyto RNA se vážou obvykle k jejich komplementárním sekvencím v 3‘ nepřekládaných oblastech (3‘UTRs) mediátorové RNA (mRNA). V současnosti známe okolo 2 000 miRNA a jejich počet stále narůstá. MiRNA jsou funkční primárně intracelulárně (kde jejich koncentrace řádově vyšší než v mezibuněčném prostoru), ale z tkáně orgánů jsou za různých podmínek uvolňovány do prakticky všech extracelulárních tekutin, především do séra a moči.
Byla popsána řada miRNA jejichž koncentrace hrají roli v udržení homeostázy cholesterolu (miRNA-33a/b, miRNA-122, miRNA-370, miRNA-27a, miRNA-185/342), viz např. Dlouhá a Hubáček, připravováno k publikaci.
Mezi „dlouhé“ nekódující RNA (long-non coding RNA – lncRNA) řadíme molekuly obsahující alespoň 200 nukleotidů. LncRNA obecně ovlivňují expresi cílových genů identickým mechanizmem jako miRNA, tedy navázáním na cílovou regulační sekvenci, a to obvykle ne pouze jednoho, ale celé řady genů.
Závěr
Celogenomové screeningy potvrdily vliv řady genů v determinaci plazmatických lipidů. Pomohly současně objevit i několik zcela nových genů a důležitých proteinů (sortilin), jejichž ovlivnění může být cílem v případě hledání nových léčiv. V jiných případech dosud přesně nevíme, jakým mechanizmem je lipidový metabolizmus genem ovlivněn, i když jeho vliv je nepopiratelný. Podrobnější analýzy detekovaných lokusů pomohly nalézt varianty s nejsilnějším vlivem, přesto prozatím neznáme ani polovinu z předpokládané genetické determinace dyslipidemií. Další analýzy tak budou velice pravděpodobně zaměřeny na sestavování genetických skóre, analýzy gen – prostředí interakcí či na analýzy mezigenových interakcí a konečně na epigenetické determinanty (metylace DNA, modifikace histonů a regulační RNA molekuly).
Podpořeno programovým projektem Ministerstva zdravotnictví ČR s reg. č. 15–28876A. Veškerá práva podle předpisů na ochranu duševního vlastnictví jsou vyhrazena.
Ing. Jaroslav Alois Hubáček, CSc., DSc.
jahb@ikem.cz
Centrum experimentální medicíny IKEM,
Praha
www.ikem.cz
Doručeno do redakce 9. 9. 2016
Přijato po recenzi 27. 10. 2016
Sources
1. Vítek L. Je cholesterol „good“ nebo opravdu „bad and ugly“? Hyper Kardiovask Prevence 2015; 4(1): 64–65.
2. Soška V, Vaverková H, Vráblík M et al. Stanovisko výboru ČSAT k doporučením ESC/EAS pro diagnostiku a léčbu dyslipidemií z roku 2011. Vnitř Lék 2013; 59(2): 120–126.
3. Ledford H. Cholesterol limits lose their lustre. Nature 2013; 494(7438): 410–411. Dostupné z DOI: <http://dx.doi.org/10.1038/494410a>. Erratum in Nature 2013; 495(7440): 155.
4. Windler E, Zyriax BC. Cholesterol-lowering therapy: Old evidence, new guidelines – Which one to follow? A critical appraisal. Atheroscler Suppl 2015; 18 : 176–179. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosissup.2015.02.027>.
5. Vrablík M. Jak hluboko má cholesterol klesnout? Hypert Kardiovask Prevence 2014; 3(2): 32–36.
6. Češka R. Ateroskleróza, lipidy a interní medicína. Vnitř Lék 2015; 61(11): 919.
7. Liu J, Zeng FF, Liu ZM et al. Effect of blood triglycerides on cardiovascular and all-cause mortality – a systematic review and meta-analysis of 61 prospective studies. Lipids Health Dis 2013; 12 : 159. Dostupné z DOI: <http://dx.doi.org/10.1186/1476–511X-12–159>.
8. Pikhart H, Hubáček JA, Persey A et al. Association between fasting plasma triglycerides, all-cause and cardiovascular mortality in Czech population. Results from the HAPIEE study. Physiol Res 2015; 64(Suppl 3): S355-S361.
9. Hubáček JA. Vysoký cholesterol není spojen s vyšší celkovou úmrtností. Hyper Kardiovask Prevence 2015; 4(1): 58–62.
10. Bruthans J, Cífková R, Lánská V et al. Explaining the decline in coronary heart disease mortality in the Czech Republic between 1985 and 2007. Eur J Prev Cardiol 2014; 21(7): 829–839. Dostupné z DOI: <http://dx.doi.org/10.1177/2047487312469476>.
11. Roberts R. Genetic of premature myocardial infarction. Curr Atheroscler Rep 2008; 10(3): 186–193.
12. Foulkes AS. Applied statistical genetics. For population-based association studies. Use R. Springer Science+Business Media LLC: 2009. ISBN 978–0-387–89554–3_1. Dostupné z DOI: <http://dx.doi.org/10.1007/978–0-387–89554–3_1>
13. Hubáček JA, Poledne R. Apolipoprotein E a jeho role v lipidovém metabolismu, kardiovaskulárních onemocněních a Alzheimerově chorobě. Prakt Lék 1998; 78(4): 162–165.
14. Hubacek JA. Apolipoprotein A5 and triglyceridemia. Focus on the effects of the common variants. Clin Chem Lab Med 2005; 43(9): 897–902.
15. Pennacchio LA, Olivier M, Hubacek JA et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 2001; 294(5540): 169–173.
16. Pennacchio LA, Olivier M, Hubacek JA et al. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet 2002; 11(24): 3031–3038.
17. Ramasamy I. Update on the molecular biology of dyslipidemias. Clin Chim Acta 2016; 454 : 143–185. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cca.2015.10.033>.
18. Vrablík M, Schwarzová L, Freiberger T et al. Familiární hypercholesterolemie: klinické nálezy, molekulární genetika a diferenciální diagnostika. Ather Rew 2016; 1(1): 21–29.
19. Češka R, Vrablík M, Altschmiedová T et al. Familiární hypercholesterolemie včera a dnes. Vlastní zkušenosti a nálezy u našeho souboru nemocných s familiární hypercholesterolemií. Vnitř Lék 2014; 60(11): 963–969.
20. Soška V, Vrablík M, Bláha V et al. PCSK9 inhibitory – nové možnosti v léčbě hypercholesterolemie: u koho budou indikovány? Stanovisko České společnosti pro aterosklerózu. Vnitř Lék 2016; 62(4): 329–333.
21. Pitha J. Dietní zvyklosti a kardiovaskulární onemocnění. Vnitř Lék 2015; 61(9): 767–768.
22. Bláha V, Bláha M, Lánská M et al. Postavení lipoproteinové aferézy v současnosti. Vnitř Lék 2015; 61(11): 958–964.
23. Bláha V, Bláha M, Lánská M et al. LDL-aferéza v léčbě familiárních hyperlipoproteinemií. Vnitř Lék 2014; 60(1): 970–976.
24. Tveten K, Strøm TB, Cameron J et al. Mutations in the SORT1 gene are unlikely to cause autosomal dominant hypercholesterolemia. Atherosclerosis 2012; 225(2): 370–375. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2012.10.026>.
25. Tichý L, Freiberger T, Zapletalová P et al. The molecular basis of familial hypercholesterolemia in the Czech Republic: spectrum of LDLR mutations and genotype-phenotype correlations. Atherosclerosis 2012; 223(2): 401–408. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2012.05.014>.
26. Talmud PJ, Shah S, Whittall R et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013; 381(9874): 1293–1301. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(12)62127–8>.
27. Rosenthal E, Blue A, Jarvik GP. Next-generation gene discovery for variants of large impact on lipid traits. Curr Opin Lipidol 2015; 26(2): 114–119. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000156>.
28. Hubáček JA, Vrablík M. Genetika dyslipidemií – včera, dnes a zítra. Vnitř Lék 2007; 53(4): 371–376.
29. Uitterlinden AG. An introduction to Genome-wide association studies: GWAS for dummies. Semin Reprod Med 2016; 34(4): 196–204. Dostupné z DOI: <http://dx.doi.org/10.1055/s-0036–1585406>.
30. Kooner JS, Chambers JC, Aguilar-Salinas CA et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet 2008; 40(2): 149–151. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.2007.61>.
31. Vrablik M, Ceska R, Adamkova V et al. MLXIPL variant in individuals with low and high triglyceridemia in white population in Central Europe. Hum Genet 2008; 124(5): 553–555. Dostupné z DOI: <http://dx.doi.org/10.1007/s00439–008–0577–6>.
32. Nakayama K, Bayasgalan T, Yamanaka K et al. Large scale replication analysis of loci associated with lipid concentrations in a Japanese population. J Med Genet 2009; 46(6): 370–374. Dostupné z DOI: <http://dx.doi.org/10.1136/jmg.2008.064063>. Erratum in J Med Genet 2009; 46(12): 861.
33. Swerdlow DI, Holmes MV, Harrison S et al. The genetic of coronary heart disease. Br Med Bull 2012; 102 : 59–77. Dostupné z DOI: <http://dx.doi.org/10.1093/bmb/lds009>.
34. Sanna S, Li B, Mullas A et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double explained heritability. PLoS Genet 2011; 7(7): e1002198. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pgen.1002198>.
35. Kathiresan S, Melander O, Guiducci C et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 2008; 40(2): 189–197. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.75>.
36. Sandhu MS, Waterworth DM, Debenham SL et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet 2008; 371(9611): 483–491. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(08)60208–1>.
37. Willer CJ, Sanna S, Jackson AU et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008; 40(2): 161–169. 40(2): 161–9. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.76>.
38. Kathiresan S, Willer CJ, Peloto GM et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 2009; 41(1): 56–65. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.291>.
39. Aulchenko YS, Ripatti S, Lindquist I et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet 2009; 41(1): 47–55. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.269>.
40. Teslowich TM, Musunuru K, Smith AV et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466(7307): 707–713. Dostupné z DOI: <http://dx.doi.org/10.1038/nature09270>.
41. Swerdlow DI, Hingorani AD, Humphries SE. Genetic risk factors and Mendelian randomization in cardiovascular disease. Curr Cardiol Rep 2015; 17(5): 33. Dostupné z DOI: <http://dx.doi.org/10.1007/s11886–015–0584-x>.
42. Haase CL, Tybjærg-Hansen A, Qayyum AA et al. LCAT, HDL cholesterol and ischemic cardiovascular disease: a Mendelian randomization study of HDL cholesterol in 54,500 individuals. J Clin Endocrinol Metab 2012; 97(2): E248-E256. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2011–1846>.
43. Weissglas-Volkov D, Pajukanta P. Genetic causes of high and low serum HDL-cholesterol. J Lipid Res 2010; 51(8): 2032–2057. Dostupné z DOI: <http://dx.doi.org/10.1194/jlr.R004739>.
44. Johansen CT, Hegele RA. The complex genetic basis of plasma triglycerides. Curr Atheroscler Rep 2012; 14(3): 227–234. Dostupné z DOI: <http://dx.doi.org/10.1007/s11883–012–0243–2>.
45. Hegele RA, Ginsberg HN, Chapman MJ et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol 2014; 2(8): 655–666. Dostupné z DOI: <http://dx.doi.org/10.1016/S2213–8587(13)70191–8>.
46. Schwarzova L, Hubacek JA, Vrablik M. Genetic predisposition to human plasma triglyceride concentration. Physiol Res 2015; 64(Suppl 3): S341-S354.
47. Hubacek JA. Apolipoprotein A5 fifteen years anniversary. Lessons from genetic epidemiology. Gene 2016; 592(1): 193–199. Dostupné z DOI: <http://dx.doi.org/10.1016/j.gene.2016.07.070>.
48. Sarwar N, Sandhu MS, Ricketts SL et al. [Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration]. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet 2010; 375(9726): 1634–1639. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(10)60545–4>. Erratum in: Lancet 2010; 376(9735): 90.
49. Futema M, Shah S, Cooper JA et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem 2015; 61(1): 231–238. Dostupné z DOI: <http://dx.doi.org/10.1373/clinchem.2014.231365>.
50. Talmud PJ, Drenos F, Shah S et al. Gene-centric association signals for lipid and apolipoproteins identified via the Human CVD Bead Chip. Am J Hum Genet 2009; 85(5): 628–642. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ajhg.2009.10.014>.
51. Willer CJ, Mohlke KL. Finding genes and variants for lipid levels after genome-wide association analysis. Curr Opin Lipidol 2012; 23(2): 98–103. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0b013e328350fad2>.
52. Willer CJ, Schmidt EM, Sengupta S et al. [Global Lipids Genetics Consortium]. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013; 45(11): 1274–1283. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.2797>.
53. Neřoldová M, Stránecký V, Hodaňová K et al. Rare variants in known and novel candidate genes predisposing to statin-associated myopathy. Pharmacogenomics 2016; 17(13): 1405–1414. Dostupné z DOI: <http://dx.doi.org/10.2217/pgs-2016–0071>.
54. Garcia-Rios A, Perez-Martinez P, Delgado-Lista J et al. Nutrigenetics of the lipoprotein metabolism. Mol Nutr Food Res 2012; 56(1): 171–183. Dostupné z DOI: <http://dx.doi.org/10.1002/mnfr.201100513>.
55. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013; 153(1): 56–69. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cell.2013.03.004>.
56. Blackmoreová S, Dawkins R. Teorie memů: kultura a její evoluce. Portál: Praha 2001. ISBN 80–7178–394–3.
57. Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA 2008; 299(11): 1345–1350. Dostupné z DOI: <http://dx.doi.org/10.1001/jama.299.11.1345>.
58. Yet I, Tsai PC, Castillo-Fernandez JE et al. Genetic and environmental impacts on DNA methylation levels in twins. Epigenomics 2016; 8(1): 105–117. Dostupné z DOI: <http://dx.doi.org//10.2217/epi.15.90>.
59. Sharma P, Kumar J, Garg G et al. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol 2008; 27(7): 357–365. Dostupné z DOI: <http://dx.doi.org/10.1089/dna.2007.0694>.
60. Zhang Y, Zeng C. Role of DNA methylation in cardiovascular diseases. Clin Exp Hypert 2016; 38(3): 261–267. Dostupné z DOI: <http://dx.doi.org/10.3109/10641963.2015.1107087>.
61. Muka T, Koromani F, Portilla E et al. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int J Cardiol 2016; 212 : 174–183. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2016.03.062>.
62. Chen H, Wang L, Jiang J. Transcriptome and miRNA network analysis of familial hypercholesterolemia. Int J Mol Med 2014; 33(3): 670–676. Dostupné z DOI: <http://dx.doi.org/10.3892/ijmm.2013.1610>.
63. Najafi-Shoushtari SH. MicroRNAs in cardiometabolic disease. Curr Atheroscler Rep 2011; 13(3): 202–207. Dostupné z DOI: <http://dx.doi.org/10.1007/s11883–011–0179-y>.
64. Kaikkonen MU, Lam MT, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res 2011; 90(3): 430–440. Dostupné z DOI: <http://dx.doi.org/10.1093/cvr/cvr097>.
65. Novák J, Souček M. MikroRNA a vnitřní lékařství: od patofyziologie k novým diagnostickým a terapeutickým postupům. Vnitř Lék 2016; 62(6): 477–485.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 11
Most read in this issue
- Dieta při dyslipidemii a metabolickém syndromu
- Hyperlipoproteinemie u dětí
- Hyperlipoproteinemie a dyslipidemie jako vzácná onemocnění: diagnostika a léčba
- Kyselina močová jako rizikový faktor kardiovaskulárních onemocnění