
Přínos magnetické rezonance pro diagnostiku kardiomyopatií a myokarditidy (2. část)
The benefit of magnetic resonance for diagnosing cardiomyopathy and myocarditis
Magnetic resonance is becoming an increasingly used examination in cardiology, since it greatly improves the accuracy of diagnosing of many heart diseases. At present magnetic resonance is the gold standard in assessing the volumes of the heart chambers and the systolic function of both ventricles. The possibility of detecting tissue characteristics to refine the diagnostics of different types of myocardial pathology is of essential importance. The authors summarize in the article the present knowledge about the use of magnetic resonance of the heart in the field of myocardial disease, i.e. cardiomyopathy and myocarditis. In the first of this article, a general overview of cardiac magnetic resonance examination has been given, followed by detailed description of its usefulness in dilated cardiomyopathy and myocarditis, in hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. The second part of the review summarizes the benefits of cardiac magnetic resonance examination in cardiac amyloidosis and other less common cardiomyopathies.
Key words:
fibrosis – cardiomyopathy – magnetic resonance – myocarditis – late contrast agent saturation
Authors:
Michal Fikrle 1,2; Petr Kuchynka 2; Martin Mašek 3; Jana Podzimková 2; Jan Kuchař 2; Aleš Linhart 2; Tomáš; Paleček 2
Published in:
Vnitř Lék 2016; 62(12): 976-984
Category:
Reviews
První část článku byla uveřejněna ve Vnitř Lék 2016; 62(10): 795–803.
Overview
Magnetická rezonance se stává v kardiologii stále více využívaným vyšetřením, neboť nabízí zásadní zpřesnění diagnostiky mnoha srdečních onemocnění. V současnosti představuje magnetická rezonance zlatý standard pro hodnocení objemů srdečních oddílů a systolické funkce obou komor. Zcela zásadní je možnost tkáňové charakteristiky zpřesňující diagnostiku řady myokardiálních patologií. V tomto článku autoři předkládají souhrn současných poznatků o využití magnetické rezonance srdce na poli onemocnění myokardu, tj. kardiomyopatií a myokarditidy. V prvé části sdělení byla nejprve obecně rozebrána problematika vyšetření magnetickou rezonancí u myokardiálních chorob s následným podrobným rozborem přínosu této zobrazovací metody u dilatační kardiomyopatie a myokarditidy, u hypertrofické kardiomyopatie a arytmogenní kardiomyopatie pravé komory. Nyní předkládaná druhá část přehledného článku je věnována přínosu vyšetření magnetickou rezonancí u srdeční amyloidózy a dalších, méně častých kardiomyopatií.
Klíčová slova:
fibróza – kardiomyopatie – magnetická rezonance – myokarditida – pozdní sycení kontrastní látkou
Nonkompaktní kardiomyopatie
Nonkompaktní kardiomyopatie (left ventricular noncompaction cardiomyopathy – LVNC) je pravděpodobně způsobena poruchou embryogeneze srdeční stěny a je definována výraznou trabekulizací volné stěny levé komory s hlubokými intertrabekulárními recesy. Fenotyp LVNC bývá ale současně někdy nalézán i u nemocných s jinými kardiomyopatiemi (např. hypertrofická kardiomyopatie – HKMP), u vrozených vývojových srdečních vad, či u jiných komplexních syndromů postihujících srdce. Nezodpovězenou otázkou tak zatím zůstává, zda je LVNC pouze samostatným onemocněním, či spíše jen určitým fenotypovým obrazem sdíleným různými srdečními onemocněními [90]. Fenotyp LVNC se patrně též může manifestovat i v odpovědi na zvýšenou mechanickou zátěž, jakou může být např. těhotenství či extrémní dlouhodobá sportovní aktivita [91,92]. Není však zcela jasné, zda v těchto případech nedochází k hypertrabekulizaci jen u jedinců s genetickým podkladem pro LVNC. Popsané abnormality charakteristické pro LVNC – hypertrabekulizace, hluboké intertrabekulární recesy a relativní či absolutní ztenčení kompaktní myokardiální vrstvy – se nejčastěji nacházejí v apikálních a midventrikulárních oblastech spodní a laterální stěny. Přední stěna a především septum bývají postiženy v menšině případů, čehož lze využít v diferenciální diagnostice vůči normálnímu nálezu zvýrazněné trabekulizace levé komory u zdravých jedinců [93].
Klinický průběh LVNC může být asymptomatický či dochází k postupující remodelaci levé komory doprovázené její systolickou dysfunkcí a manifestující se srdečním selháním, arytmiemi či tromboembolickými příhodami [94]. Diagnóza LVNC je výhradně postavena na zobrazovacích metodách. Bylo publikováno několik echokardiografických kritérií, z nichž žádné se dosud nestalo jasným zlatým standardem. Autority v dané oblasti považují dnes za nejdůležitější původní Jenniho kritérium poměru síly nonkompaktní vrstvy ke kompaktní vrstvě nad 2,0 v end-systole při hodnocení v parasternální krátké ose [95]. Současně je pomocí barevného dopplerovského mapování patrný tok krve do intertrabekulárních recesů.
Magnetická rezonance posuzuje detailněji než echokardiografie hůře zobrazitelné segmenty levé komory, především oblast hrotu, na němž je postižení často nejvíce manifestní. Za signifikantní pro diagnózu LVNC je na základě práce Petersena et al v současnosti brán poměr nonkompaktní vrstvy ke kompaktní nad 2,3; hodnocení probíhá v end-diastole [96] (obr. 8). Recentně se objevily i další práce snažící se definovat přesnější CMR kritéria (cardiac magnetic resonance) hodnocení LVNC. Francouzští autoři navrhují porovnávat souhrnnou hmotnost trabekul vůči mase celého levokomorového myokardu: hodnota přesahující 20 % s vysokou senzitivitou i specificitou svědčí pro LVNC [97]. Jiný přístup v CMR diagnostice nonkompakce zvolili angličtí autoři, kteří využili k popisu komplexních a nepravidelných trabekul tzv. fraktální analýzu [98]. Jedná se o matematický postup využívající popis komplexity útvarů pomocí tzv. fraktální dimenze, což je bezrozměrná míra komplexity obrazců. S narůstající strukturální komplexitou útvarů narůstá i hodnota této fraktální dimenze. Tato metoda prokázala signifikantně větší komplexnost trabekul u LVNC ve srovnání se zdravými kontrolami, při větší přesnosti a s lepší reproducibilitou než dosavadní CMR kritéria nonkompakce. Dosud největší soubor pacientů s LVNC vyšetřených pomocí CMR byl analyzován ve studii Dawsonové et al [99]. V této práci je navrhován poměrně robustní postup pro stanovení pravděpodobnosti přítomnosti LVNC na základě hodnocení poměrů end-diastolických rozměrů nonkompaktní a kompaktní vrstvy integrovaně v bazálních, středních a apikálních segmentech levé komory.
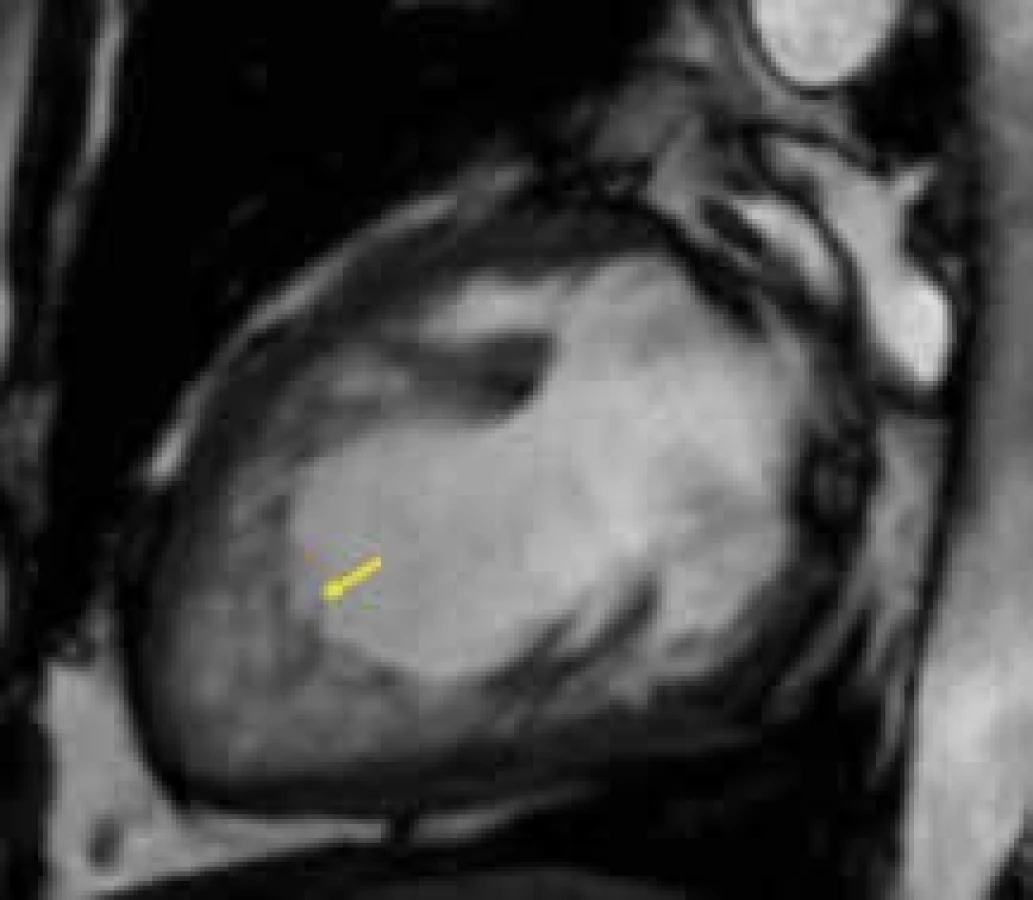
Kromě samotné diagnostiky LVNC je provedení CMR u této kardiomyopatie též vhodné u nemocných obtížně vyšetřitelných k přesnému posouzení velikosti a systolické funkce levé komory a vyloučení přítomnosti intrakavitární trombózy.
Amyloidóza
Amyloidóza je onemocnění charakterizované ukládáním patologického bílkovinného substrátu, amyloidu, do intersticiálního prostoru a v našich geografických podmínkách představuje nejčastější příčinu restriktivní kardiomyopatie. S kardiálním postižením je spojena především AL-amyloidóza, dříve označovaná jako primární, která je podmíněna patologickou B lymfocytární proliferací s nadprodukcí volných imunoglobulinových řetězců k nebo l. Srdeční postižení je také typické pro méně častou ATTR-amyloidózu charakterizovanou ukládáním proteinu transtyretinu v jeho divoké (senilní ATTR-amyloidóza) či mutované (familiární ATTR-amyloidóza) formě [100–103].
Amyloidóza srdeční je definována buď přímo pozitivním nálezem endomyokardiální biopsie, či nepřímo jako echokardiografický nález zesílení stěny srdeční nad 12 mm při nepřítomnosti jiného onemocnění vysvětlujícího tuto hypertrofii a současném pozitivním histologickém nálezu amyloidózy v jiné nekardiální tkáni [104]. Obraz restriktivní kardiomyopatie se vyvíjí až v pokročilejších fázích amyloidózy srdce, proto v časnějších stadiích onemocnění nemusí být vyjádřeny všechny morfologické a funkční diagnostické znaky této kardiomyopatie, především restriktivní typ komorového plnění či biatriální dilatace [105,106]. Žádná z neinvazivních diagnostických metod není schopna zcela stoprocentně odlišit jednotlivé subtypy amyloidu a diagnózu amyloidózy je takto nutno vždy opřít o pozitivní bioptický nález amyloidu v některé tkáni, jež pak umožní i přesné imunohistochemické určení amyloidu.
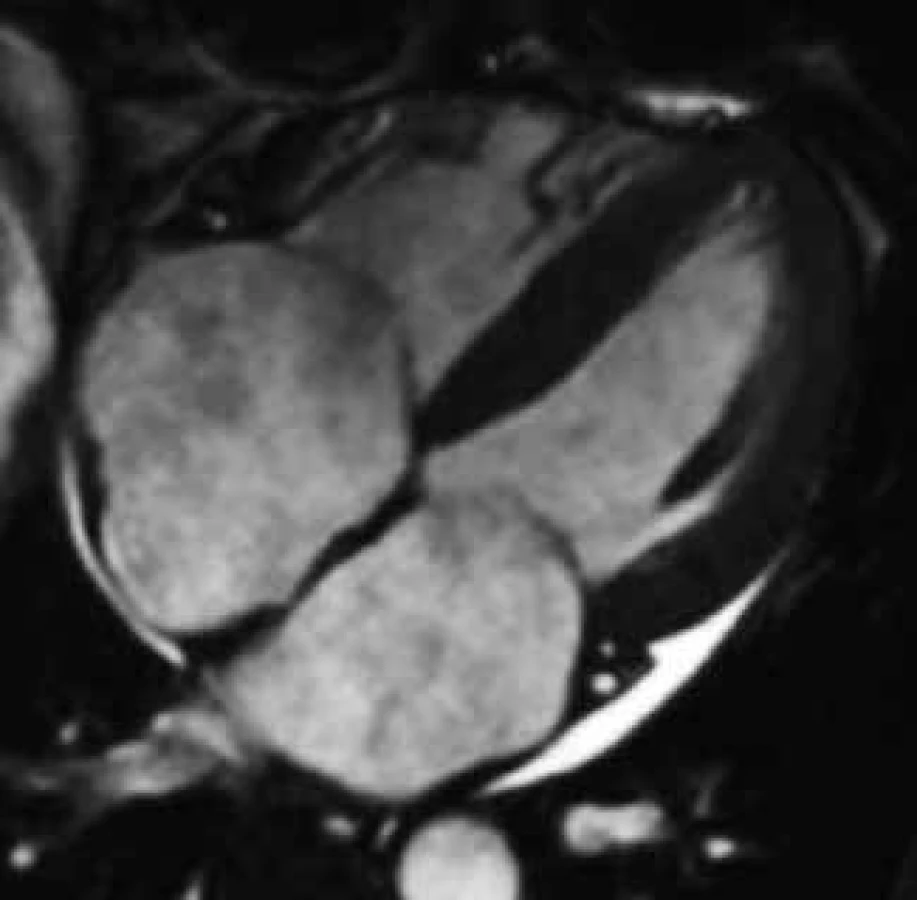
Využití CMR v diagnostice amyloidózy srdeční nabývá stále většího významu, neboť se zdá, že CMR obraz nejen poskytuje velmi přesné anatomické informace o síle stěn srdce a velikosti jeho oddílů, ale navíc pomocí LGE umožňuje i tkáňovou charakteristiku, jež ve většině případů amyloidové kardiomyopatie poskytuje poměrně specifický a unikátní nález. Základním morfologickým CMR nálezem u amyloidové kardiomyopatie je, podobně jako při echokardiografickém vyšetření, obraz zesílení stěn nezvětšené levé komory, typicky koncentrického charakteru, méně často v podobě izolovaného zesílení septa komor či volné stěny. Často bývá konkomitantně patrno i zesílení volné stěny pravé komory. V pokročilejších fázích onemocnění je možné detekovat i zesílení stěn síní a interatriálního septa, v některých případech i chlopenních cípů [97]. Toto zesílení srdečních stěn ale není obrazem pravé hypertrofie myokardu, ale jeho patologické infiltrace amyloidem [107–109]. Většinou je již v časných stadiích vyjádřena dilatace levé síně odrážející poruchu plnění levé komory, posléze dochází k dilataci i síně pravé (obr. 9). Celková systolická funkce levé komory bývá zpočátku zachována, nicméně již v iniciálních fázích amyloidové kardiomyopatie je i kvalitativně patrna porucha kontrakce stěn levé komory v longitudinální ose [110]. S progresí onemocnění dochází následně i k poklesu původně normální ejekční frakce. Perikardiální výpotek je charakteristickou známkou, která je ale opět odrazem spíše pokročilejšího onemocnění [91].
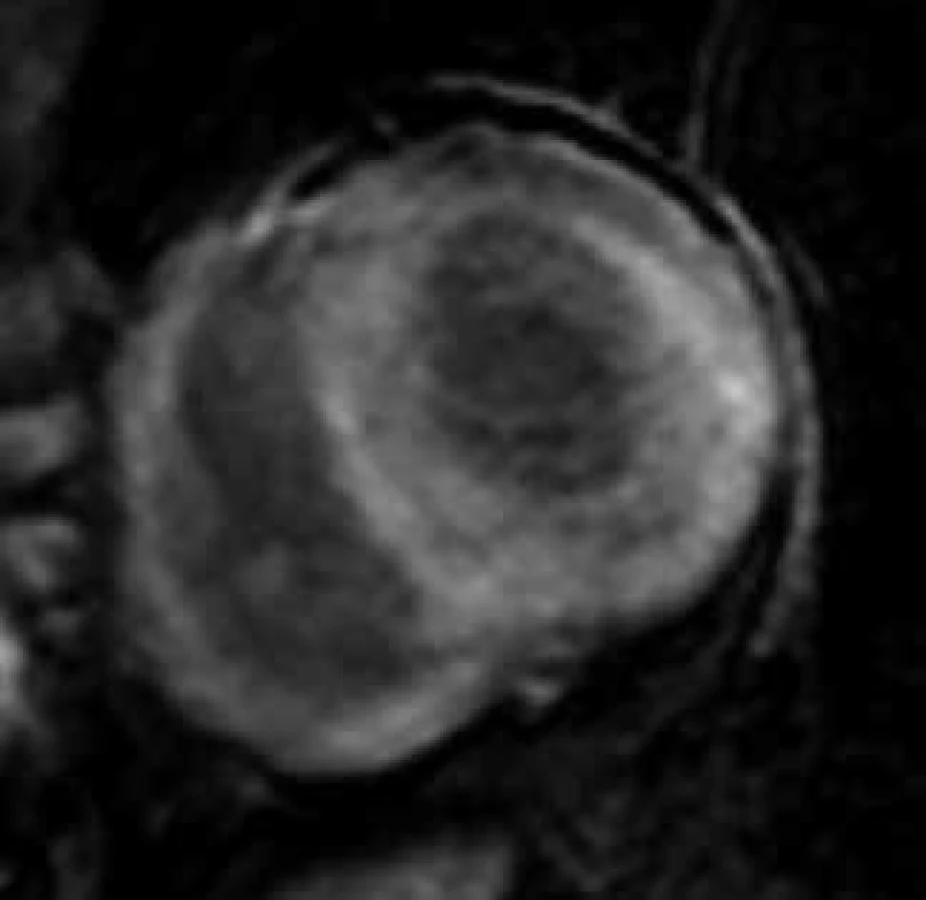
Jedinečným přínosem CMR v diagnostice srdeční amyloidózy je možnost tkáňové charakteristiky metodou LGE, která ve většině případů umožňuje jasně odlišit amyloidovou kardiomyopatii od jiných příčin vedoucích k obrazu zesílení stěn komorového myokardu, typicky arteriální hypertenze či hypertrofické kardiomyopatie, sarkomerické i nesarkomerické etiologie. Využitím LGE v diagnostice amyloidové kardiomyopatie se zabývala celá řada prací, včetně studie autorů tohoto sdělení [111–114]. Výsledky těchto prací přesvědčivě ukázaly, že až u 85 % jedinců s biopticky verifikovanou srdeční amyloidózou je přítomen výrazně specifický globální typ LGE zahrnující buďto jen subendokardiální oblast či zasahující dále transmurálně, homogenně či heterogenně (obr. 10). Podkladem tohoto typu LGE se dle autoptických studií jeví expanze intersticia myokardu dominantně amyloidovou infiltrací, možnou spoluúčast intersticiální fibrózy ale nelze vyloučit [115]. Jestliže u pacienta s extrakardiálně biopticky verifikovanou amyloidózou a suspektním, nikoli však jednoznačným echokardiografickým nálezem detekujeme pomocí CMR výše uvedený specifický globální typ LGE, je možné považovat diagnózu amyloidové kardiomyopatie za prokázanou. Díky abnormálním vlastnostem infiltrovaného myokardu je pro srdeční amyloidózu charakteristické i obtížné nastavení inverzního času pro hodnocení LGE, a i toto tzv. obtížné „nulování myokardu“ bývá pokládáno za CMR známku suspektní z amyloidové kardiomyopatie [104,116].
Podání gadolinia však může být u pacientů s amyloidózou rizikové vzhledem k často přítomnému ledvinnému postižení. I v těchto případech se zdá být nadějné kvantitativní hodnocení nativního T1 relaxačního času, které se jeví být pro detekci infiltrace myokardu senzitivnější než přítomnost LGE, a to i v časných fázích amyloidové kardiomyopatie [117,118]. Bylo též demonstrováno, že nárůst průměrného T1 relaxačního času koreluje s množstvím deponovaného amyloidu a taktéž s parametry systolické i diastolické dysfunkce levé komory [116]. Poměrně specifickým pro infiltraci myokardu AL-amyloidem v porovnání s ostatními kardiomyopatiemi se ukazuje i relativně větší nárůst extracelulárního objemu myokardu kvantifikovatelný porovnáním nativního a postkonstrastního T1 relaxačního času [3,119,120]. Hodnota nativního T1 relaxačního času a velikost extracelulárního objemu bývají dokonce označovány za „biomarkery“ u pacientů s AL-amyloidózou srdeční, které pozitivně korelují s mortalitou u tohoto onemocnění [121]. U nemocných s transtyretinovou amyloidózou je T1 relaxační čas též zvýšen, a to podstatně více než u myokardiálních patologií spojených s fibrotizací intersticiálního prostoru, jako jsou jako např. aortální stenóza či hypertrofická kardiomyopatie (HKMP) [122,123]. Nárůst T1 relaxačního času sice bývá u transtyretinové amyloidózy menší než u AL-amyloidové kardiomyopatie, nicméně i tak umožňuje odlišit hypertrofii levé komory při aortální stenóze či HKMP se stejnou diagnostickou přesností jako u AL-amyloidózy; podobně jako u AL-amyloidózy může hrát nativní T1 mapování i při detekci časného stadia transtyretinové kardiomyopatie [123]. Jak bylo uvedeno výše, žádná neinvazivní metoda, ani CMR a typ LGE ani T1 mapování, však nejsou specifické pro některou z forem amyloidózy, a proto ke stanovení jejího definitivního typu (AL, ATTR, event. jiné) je nutné histologické vyšetření extrakardiální tkáně či myokardiální biopsie.
Sarkoidóza
Sarkoidóza je systémové granulomatózní onemocnění nejasné etiologie, které může postihnout jakýkoli orgán včetně srdce. Incidence je velmi rozdílná dle sledované populace, od 3 do asi 100 jedinců na 100 000. Zajímavostí je, že se sarkoidóza vyskytuje častěji v některých populacích, např. u afro-americké, ve Skandinávii a Irsku, na Islandu a v Japonsku [124]. Ač bývají kardiální symptomy přítomny pouze u 5 % nemocných se sarkoidózou, autopticky lze kardiální postižení dle různých prací prokázat až u 25–50 % nemocných [125–128]. Postižení srdce výrazně zhoršuje prognózu pacientů se sarkoidózou, neboť dle japonské studie téměř 80 % nemocných umírá právě z kardiálních příčin [129]. Srdeční sarkoidóza se může projevovat symptomy a známkami srdečního selhávání, nebo četnými arytmickými manifestacemi zahrnujícími jak různé typy poruch srdečního vedení včetně pokročilé AV blokády, taktéž supraventrikulární a především komorové tachyarytmie, jež mohou být i podkladem náhlého úmrtí. Histologicky jsou u srdeční sarkoidózy popisována 3 postupná stadia: edém myokardu, jeho granulomatózní infiltrace a konečně fibrotizace myokardu [130]. Ač je nález nekaseifikujících granulomů v srdeční biopsii téměř patognomonickým nálezem, je bohužel výtěžnost necílené endomyokardiální biopsie nízká, a to nejen kvůli ložiskovému výskytu granulomů, které tak nemusí být biopsií zachyceny, ale i v důsledku přirozeného průběhu onemocnění, během kterého dochází k postupné fibrotizaci i v místech předtím infiltrovaných granulomy [131]. Jelikož diagnostika srdeční sarkoidózy není jednoduchá, jsou v současnosti používána diagnostická kritéria navržená japonskými odborníky, která se opírají buďto o přímý histologický průkaz nekaseifikujících epiteloidních granulomů ve vzorcích myokardu či v případě negativní srdeční biopsie o splnění alespoň 2 ze 4 velkých či jednoho velkého a alespoň 2 malých klinických kritérií (jejich podrobné rozvedení viz příslušná literatura) [108].
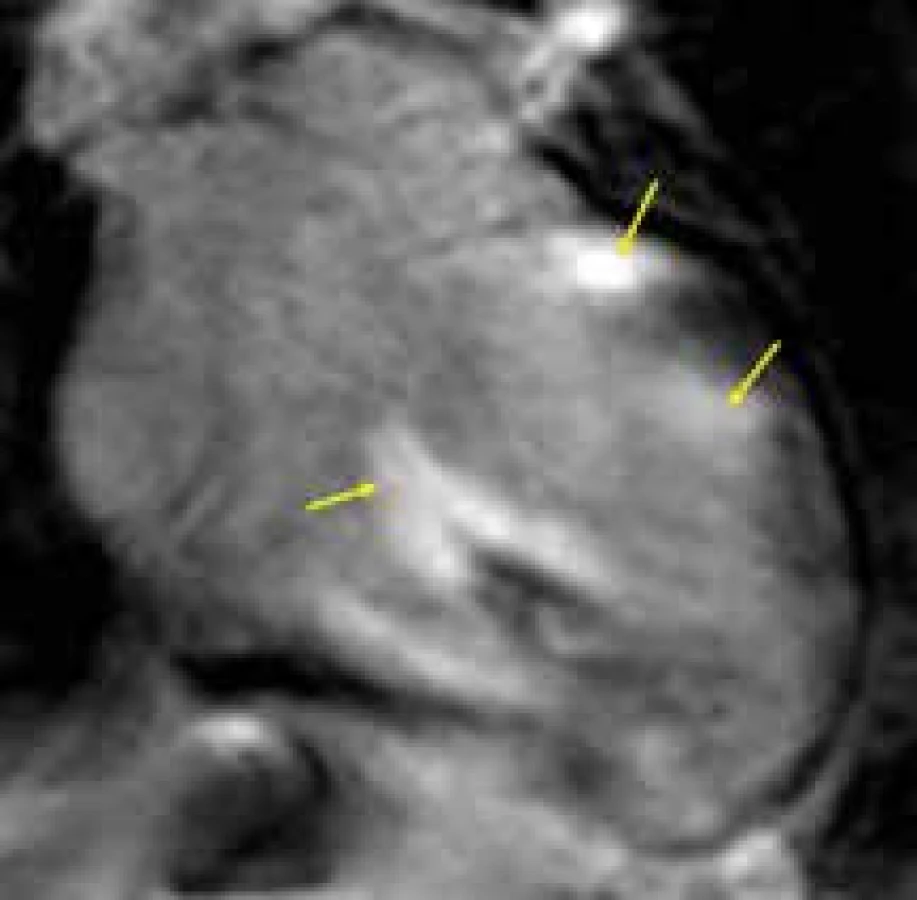
Zobrazovací metody hrají v diagnostickém procesu srdeční sarkoidózy samozřejmě kruciální úlohu. Echokardiograficky je nejčastěji nalézán obraz dilatační kardiomyopatie s dilatací a difuzní hypokinezou levé komory, někdy bývají nacházeny ložiskové poruchy kinetiky odpovídající ložiskové distribuci sarkoidové infiltrace. Edém i myokardiální infiltrace mohou způsobovat lehké zesílení stěny srdeční. Při fibrotizaci, jako dalším stadiu onemocnění, dochází naopak k zeslabení stěny srdeční, které se typicky projevuje v oblasti interventrikulárního septa bazálně [132], někdy bývají přítomna i aneuryzmata, lokalizovaná nejčastěji bazálně posteriorně [133,134]. Tyto morfologické změny jsou samozřejmě zobrazitelné i pomocí CMR [135]. Opět zcela zásadní je ovšem schopnost bližší specifikace tkáňových změn pomocí CMR. V T2 váženém obraze je dobře diagnostikovatelný edém myokardu vyskytující se v časném stadiu srdeční sarkoidózy, společně i s časným sycením gadoliniem [136]. Granulomy mohou být při CMR studii někdy zobrazitelné jako ložiska s centrální fibrózou (ložiska LGE) a periferním edémem [137]. Jizvení myokardu charakteristické pro pokročilejší stadia sarkoidózy je pomocí LGE techniky nacházeno převážně subepikardiálně nebo midmyokardiálně, v některých případech až transmurálně, vzácně též subendokardiálně, a to predilekčně v bazálních segmentech anteroseptální a anterolaterální stěny levé komory; současně je LGE často vyjádřen v papilárních svalech [138–140] (obr. 11). Vzhledem k vysoké diagnostické výpovědní hodnotě CMR je její provedení doporučováno jako metoda prvé volby u nemocných se suspekcí na srdeční sarkoidózu. V případě pozitivního CMR nálezu je pak indikováno provedení PET-CT hrudníku a následně bioptické vyšetření k definitivnímu potvrzení diagnózy. Magnetická rezonance může být též využita pro sledování účinku terapie sarkoidózy srdce [141].
Endomyokardiální fibróza
Endomyokardiální fibróza je hlavní příčinou restriktivní kardiomyopatie v rozvojových zemích, především v tropických oblastech [142]. Etiologie tohoto onemocnění není známa, i když je obviňována řada faktorů – infekčních, autoimunitních i genetických – finálně vedoucích k hypereozinofilii. Endomyokardiální fibróza je charakterizována progredující fibrotizací subendokardu hlavně vtokové části a apexu jedné či obou srdečních komor. Dochází k narušení morfologie srdečních oddílů, objem komor se zmenšuje díky obliteraci hrotové části narůstající fibrózou, síně naopak postupně dilatují s postupným vývojem diastolické dysfunkce a navýšením plnících tlaků. Echokardiograficky je toto onemocnění často obtížně postihnutelné, a činí diagnostické obtíže v odlišení apikální hypertrofie, či apikálního trombu, popř. tumoru [6]. Magnetická rezonance díky lepší vizualizaci tkáňových rozhraní i možnosti tkáňové charakteristiky nabízí daleko přesnější zobrazení, a tím i odlišení této choroby od zmiňovaných nozologických jednotek. Typickým CMR nálezem u endomyokardiální fibrózy je normální velikost srdeční komory se zesílením až obstrukcí její hrotové oblasti, které je podmíněno fibrotickým zesílením endokardu prokazatelným technikou LGE, často s nasedajícím hyposignálním trombem [143] (obr. 12). Výše uvedené postižení může být univentriklulární či biventrikulární.
Fabryho choroba
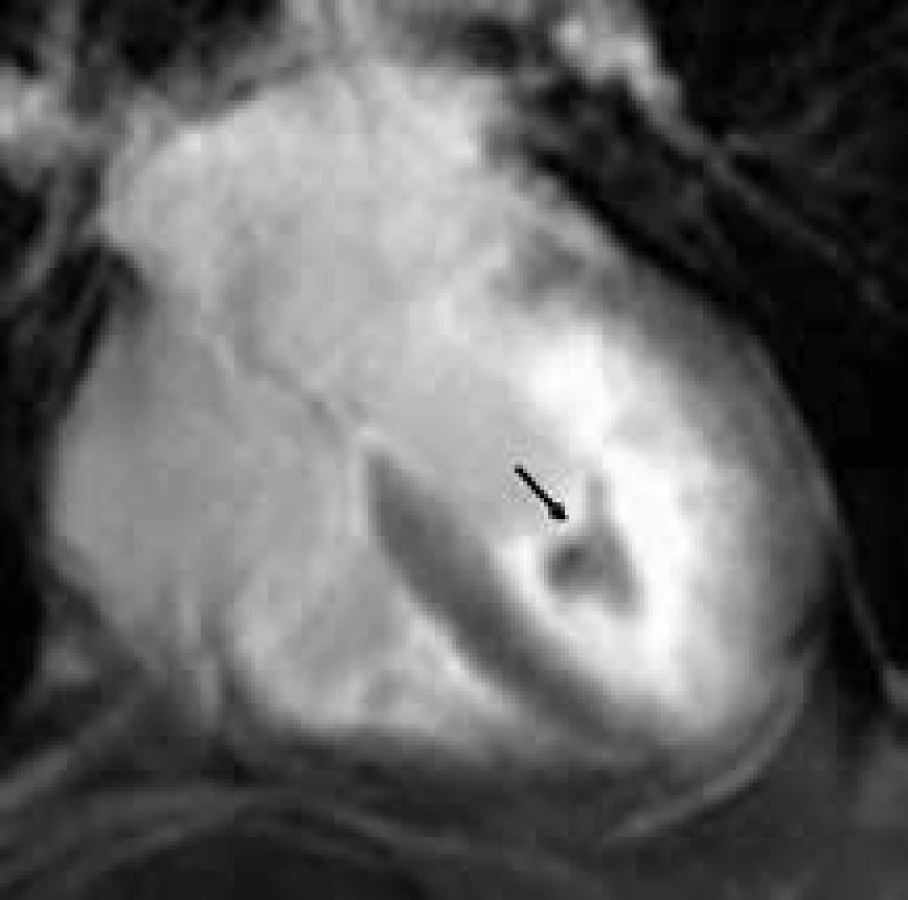
Fabryho choroba (FCH) je na chromozom X vázané onemocnění způsobené nedostatečnou funkcí lyzosomálního enzymu α-galaktosidázy A vedoucí k progedující intracelulární akumulaci glykosfingolipidů v řadě tkání a orgánů. Hlavní prognostický dopad pro nemocné má postižení srdce a ledvin [39]. Fabryho choroba je jednou z nejčastějších nesarkomerických forem hypertrofické kardiomyopatie v dospělosti. Vzhledem k tomu, že u tohoto onemocnění máme k dispozici možnost specifické terapie v podobě substituce chybějícího enzymu, je správná diagnostika FCH zcela zásadní. U mužů hemizygotů je diagnostika jednoduchá a spočívá v průkazu výrazně snížené aktivity α-galaktosidázy A v plazmě či periferních leukocytech; u žen heterozygotek, které mohou mít normální až velmi nízkou hladinu α-galaktozidázy A, je diagnostika FCH obtížnější, s nutností genetické konfirmace přítomnosti kauzální mutace. Nejčastějším morfologickým nálezem je u FCH koncentrická hypertrofie jedné, resp. obou komor; asymetrická hypertrofie septa komor je vzácnějším nálezem. K rozvoji kardiomyopatie při FCH dochází u mužů charakteristicky na přelomu 3. a 4. dekády života, u žen později. Ejekční frakce levé komory je krom pokročilých forem onemocnění zachovaná a k jejímu zhoršování dochází v souvislosti s rozvojem poruchy kinetiky v bazálním segmentu posterolaterální stěny levé komory, což souvisí s rozvojem nahrazující fibrózy predilekčně v této oblasti. Diastolická dysfunkce je typicky mírně až středně těžce postižena, restriktivní typ plnění se vyvíjí až ve finálních stadiích kardiomyopatie. U řady nemocných je přítomna dilatace bulbu aorty a zesílení chlopenních cípů aortální a mitrální chlopně podmiňující většinou jen málo závažné regurgitační vady.
Magnetická rezonance má velmi důležitou úlohu v diferenciální diagnostice FCH vůči ostatním příčinám hypertrofie levé komory, především vůči sarkomerické HKMP. Ve studii Moona et al byla zjištěna přítomnost LGE u poloviny pacientů s verifikovanou FCH, především však 92 % těchto nálezů vykazovalo lokalizaci midmyokardiálně v bázi až středním segmentu posterolaterální stěny [144] (obr. 13). Na základě prací autorů z jiných pracovišť, potvrzujících výsledky této studie, se nález LGE v bazálním posterolaterálním segmentu midmyokardiálně v terénu hypertrofie levé komory zdá být poměrně typickou známkou hypertrofické kardiomyopatie při FCH a odráží přítomnost nahrazující fibrózy v dané lokalizaci [145]. Přítomnost LGE v této lokalizaci je možné detekovat již před rozvojem regionální poruchy kinetiky v této oblasti. Vzhledem ke své vysoké, byť ne 100% specificitě by nález midmyokardiálního LGE v bazálním segmentu posterolaterální stěny hypertrofické levé komory měl vždy vzbudit podezření na možnost FCH. Zvláště u žen heterozygotek, bez jasné rodinné anamnézy FCH, může jít o důležitý klinický ukazatel implikující indikaci genetického testování této choroby.
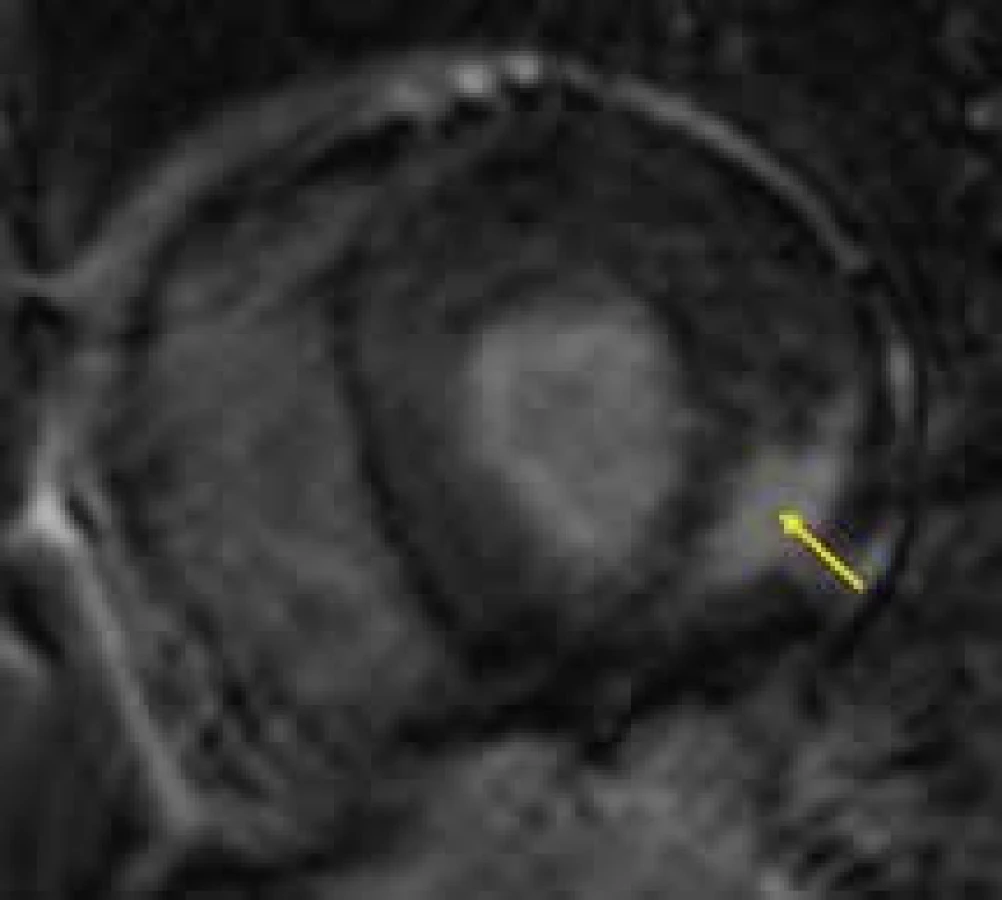
Vzhledem k častému postižení ledvin při FCH je podání gadoliniového kontrastu spojeno s vyšším rizikem systémové nefrogenní sklerózy [146]. I těchto případech je proto snaha o nalezení klinicky relevantních možností tkáňové CMR charakteristiky nevyžadujících podání kontrastní látky. Imbriaco et al [147] popsali možnost využití CMR v diagnostice FCH i bez využití gadoliniového kontrastu pouze měřením T2 relaxačního času, který se jeví prodloužený oproti kontrolní skupině, což autoři vysvětlují jinými biofyzikálními vlastnostmi tkáně s akumulovanými glykosfingolipidy. Nativním T1 mapováním se u nemocných s FCH ve své práci zabývali Pica et al [148]. Signifikantní redukce T1 relaxačního času, opět podmíněná intracelulárním střádáním glykosfingolipidů, byla v porovnání se zdravými kontrolami zjištěna jak u pacientů s fenotypicky vyjádřenou kardiomyopatií, tak i u jedinců geneticky pozitivních pro FCH, ale dosud bez vyjádřené hypertrofie levé komory. I v případě FCH by tak nativní T1 mapování a T2 mapování mohlo přispět k detekci preklinických stadií myokardiálního postižení a v důsledku toho k časnému zahájení enzymatické substituční terapie, resp. i monitorování jejího efektu [149].
Danonova choroba
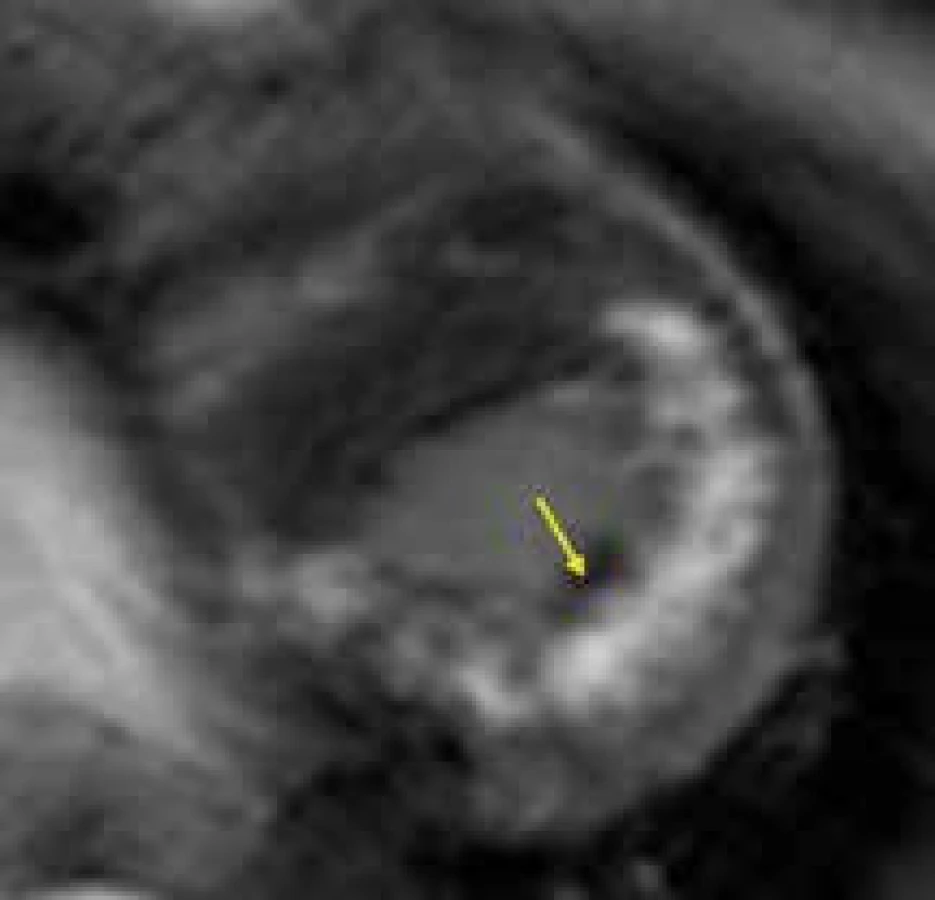
Danonova choroba je vzácným, na chromosom X vázaným metabolickým onemocněním způsobeným deficiencí lyzosomálního LAMP2 proteinu. Tento protein funguje jako membránový transportér na membráně lyzosomu, a nejedná se tedy o typický enzymatický defekt jako u většiny jiných lyzosomálních střádavých chorob. Onemocnění se projevuje hlavně hypertrofickou kardiomyopatií, myopatií kosterního svalstva a různým stupněm mentální retardace. Jelikož se jedná o systémové onemocnění, mohou být postiženy i další orgány, nejčastěji sítnice či játra. Danonova choroba se vzhledem k vazbě na X chromosom časněji a agresivněji projevuje u mužů. Postupně, v 2. dekádě života, dochází ke koncentrickému zesílení stěn srdečních levé i pravé komory dosahujících až excesivních hodnot, posléze se velmi rychle rozvíjí systolická dysfunkce a dilatace levé komory. Kardiomyocyty naplněné glykogenem často narušují elektricky nevodivý fibrózní anulus srdeční a vytvářejí akcesorní vodivá síňo-komorová spojení, což je podkladem velmi častého obrazu preexcitace na EKG. Pokud není včas indikována transplantace srdce, umírají jedinci s Danonovou chorobou ve 3. dekádě života na srdeční selhání či maligní arytmie [150].
Vyšetření CMR může být přínosem i u Danonovy choroby, znovu především díky tkáňové charakteristice. Vzhledem k raritnosti výskytu tohoto onemocnění jsou zatím v literatuře popsány jen kazuistiky pacientů vyšetřených pomocí CMR. Na základě těchto sdělení se zatím zdá, že pro hypertrofickou kardiomyopatii při Danonově chorobě by mohla být typická přítomnost intramurálního LGE v oblastech volných stěn levé komory či hrotu, s vynecháním interventrikulárního septa [151,152] (obr. 14). Tento nález by v případě potvrzení své specificity pro Danonovu chorobu na větších souborech pacientů mohl být velmi dobrým nástrojem v diferenciální diagnostice vůči jiným typům HKMP.
Kardiální postižení u svalových dystrofií
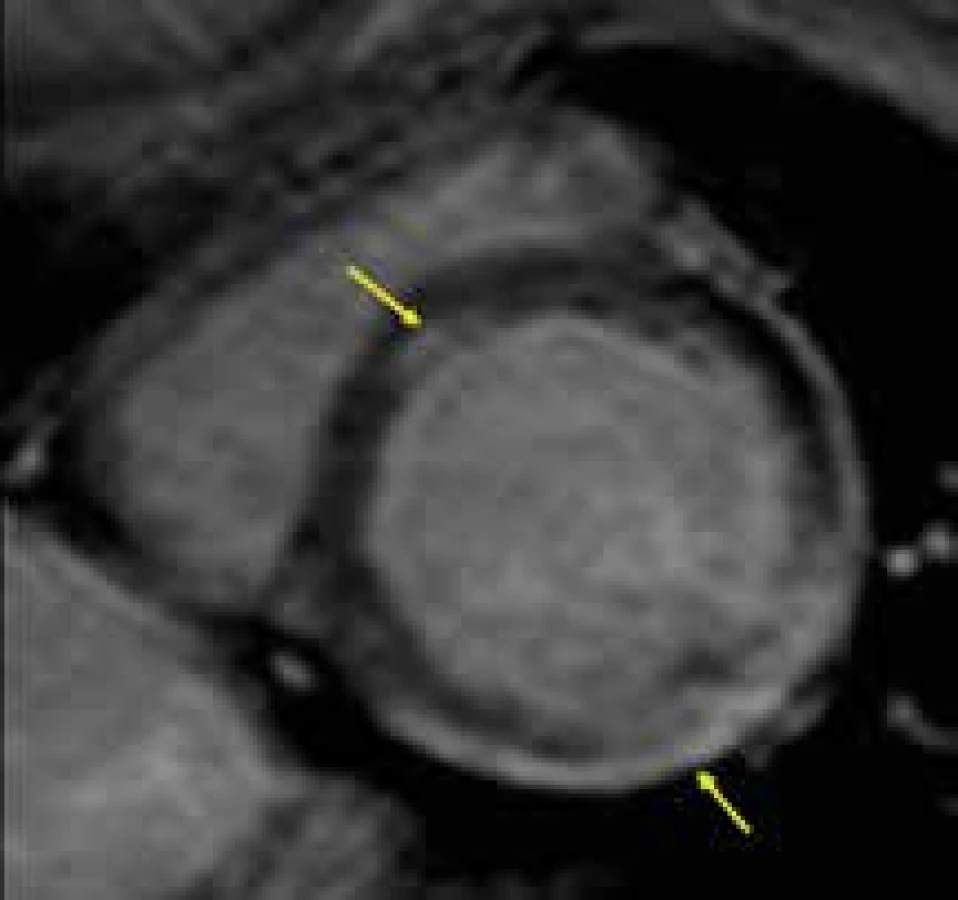
Svalové dystrofie představují skupinu onemocnění způsobenou většinou X vázaným genetickým defektem genů dystrofinového komplexu. Dystrofinový komplex je zapojen do strukturální vazby mezi cytoskeletem svalového vlákna a extracelulární matrix a jeho postižení se projevuje progredující svalovou slabostí. K postižení myokardu v podobě dilatační kardiomyopatie dochází bez výjimky u Duchenneovy svalové dystrofie, a to již v období dospívání až časné dospělosti. U ostatních forem svalových dystrofií je vývoj myokardiální dysfunkce také možný, především u Beckerovy dystrofie, manifestuje se ale až v dospělosti [153,154]. I zde mohou být kromě mužů postiženy i ženy heterozygotky, ovšem v pozdějším věku a obvykle méně výrazně. Dle prací francouzských autorů se zdá, že rychlost progrese dysfunkce levé komory a srdečního selhání může být zpomalena podáváním perindoprilu již u chlapců s ještě normální systolickou funkcí LK [155].
Magnetická rezonance umožňuje přesné vyhodnocení velikosti srdečních oddílů a systolické funkce levé komory, může být využita i k přesnějšímu popisu mechanizmu relativně často zjišťované mitrální regurgitace, která u Duchenneovy choroby bývá dávána do souvislosti s dysfunkcí zadního papilárního svalu, kdežto u Beckerovy dystrofie spíše s dilatací mitrálního anulu [156]. Velmi důležité je, že pomocí techniky LGE lze pravděpodobně detekovat již časné fibrotické změny myokardu, které jsou přítomny ještě před rozvojem manifestní dilatační kardiomyopatie, a to u obou pohlaví [157–159]. U Duchenneovy svalové dystrofie je přítomnost LGE v iniciálních fázích kardiálního postižení typicky lokalizována subepikardiálně, většinou v oblasti posterolaterální stěny (obr. 15) [160]. Výše uvedené skutečnosti mají velkým význam především k časné detekci kardiálního postižení u žen heterozygotek, u kterých může dojít k rozvoji myokardiální dysfunkce v dospělém věku. Vzhledem k současné strategii preventivní léčby ACE inhibitory u všech chlapců s Duchenneovou chorobou lze očekávat, že v brzké budoucnosti budou provedeny studie zkoumající efekt jejich preventivního podávání i u starších žen heterozygotek s ještě normální velikostí a funkcí levé komory, ale s CMR nálezem počínající fibrózy myokardu v podobě pozitivního nálezu LGE.
Hemochromatóza
Hemochromatóza je onemocnění způsobené nadměrnou intracelulární akumulací železa v řadě tkání včetně myokardu, ať už v rámci primární hemochromatózy, geneticky podmíněné poruchy metabolizmu železa, či při její sekundární etiologii, typicky jako důsledek opakovaných krevních transfuzí, např. u nemocných s myelodysplastickým syndromem, talasemií, či srpkovitou anémií. V myokardu se železo ukládá především subepikardiálně a jeho průkaz pomocí endomyokardiální biopsie tak může být obtížný [161]. Železo vzhledem ke svému toxickému působení cestou volných radikálů zapříčiňuje oxidativní stres, který má za následek nekrózu kardiomyocytů. To může vést nejen ke vzniku řady arytmií, ale i k rozvoji srdečního selhání. Srdeční selhání v důsledku srdeční hemochromatózy je hlavní příčinou úmrtí pacientů trpících β-talasemií major [162]. Vzhledem k možnosti podávání účinných chelatačních látek je stanovení diagnózy myokardiálního postižení i následné sledování účinku této terapie pro pacienta s hemochromatózou velmi důležité. Echokardiograficky i při necíleném CMR vyšetření je nález nespecifický. Nacházíme mírné difuzní zesílení stěn myokardu, dilataci levé komory, postupně se zhoršující diastolickou dysfunkci až do obrazu restrikce a progredující systolickou dysfunkci [163–166].
Přítomnost paramagnetických železitých iontů mění magneticko-rezonanční vlastnosti myokardu. Konkrétně narůstá inhomogenita magnetického pole, čímž se ztrácí signál jak v T1, tak i v T2 váženém obraze a tkáně se tak jeví hypointenzivnější, než je obvyklé. To umožňuje poměrně spolehlivou detekci myokardiální depozice železa pomocí speciální sekvence hodnotící relaxační čas T2*. T2* relaxační čas je závislý na T2 relaxaci a bývá též označován jako efektivní T2 relaxační čas, neboť zohledňuje rychlejší ztrátu fázové koherence signálu v transverzální rovině danou magnetickou inhomogenitou [167]. V přítomnosti depozice železa v myokardu se tento čas zřetelně zkracuje z obvyklých asi ≥ 50 ms až k hodnotám < 20 ms [168]. Velikost poklesu T2* relaxačního času odpovídá množství naakumulovaného železa [169]. Vyšetření CMR s vyhodnocením T2* času tak umožňuje časnou detekci akumulace železa v myokardu ještě před vznikem zjevné kardiomyopatie a včasné zahájení chelační terapie i monitoraci jejího efektu. Bylo totiž ukázáno, že stanovení T2* relaxačního času odráží množství naakumulovaného železa v myokardu přesněji než hladina sérového ferritinu či množství železa obsaženého v jaterním parenchymu, což byly parametry dříve užívané k hodnocení efektu léčby hemosiderózy [170]. Pravidelné vyšetřování CMR s hodnocením T2* relaxačního času by proto v současnosti mělo být rutinní součástí diagnostického procesu i sledování efektu chelační léčby u nemocných s hemochromatózou.
Takotsubo kardiomyopatie
Takotsubo kardiomyopatie je řazena mezi neklasifikované kardiomyopatie a je charakterizována akutně vzniklou reverzibilní poruchou kinetiky komorového myokardu charakteru akinezy až dyskinezy, typicky postihující hrotovou část levé komory (klasická forma, tzv. apical ballooning), méně často její střední či bazální segmenty (atypické formy), a to v nepřítomnosti koronárního postižení, jež by mohlo danou poruchu kinetiky způsobit. Laboratorně je vyjádřena jen mírná elevace troponinu, která kontrastuje s poměrně rozsáhlou ložiskovou poruchou stažlivosti myokardu, a odráží skutečnost, že předpokládaným patofyziologickým podkladem takotsubo kardiomyopatie není nekróza myokardu, ale jeho omráčení pravděpodobně podmíněné výrazně zvýšenými hladinami endogenních katecholaminů, nejčastěji v důsledku psychického či fyzického stresu. V diferenciální diagnostice jsou nejčastěji zvažovány akutní koronární syndrom a akutní myokarditida [171].
Kromě typické poruchy kinetiky v oblasti hrotu, či při atypických formách midventrikulárně, resp. bazálně, nachází CMR v akutní fázi kardiomyopatie, na rozdíl od akutního infarktu myokardu, pouze obraz edému myokardu v T2 váženém obraze [172], charakteristicky bez současné přítomnosti LGE, tj. známky nekrózy myokardu [173,174]. Tím se akutní CMR obraz takotsubo kardiomyopatie zásadně liší od nálezů u akutního infarktu myokardu či akutní myokarditidy, u nichž jsou kromě edému myokardu patrna též ischemická, resp. u myokarditidy neischemická ložiska LGE. Je však nutno zmínit, že raritně byl u takotsubo kardiomyopatie nález nevelkých a spíše hyposignálních ložisek LGE též popsán v oblasti srdečního hrotu a jeho průkaz byl markerem horší prognózy [175–177]. Diagnózu takotsubo kardiomyopatie pak v dalším průběhu potvrzuje kompletní restituce normální kinetiky levé komory, nejčastěji v průběhu 2–3 týdnů, kterou lze dobře verifikovat kontrolním CMR vyšetřením, zároveň prokazujícím ústup edému myokardu a trvající absenci LGE, tj. reparační fibrózy myokardu.
Peripartální kardiomyopatie
Peripartální kardiomyopatie (PPKMP) je definována jako nově vzniklá systolická dysfunkce levé komory prezentující se známkami srdečního selhání koncem těhotenství či v prvních měsících po porodu, za předpokladu, že není zjištěna jiná příčina myokardiální dysfunkce. Levá komora nemusí být dilatovaná, ale její ejekční frakce je téměř vždy snížena pod 45 % [178]. O incidenci PPKMP je známo málo, podle studií provedených na území USA by měla činit 1 : 2 500–4 000 porodů [160]. Etiologie onemocnění zůstává neobjasněna, v současnosti je nejvíce zkoumána teorie kardiodepresivního působení oxidativního stresu vznikajícího v rámci osy katepsin-prolaktin [160]. Asi u 30–50 % pacientek dochází k plnému zotavení, s návratem ejekční frakce levé komory k normálním hodnotám. Pokud však nedojde k normalizaci systolické funkce do 6 měsíců po porodu, jedná se většinou o ireverzibilní postižení, které je spojeno s horší prognózou [6]. Diagnóza PPKMP je většinou opřena o echokardiografické vyšetření prokazující globální systolickou dysfunkci hypokinetické levé komory [179]. Magnetická rezonance může u hůře echokardiograficky vyšetřitelných žen pomoci s přesným hodnocením velikosti levé komory, její celkové i regionální systolické funkce. Tkáňová CMR charakteristika myokardu u PPKMP má svůj význam z prognostických důvodů, neboť u některých žen lze detekovat midmyokardiální ložiska LGE, jejichž nález je považován za predikční faktor perzistující dysfunkce levé komory a horší prognózy. Naopak absence LGE ukazuje na větší pravděpodobnost normalizace ejekční frakce levé komory [180,181].
Závěr
Magnetická rezonance srdce se v posledních letech stala integrální součástí diagnostiky mnoha kardiálních onemocnění včetně kardiomyopatií a myokarditidy. Představuje zlatý standard pro kvantifikaci velikosti srdečních oddílů a posouzení systolické funkce obou komor. V oblasti onemocnění myokardu se výrazně uplatňuje též její unikátní schopnost tkáňové charakteristiky. Magnetická rezonance srdce tak nabízí zásadní diagnostické vodítko v diagnóze akutní myokarditidy a specifické etiologie všech typů kardiomyopatií. Z prognostického hlediska se jako velmi významná ukazuje u celé řady chorob myokardu možnost hodnotit přítomnost a množství nahrazující fibrózy myokardu. Vyšetření magnetickou rezonancí již nyní představuje výrazný přínos v diagnostickém algoritmu myokardiálních patologií a její úloha bude v blízké budoucnosti jistě dále narůstat.
Práce byla podpořena projektem PRVOUK P35/LF1/5.
prof. MUDr. Tomáš Paleček, Ph.D.
tpalec@lf1.cuni.cz
II. interní klinika – klinika kardiologie a angiologie 1. LF UK a VFN Praha
www.vfn.cz
Doručeno do redakce 19. 6. 2016
Přijato po recenzi 1. 8. 2016
Sources
Seznam literatury obsahuje pouze výběr recentních literárních odkazů. Úplný seznam litertury naleznete na www.vnitrnilekarstvi.eu
90. Arbustini E, Weidemann F, Hall JL. Left Ventricular Noncompaction A Distinct Cardiomyopathy or a Trait Shared by Different Cardiac Diseases? J Am Coll Cardiol 2014; 64(17): 1840–1850.
95. Jenni R, Oechslin E, Schneider J et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001; 86(6): 666–671.
96. Petersen SE, Selvanayagam JB, Wiesmann F et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005; 46(1): 101–105.
97. Jacquier A, Thuny F, Jop B et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J 2010; 31(9): 1098–1104.
98. Captur G, Muthurangu V, Cook C et al. Quantification of left ventricular trabeculae using fractal analysis. J Cardiovasc Magn Reson 2013; 15 : 36. Dostupné z DOI: <http://dx.doi.org/10.1186/1532–429X-15–36>.
99. Dawson DK, McLernon DJ, Raj VJ et al. Cardiovascular Magnetic Resonance Determinants of Left Ventricular Noncompaction. Am J Cardiol 2014; 114(3): 456–462.
101. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 2011; 97(1): 75–84.
111. Fikrle M, Palecek T, Masek M et al. The diagnostic performance of cardiac magnetic resonance in detection of myocardial involvement in AL amyloidosis. Clin Physiol Funct Imaging 2016; 36(3): 218–224.
117. Karamitsos TD, Piechnik SK, Banypersad SM et al. Noncontrast T1 Mapping for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging 2013; 6(4): 488–497.
124. Dubrey SW, Falk RH. Diagnosis and Management of Cardiac Sarcoidosis. Prog Cardiovasc Dis 2010; 52(4): 336–346.
139. Smedema JP, Snoep G, van Kroonenburgh MP et al. Evaluation of the accuracy of gadolinium-enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol 2005; 45(10): 1683–1690.
143. Salemi VM, Rochitte CE, Shiozaki AA et al. Late gadolinium enhancement magnetic resonance imaging in the diagnosis and prognosis of endomyocardial fibrosis patients. Circ Cardiovasc Imaging 2011; 4(3): 304–311.
144. Moon JC, Sachdev B, Elkington AG et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J 2003; 24(23): 2151–2155.
147. Imbriaco M, Spinelli L, Cuocolo A. MRI characterization of myocardial tissue in patients with Fabry’s disease. Am J Roentgenol 2007; 188(3): 850–853.
148. Pica S, Sado DM, Maestrini V et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2014; 16 : 99. Dostupné z DOI: <http://dx.doi.org/10.1186/s12968–014–0099–4>.
150. Boucek D, Jirikowic K, Taylor M. Natural history of Danon disease. Genet Med 2011; 13(6): 563–568.
151. Majer F, Pelak O, Kalina T et al. Mosaic tissue distribution of the tandem duplication of LAMP2 exons 4 and 5 demonstrates the limits of Danon disease cellular and molecular diagnostics. J Inherit Metab Dis 2014; 37(1): 117–124.
160. Verhaert D, Richards K, Rafael-Fortney JA et al. Cardiac Involvement in Patients with Muscular Dystrophies, Magnetic Resonance Imaging Phenotype and Genotypic Considerations. Circ Cardiovasc Imaging 2011; 4(1): 67–76.
167. Anderson LJ, Holden S, Davis B et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J 2001; 22(23): 2171–2179.
170. Kirk P, Roughton M, Porter JB et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation 2009; 120(20): 1961–1968.
172. Abdel-Aty H, Cocker M, Friedrich MG. Myocardial edema is a feature of Tako-Tsubo cardiomyopathy and is related to the severity of systolic dysfunction: insights from T2-weighted cardiovascular magnetic resonance. Int J Cardiol 2009; 132(2): 291–293.
173. Deetjen AG, Conradi G, Mollmann S et al. Value of gadolinium-enhanced magnetic resonance imaging in patients with Tako-Tsubo-like left ventricular dysfunction. J Cardiovasc Magn Reson 2006; 8(2): 367–372.
178. Sliwa K, Hilfiker-Kleiner D, Petrie MC et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Failure 2010; 12(8): 767–778.
181. Mouquet F, Lions C, de Groote P et al. Characterisation of peripartum cardiomyopathy by cardiac magnetic resonance imaging. Eur Radiol 2008; 18(12): 2765–2769.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 12
Most read in this issue
- Trombofília
- Káva ako hepatoprotektívny faktor
- Perorální antidiabetika v léčbě diabetes mellitus 1. typu
- Takotsubo syndrom: incidence, etiologie, komplikace, léčba a prognóza