
Von Hippelov-Lindauov syndróm – dve strany jednej mince
Von Hippel-Lindau syndrome – two sides of the same coin
Von Hippel-Lindau syndrome (VHL) is a rare genetic disease. Its incidence is 1 : 36,000, there is the familial occurrence in 80 % of cases , the remaining cases are de novo mutations. The disease is caused by the highly penetrant mutations in the VHL gene (3p25.3) and is characterized by the occurrence of benign and malignant neoplasms. The most common VHL tumors are the tumors of the retina, brain and spinal hemangioblastomas, renal cell carcinoma, pheochromocytoma, endolymfatic sac tumors and pancreatic tumors and cysts. The mean age of the VHL patients during the diagnosis is 20–40 years. The diagnosis can be confirmed by a positive family history and the presence of one of the typical tumor. In case of no family history, the diagnosis has to be assessed by the presence of the multiple tumors. The clinical signs and prognosis of VHL depend on the location and extent of the tumors. The life expectancy is 50 years. The most common causes of death are complications of the renal cancer and the brain tumors. The treatment requires a multidisciplinary collaboration through the whole life of patients. This 2 cases report we demonstrate the differences among the patients with de novo mutations disease and the patient with familial incidence.
Key words:
pheochromocytoma – renal cell carcinoma – von Hippel-Lindau syndrome
Authors:
Patrícia Páleníková 1; Monika Adamcová 1; Igor Šturdík 1; Beáta Ftáčniková 2; Lucia Copáková 3; Juraj Payer 1
Authors‘ workplace:
V. interná klinika LF UK a UNB, Nemocnica Ružinov, Bratislava, Slovenská republika
1; I. rádiologická klinika LF UK, SZU a UNB, Nemocnica Ružinov, Bratislava, Slovenská republika
2; Oddelenie lekárskej genetiky Národného onkologického ústavu, Bratislava, Slovenská republika
3
Published in:
Vnitř Lék 2016; 62(12): 1004-1008
Category:
Reviews
Overview
Von Hippelov-Lindauov syndróm (VHL) je vzácne, hereditárne autozomálne dominantné, geneticky podmienené ochorenie. Výskyt ochorenia je 1 : 36 000, pričom v 80 % prípadov ide o familiárny výskyt a vo zvyšných prípadoch vzniká mutácia de novo. VHL je spôsobený vysoko penetračnými mutáciami v géne VHL (3p25.3). VHL je charakterizovaný výskytom malígnych a benígnych novotvarov. Medzi najčastejšie VHL tumory patria nádory sietnice, mozgové a spinálne hemangioblastómy, karcinómy obličiek, feochromocytóm, tumory endolymfatického vaku a nádory a cysty pankreasu. Priemerný vek pri stanovení diagnózy je 20–40 rokov. Diagnóza môže byť potvrdená pozitívnou rodinnou anamnézou a prítomnosťou jedného typického nádoru. Pri negatívnej rodinnej anamnéze je na potvrdenie diagnózy potrebná prítomnosť viacerých nádorov. Klinické príznaky aj prognóza závisia od lokalizácie a rozsahu nádoru. Priemerná dĺžka života dosahuje 50 rokov. Najčastejšími príčinami úmrtia sú karcinóm obličky a komplikácie mozgových tumorov. Liečba vyžaduje multidisciplinárnu spoluprácu s nutnosťou sledovania počas celého života. Na kazuistikách 2 pacientov demonštrujeme odlišnosti u pacienta so získanou mutáciou de novo a pacienta s familiárnym výskytom, pričom rozdiely nachádzame v priebehu ochorenia ako aj v klinickom obraze.
Kľúčové slová:
feochromocytóm – karcinóm obličky – von Hippelov-Lindauov syndróm
Úvod
Von Hippelov-Lindauov syndróm (VHL) je vzácne geneticky podmienené ochorenie s autozomálne dominantným typom dedičnosti. Je charakterizovaný výskytom malígnych a benígnych novotvarov.
Najčastejšie asociovanými tumormi VHL syndrómu sú nádory sietnice, mozgové a spinálne hemangioblastómy, svetlobunkový renálny karcinóm, bilaterálny feochromocytóm, tumory endolymfatického vaku a nesekretorické tumory Langerhansových ostrovčekov spolu s ďalšími pankreatickými léziami, ako sú cysty a mikrocystické adenómy.
Prevalencia ochorenia je 1 : 53 000, incidencia sa pohybuje v rozmedzí 1 : 35 000–40 000. Priemerný vek vzniku ochorenia alebo stanovania diagnózy je 20–40 rokov [1,2].
Prejavy, liečba aj prognóza ochorenia závisia od lokalizácie a veľkosti nádoru alebo cysty.
Patogenéza
VHL je spôsobený vysoko penetračnými zárodočnými mutáciami v géne VHL (3p25.3) alebo deléciou tzv. VHL tumor supresorového génu, ktorý je lokalizovaný na krátkom ramienku chromozómu 3 [3]. Produkt génu reguluje aktivitu HIF (hypoxia inducible factors). V neprítomnosti VHL proteínu je HIF kontinuálne aktívny a vedie k zvýšenej expresii niekoľkých cieľových génov, vrátane vaskulárneho endotelového rastového faktora (vascular endothelial growth factor – VEGF), čo má za následok rast vaskulárnych tumorov [4,5].
Abnormálny alebo chýbajúci gén spôsobí narušenie normálneho priebehu bunkového cyklu a excesívna angiogenéza vedie k vzniku neoplázií. K vzniku nádorových ochorení prispieva aj narušená schopnosť buniek odpovedať na hypoxické stavy organizmu [4,5].
V 80 % prípadov ide o familiárny výskyt, vo zvyšných 20 % prípadov vzniká mutácia de novo [2].
Diagnostika
Na potvrdenie diagnózy je pri pozitívnej rodinnej anamnéze potrebná prítomnosť jedného typického nádoru: hemangioblastóm sietnice alebo mozočka, bilaterálny feochromocytóm, svetlobunkový karcinóm obličky. Ak je rodinná anamnéza negatívna, je k stanoveniu diagnózy nutný výskyt viacerých nádorov. Potvrdenie diagnózy sa realizuje genetickým vyšetrením – DNA analýza VHL génu pomocou PCR (polymerase chain reaction) a priameho sekvenovania [2,6,7,9,10].
V klinickej praxi by sme mali na možnú diagnózu syndrómu VHL myslieť u pacientov, u ktorých pozorujeme skoršiu manifestáciu nádorových ochorení ako pri sporadických formách. Napr. hemangioblastóm CNS sa objavuje skôr o 10 rokov, renálny karcinóm o 25 rokov skôr. Skorší výskyt nádorových ochorení je jedným z typických prejavov VHL.
Zvyčajne sa genetické testovanie indikuje u pacienta, ktorý spĺňa klinické diagnostické kritériá, u rodinných príslušníkov pacienta so zistenou VHL mutáciou a u pacientov, u ktorých sa vyskytne v mladom veku jeden z typických VHL tumorov. Pacientom aj ich rodinám by malo byť ponúknuté genetické poradenstvo. Prediktívne vyšetrenie je možné realizovať bez ohľadu na vek vzhľadom k možnosti manifestácie závažných prejavov ochorenia u detí [8–10].
V diferenciálnej diagnostike je nutné vylúčiť MEN 2 syndróm (mnohopočetná endokrinná neoplázia), pri ktorom je prítomný feochromocytóm často bilaterálne, neurofibromatózu, polycystické obličky, dedičný feochromocytóm – paraganglióm, Carneyho syndróm (trias: extraadrenálny paraganglióm, pľúcny chondróm, GISTóm).
Priebeh ochorenia a liečba
Klinické prejavy závisia od lokalizácie a rozsahu nádoru alebo cysty. Priemerný vek v čase diagnózy nádorov u VHL je podstatne nižší než v sporadických prípadoch. Bola popísaná intrafamiliárna variabilita ochorenia. Priemerná dĺžka života je 50 rokov. Najčastejšiou príčinou úmrtia sú karcinómy obličiek, hemangioblastómy CNS, a to najmä komplikácie mozgových nádorov.
Hemangioblastómy CNS sú jedny z najčastejších VHL tumorov, vyskytujú sa u asi 60–80 % pacientov s VHL. Postihujú mozoček, mozgový kmeň a miechu. Môžu byť benígne, pričom klinické ťažkosti sú zväčša následkom útlaku priľahlého nervového tkaniva a zvýšeného intrakraniálneho tlaku. Prejavia sa cefaleou, vracaním a ataxiou. Priemerný vek pacientov v dobe ich výskytu je okolo 33 rokov [2,8].
Retinálne hemangioblastómy postihujú až 60 % pacientov s VHL, v 50 % sú obojstranné, ale môžu byť mnohopočetné aj v jednom oku. Prebiehajú od asymptomatických foriem po odlúčenie sietnice, makulárny edém, glaukóm, prípadne až slepotu. Priemerný vek pacientov v dobe výskytu je okolo 12–25 rokov [2,8].
Svetlobunkový karcinóm obličky sa vyskytuje u 40 % pacientov s VHL. Býva solitárny, mnohopočetný, v niektorých prípadoch bilaterálny. Je najčastejšou príčinou úmrtia pacientov [2,8].
Feochromocytóm (FEO) pozorujeme v 10–15 % prípadov pacientov s VHL. Podľa jeho prítomnosti rozdeľujeme VHL na 2 typy: VHL – typ 1 s FEO, VHL – typ 2 bez FEO. Medzi klinické príznaky patrí hypertenzná kríza a s ňou spojený infarkt myokardu alebo náhla cievna mozgová príhoda. Pri diagnostike sa stanovujú metabolity katecholamínov. Môže však prebiehať aj bez typických klinických príznakov hypertenzie. V takom prípade k detekcii feochromocytómu prispeje magnetická rezonancia (MR), scintigrafia s MIBG (metajodbenzylguanidín) alebo celotelová pozitrónová emisná tomografia (PET) [2,5,8,11,12].
Postihnutie pankreasu u pacientov s VHL je v 20–50 % prípadoch a zahŕňa benígne pankreatické cysty a cystadenómy, ktoré môžu byť asymptomatické alebo spôsobujú útlak tkaniva pankreasu. Pankreatické neuroendokrinné nádory nachádzame u 15 % pacientov, môžu byť asymptomatické alebo malígne s metastatickým šírením [2,8].
Epididymálne cysty a cystadenómy sa vyskytujú u 60 % pacientov mužského pohlavia s VHL.
Endolymfatické sac nádory (ELST) sú u 10 % pacientov a ich následkom je strata sluchu [2,5,8].
Liečba zahŕňa chirurgické odstránenie nádoru. Pacienti s VHL sú dlhodobo dispenzarizovaní u viacerých špecialistov (pediater, oftalmológ, neurológ, otorinolaryngológ, endokrinológ) a majú presne určený harmonogram laboratórnych a zobrazovacích vyšetrení – ako je uvedené v tab. [2,5,8,11].

Rozdiely medzi familiárnou a sporadickou formou demonštrujeme na 2 kazuistikách.
Kazuistika 1
V prvej kazuistike popisujeme prípad pacienta narodeného v roku 1975 s anamnézou bilaterálnych tumorov nadobličky, ktorý bol koncom roka 2005 prijatý na V. internú kliniku LF UK a UN Bratislava pre celkovú slabosť, ataxiu, vracanie, chudnutie, bolesti hlavy v záhlaví.
V rodine pacienta neboli zaznamenané genetické ochorenia ani vývojové anomálie.
V roku 1994 bola ultrasonografickým a CT vyšetrením brucha u pacienta diagnostikovaná cystická degenerácia pankreasu, s 2 solídnymi nádormi v oblasti hlavy a chvosta pankreasu do 4 cm, s početnými léziami do 2,5 cm. V roku 2002 bol CT vyšetrením potvrdený bilaterálny nádor nadobličiek (s priemerom 4 cm vpravo, 3 cm vľavo) s opakovane zvýšenými katecholamínmi v moči. Genetickým testovaním bola mutácia RET protoonkogénu negatívna, čím nebola potvrdená viacpočetná endokrinná neoplázia – MEN 2A syndróm. V marci roku 2003 bola realizovaná MIBG (metajodbenzylguanidín) scintigrafia, pri ktorej sa na nadobličkách potvrdil bilaterálny tumor neuroendokrinného pôvodu s chromafinnými vezikulami – suponovaný feochromocytóm. Pacient navrhovanú obojstrannú adrenalektómiu odmietol. Počas 3 rokov zmenil životný štýl, prešiel na vegetariánsku stravu a užíval homeopatiká. Stále však trpel závratmi, dyspepsiou a chudnutím.
Objektívne pri prijatí na našu kliniku bol pacient tachykardický, hodnoty TK sa pohybovali do maxima 150/90 mm Hg. Prítomná bola nestabilita pri chôdzi, subikterus sklér, hmatateľná rezistencia veľkosti 5 cm pod pravým rebrovým oblúkom a v mezogastriu. Z laboratórnych odberov boli patologicky zvýšené hepatálne parametre s prevahou obštrukčných enzýmov pri predpokladanom útlaku žlčových ciest. Plánovali sme kontrolné CT abdomenu a ďalšiu diagnostiku bilaterálnych feochromocytómov, avšak vzhľadom na výraznú ataxiu bol pacient primárne konzultovaný s neurológom, ktorý nachádza známky cerebelárneho poškodenia. Oftalmologické vyšetrenie bolo negatívne, bez nálezu na očnom pozadí. Realizované CT vyšetrenie mozgu s nálezom tumoróznych vaskularizovaných ložísk charakteru hemangioblastómu v oboch mozočkových hemisférach a v spinálnom kanáli, so sprievodným kolaterálnym edémom a 3-komorovým hydrocefalom. Vzhľadom na nález hemangioblatómov a bilaterálneho feochromocytómu sme suponovali diagnózu VHL syndrómu. Avšak pacient bol pre neurologické ťažkosti preložený na kliniku neurochirurgie, za účelom exstirpácie cerebelárnych tumorov. Po ich odstránení odozneli cererbelárne príznaky a u pacienta sme pokračovali v ďalšej diagnostike. CT vyšetrením brucha sa zachytila veľkostná progresia ložísk v pankrease (v hlave 45 × 30 mm, v tele 70 × 65 mm), potvrdila sa cholestáza s dilatáciou intrahepatálnych a extrahepatálnych žlčovodov, dilatácia v. portae, viaceré tumorózne vaskularizované ložiská v pečeni (12 mm), v oboch nadobličkách charakteru feochromocytómu (40 × 40 mm), tumorózne ložisko v ľavej obličke (12 mm), obr. 1. Magnetickou rezonanciou obličiek a MIBG scintigrafiou sa potvrdila prítomnosť bilaterálneho feochromocytómu (obr. 2), avšak tumorózne ložisko na ľavej obličke na MR nebolo dokázané.
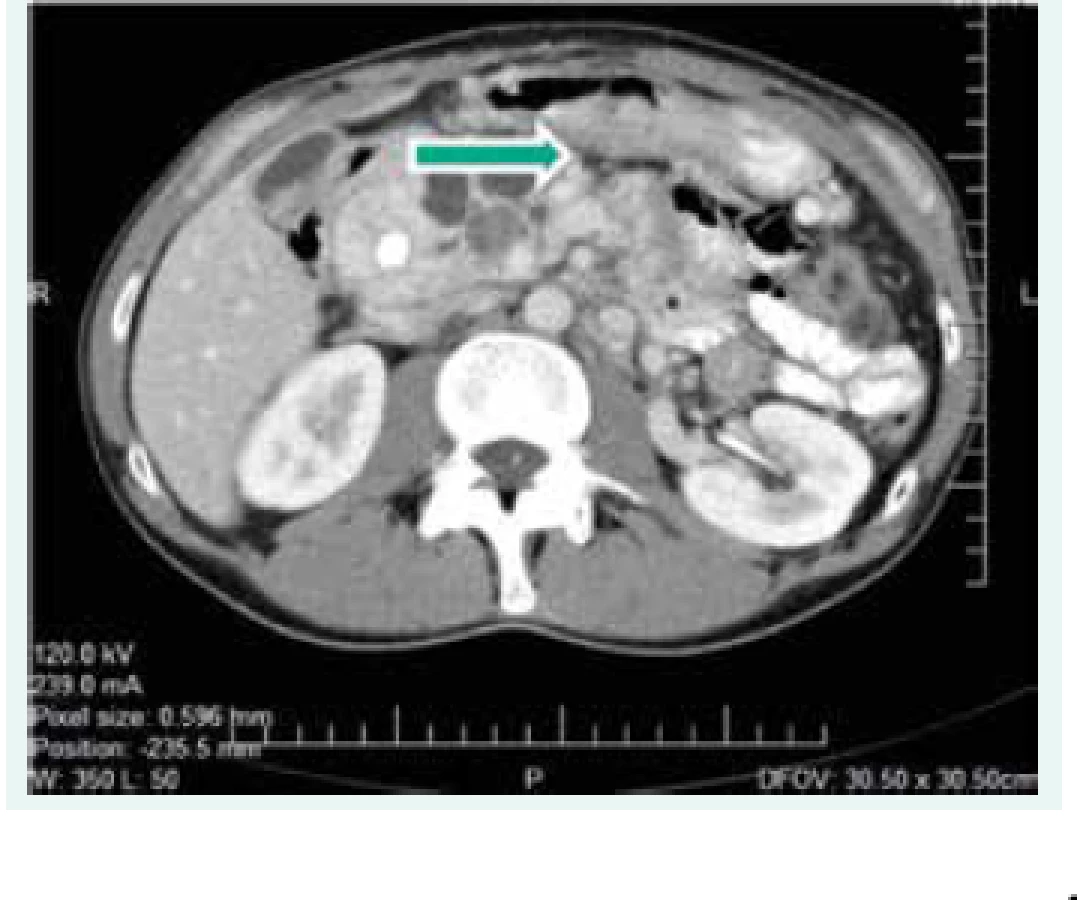
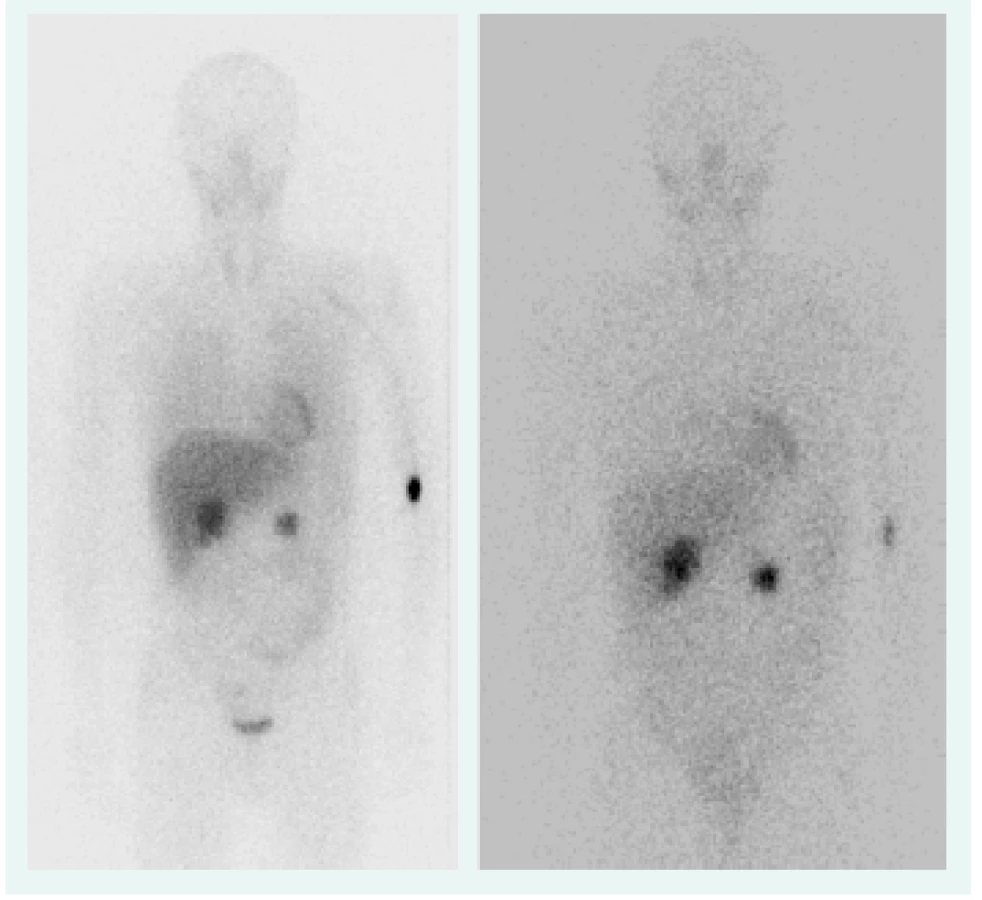
Vzhľadom na multisystémovú nádorovú manifestáciu sme realizovali genetické testovanie na mutáciu VHL génu s potvrdením syndrómu VHL typ 1 (s bilaterálnym feochromocytómom). Pri negatívnej rodinnej anamnéze ide o pacienta so zistenou mutáciou de novo (stanovenie konkrétneho typu mutácie v tej dobe nebolo možné realizovať). Pacient podstúpil obojstrannú adrenalektómiu. Histologicky sa potvrdil bilaterálny feochromocytóm. Pre cholestázu bola vykonaná endoskopická retrográdna cholangiopankreatografia (ERCP) s endoskopickou papilosfinkterotómiou a implantáciou duodenobiliárneho stentu.
Po prepustení z nemocnice bol pacient dispenzarizovaný s pravidelnými kontrolami, s dobrou kvalitou života, bez výraznejších subjektívnych ťažkostí. Počas tohto obdobia mu boli pravidelne realizované výmeny duodenobiliárnych drénov a sklerotizácia a ligácia varixov pažeráka.
V roku 2010 sa stav komplikoval akútnou cholangoitídou s rozvojom sepsy, s progresiou cystických lézií pankreasu podľa CT vyšetrenia brucha. Stav sa postupne komplikoval hemoragickým šokom pri masívnej hemateméze pri rozsiahlych fundických varixoch pri portálnej gastroduodenopatii. Po stabilizácii stavu bola na chirurgickom pracovisku vzhľadom na ťažkú portálnu hypertenziu a rozsah krvácania realizovaná azygoportálna dekonexia. Napriek tomu došlo k recidíve krvácania z hornej časti tráviaceho traktu. Pacient zomrel ako 35-ročný.
Kazuistika 2
V druhej kazuistike popisujeme prípad pacientky narodenej v roku 1965, ktorá ako 33-ročná podstúpila duodenopankreatektómiu pre neuroendokrinný tumor pankreasu, bez histologizácie nálezu. Ako 39-ročnej bol pacientke diagnostikovaný cereberálny hemangioblastóm, o 2 roky neskôr hemangióm krčnej chrbtice a benígny nádor pečene, ktoré boli kompletne chirurgicky odstránené. Rodinná anamnéza bola pozitívna – otec pacientky bol ako 41-ročný operovaný na nádor mozgu, otcov brat zomrel na karcinóm obličky diagnostikovaný vo veku 48 rokov a druhý otcov brat zomrel na nádor mozgu ako 30-ročný.
V apríli roku 2007 bola pacientka prijatá na našu kliniku za účelom diferenciálneho doriešenia stavu. U pacientky vzhľadom na výskyt viacerých nádorových ochorení v mladom veku s prihliadnutím na pozitívnu rodinnú anamnézu sme suponovali VHL syndróm a indikovali sme genetické vyšetrenie. Genetická analýza potvrdila patologickú germinatívnu mutáciu génu VHL, čím sme definitívne potvrdili syndróm VHL typ 2 (konkrétnu mutáciu laboratórium nestanovovalo). Pozitívny výsledok znamená, že pacientka zdedila patologickú zárodočnú mutáciu od svojho otca, a teda sú syndrómom VHL ohrození aj pokrvní príbuzní. Keďže najzávažnejšou komplikáciou ochorenia je vznik svetlobunkového renálneho karcinómu, bolo doplnené MR vyšetrenie, ktoré detekovalo iba viacero cystických ložísk v oboch obličkách. U pacientky sa doteraz nepotvrdili hemangiómy sietnice.
Pacientka, rovnako ako pacient z 1. kazuistiky, bola zaradená do vysokorizikovej skupiny s nevyhnutnou celoživotnou dispenzarizáciou, ktorá zahŕňa ročne abdominálnu sonografiu, sledovanie urológom, kontroly očného pozadia, MRI mozgu a miechy, CT/MRI vyšetrenie brucha a laboratórne kontroly katecholamínov pre možný vznik feochromocytómu.
Pacientka je v súčasnosti bez klinických ťažkostí, bez progresie lokálnych nálezov a nevyžaduje terapeutickú intervenciu.
Zhrnutie
VHL je zriedkavé geneticky podmienené ochorenie s rôznorodou symptomatológiou a variabilným priebehom podľa lokalizácie nádoru alebo cýst. Priemerná dĺžka života je 50 rokov. Najčastejšou príčinou úmrtia je karcinóm obličky a hemangioblastómy CNS.
Na rozdiel od literatúry ani jeden z našich pacientov nemal hemangioblastóm sietnice ani svetlobunkový karcinóm obličky. Pacient z 1. kazuistiky mal negatívnu rodinnú anamnézu, cysty a nádor pankreasu, bilaterálny feochromocytóm, hemangioblastóm mozočka a spinálneho kanála. Prvé prejavy ochorenia začali v 19. roku života. Geneticky bola potvrdená mutácia génu VHL de novo.
Pacientka z 2. kazuistiky mala pozitívnu rodinnú anamnézu, neuroendokrinný tumor pankreasu a benígny tumor pečene, hemangioblastóm mozočka a krčnej miechy. Genetickou analýzou bola zistená patologická germinatívna mutácia génu VHL génu od otca.
Z našich zistení, pacient so sporadickou formou mal závažnejší priebeh ochorenia v porovnaní s familiárnou formou u pacientky v 2. kazuistike, u ktorej je priebeh menej agresívny. V literatúre sme našli minimum porovnávaní závažnosti klinického stavu pri formách de novo alebo hereditárnych [13], pravdepodobne i vzhľadom na to, že závažnosť ochorenia je variabilná najmä v závislosti od samotnej mutácie [14]. Boli popísané genotypovo-fenotypové mutácie, napr. pri mutáciách nonsense, frameshift alebo missense mutáciách – vznik von Hippelovho-Lindauovho syndrómu typ 1, zatiaľ čo pri mutáciách truncated – vznik renálneho svetlobunkového karcinómu, alebo pri substitučných mutáciách – vznik retinálnych hemangioblastómov [15].
Je nutné myslieť na tento syndróm, hlavne pri výskyte typických VHL nádorov u pacientov v mladšom veku, dôsledne odobrať rodinnú anamnézu, v prípade potvrdenia diagnózy realizovať i genetickú konzultáciu a testovanie rodinných príslušníkov, čo môže viesť k včasnej prevencii a diagnostike nádorových ochorení. Liečba pacienta s VHL syndrómom vyžaduje multidisciplinárnu spoluprácu s nutnosťou celoživotného sledovania pacienta. Správna diagnostika a adekvátne liečebné postupy dokážu aj napriek nepriaznivej prognóze zlepšiť kvalitu a dĺžku života.
MUDr. Patrícia Páleníková, PhD.
patricia.palenikova@gmail.com
V. interná klinika LF UK a UNB, Nemocnica Ružinov,
Bratislava,
Slovenská republika
www.unb.sk
Doručeno do redakce 3. 9. 2016
Přijato po recenzi 23. 10. 2016
Sources
1. Maher ER, Yates JRW, Harries R et al. Clinical Features and Natural History of von Hippel-Lindau Disease. QJM 1990; 77(283): 1151–1163.
2. Maher ER, Kaelin WG Jr. von Hippel-Lindau Disease. Medicine (Baltimore) 1997; 76(6): 381–391.
3. Richards FM, Phipps ME, Latlf F et al. Mapping the Von Hippel-Lindau disease tumour suppressor gene: identification of germline deletions by pulsed field gel electrophoresis. Hum Mol Genet 1993; 2(7): 879–882.
4. Plate KH, Breier G, Weich HA et al. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992; 359(6398): 845–848.
5. Glasker S, Neumann HPH, Koch CA et al. Von Hippel-Lindau Disease. In: De Groot LJ (ed), Beck-Peccoz P, Chrousos G et al: Endotext. South Dartmouth (MA): MDText.com; 2000. Dostupné z WWW: <https://www.ncbi.nlm.nih.gov/books/NBK279124/>
6. Hes FJ, Höppener JWM, Lips CJ. Pheochromocytoma in Von Hippel-Lindau Disease. J Clin Endocrinol Metab 2003; 88(3): 969–974.
7. Maher ER, Neumann HPH, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet 2011; 19(6): 617–623. Dostupné z DOI: <http://dx.doi.org/10.1038/ejhg.2010.175>.
8. Plevova P, Novotny J, Křepelova A. Von Hippel - Lindauova choroba. Klinická onkologie 2009; 22(Suppl 1): S23-S24. Dostupné z WWW: <http://www.linkos.cz/files/klinicka-onkologie/149/3449.pdf>.
9. Shuin T, Yamasaki I, Tamura K et al. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol 2006; 36(6): 337–343.
10. Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol 2007; 2 : 145–173.
11. Von Hippel-Lindau Syndrome overview. American Society of Clinical Oncology. Editorial Board April 2013. Dostupné z WWW:
12. Zelinka T, Widimský J Jr. Feochromocytom – proč je jeho časná diagnóza pro pacienta důležitá? Vnitř Lék 2015; 61(5): 487–491.
13. Lee JS, Lee JH, Lee KE et al. Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-α binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med Genet 2016; 17(1): 48. Dostupné z DOI: <http://dx.doi.org/10.1186/s12881–016–0306–2.
14. Vaňuga P, Pura M, Kreze A Jr. Genetické pozadie nádorov adrenomedulárneho a extraadrenálneho chromafinného tkaniva – aktuality. Vnitř Lék 2010; 56(12): 1296–1302.
15. McNeill A, Rattenberry E, Barber R et al. Genotype-phenotype correlations in VHL exon deletions. Am J Med Genet A 2009; 149A(10): 2147–2151. Dostupné z DOI:
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 12
Most read in this issue
- Trombofília
- Káva ako hepatoprotektívny faktor
- Perorální antidiabetika v léčbě diabetes mellitus 1. typu
- Takotsubo syndrom: incidence, etiologie, komplikace, léčba a prognóza