
Schnitzlerové syndrom
Diferenciální diagnostika, přehled léčebných možností a popis 5 případů léčených anakinrou
Schnitzler’s Syndrome
Differential diagnostics, an overview of therapeutic options and description of 5 cases treated with anakinra
Schnitzler’s syndrome is an acquired auto-inflammatory disease of still unclear origin. The Strasbourg criteria were adopted (non-infectious fever, chronic urticaria, changes in the bone structure, leukocytosis and higher values of inflammatory markers – CRP and presence of monoclonal immunoglobulin mostly of type IgM, very rarely of IgG) to establish this diagnosis. The first-choice therapy for this disease is the blocking of interleukin-1 effects. In practice, the interleukin-1 receptor antagonist, anakinra, is the most commonly used. Currently reports also appear of the use of other medicines blocking the effect of interleukin-1, namely canakinumab and rilonacept. We have been treating 5 patients with anakinra (108, 72, 33, 32 and 1 months) on a long-term basis. In all the patients, we commenced administration of anakinra in a dose of 100 mg once a day. As a result of 100 mg being administered once a day, all symptoms went away completely in 4 patients, while they receded by about 75 % in 1 patient, without disappearing completely. This patient needs an increased dose of 2 ampoules per day on the days of spontaneously intensified medical ailments. After one year of treatment it turned out for one of the four patients whose symptoms had completely disappeared when administered the 100mg daily dose, that he only needed the respective dose of anakinra at 48-hour intervals. However this patient does not tolerate further extension of the intervals between dose administrations. We have not recorded any adverse effects of anakinra in the course of the treatment, and no decline in the efficiency of anakinra has been observed: it acts as effectively now as it did at the beginning of the treatment. The text discusses the differential diagnostics of the Schnitzler syndrome.
Key words:
anakinra – auto-inflammatory diseases – canakinumab – fever of unknown origin – FUO – interleukin 1 – cryopyrin-associated autoinflammatory syndrome (CAPS) – monoclonal gammopathy – rilonacept – Schnitzler’s Syndrome – Adult Still’s disease
Authors:
Zdeněk Adam 1; Anna Šedivá 2; Renata Koukalová 3; Zdeněk Řehák 3; Hana Petrášová 4; Petr Szturz 1; Zdenka Adamová 5; Eva Vetešníková 1; Luděk Pour 1; Marta Krejčí 1; Viera Sandecká 1; Eva Pourová 6; Zdeňka Čermáková 7; Sabina Ševčíková 8; Zdeněk Král 1; Jiří Mayer 1
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno, pracoviště Bohunice
1; Ústav imunologie 2. LF UK a FN v Motole, Praha
2; Oddělení nukleární medicíny, centrum PET, RECAMO, Masarykova onkologického ústavu Brno
3; Radiologická klinika LF MU a FN Brno, pracoviště Bohunice
4; Ambulance pro děti a dorost, Obilní trh 9, Brno
5; Ambulace praktického lékaře pro dospělé, Pustiměř
6; Oddělení klinické biochemie FN Brno a Katedra laboratorních metod LF MU Brno
7; Ústav patologické fyziologie LF MU Brno
8
Published in:
Vnitř Lék 2016; 62(9): 713-727
Category:
Reviews
Overview
Schnitzlerové syndrom je získané autoinflamatorní onemocnění zatím nejasné etiologie. Pro stanovení této diagnózy byla přijata Štrasburská kritéria (neinfekční horečka, chronická kopřivka, změny kostní struktury, leukocytóza a zvýšené hodnoty zánětlivých markerů – CRP a přítomnost monoklonálního imunoglobulinu většinou typu IgM, zcela výjimečně IgG). Léčbou volby pro tuto nemoc je blokáda účinků interleukinu 1. V praxi je nejčastěji využíván antagonista receptoru pro interleukin 1, anakinra. V současnosti se objevují zprávy i o použití dalších léků blokujících účinek interleukinu 1, kanakinumab a rilonacept. Dlouhodobě léčíme 5 pacientů preparátem anakinra (108, 72, 33, 32 a 1 měsíc). U všech nemocných jsme začali s aplikací anakinry v dávce 100 mg 1krát denně. Při dávkování 100 mg 1krát denně vymizely kompletně všechny příznaky u 4 nemocných, pouze u 1 nemocného došlo k ústupu příznaků asi o 75 %, nikoliv však k úplnému vymizení. Tento pacient potřebuje navýšení dávky ve dnech se spontánním zintenzivněním potíží na 2 ampulky denně. U jednoho ze 4 pacientů, u nichž příznaky při dávkování 1krát denně zcela vymizely, se po roce léčby ukázalo dostačující podávat anakinru v 48hodinových intervalech. Delší prodloužení intervalu mezi aplikacemi však netoleruje. V průběhu léčby jsme nezaznamenali žádné nežádoucí účinky anakinry a v průběhu léčby nedochází k poklesu účinnosti, aplikace anakinry jsou stejně účinné jako na začátku léčby. V textu rozvádíme diferenciální diagnostiku Schnitzlerové syndromu.
Klíčová slova:
anakinra – autoinflamatorní choroby – kanakinumab – horečka nejasné etiologie (Fever of unknown origin – FUO) – interleukin 1 – kopřivka – kryopyrin asociovaný periodický syndrom (CAPS) – monoklonální gamapatie – rilonacept – Schnitzlerové syndrom – Stillova choroba dospělých
Úvod
V roce 1972 a 1974 poprvé popsala francouzská kožní lékařka Liliane Schnitzler 5 pacientů s chronickými neinfekčními horečkami, chronickou kopřivkou (urtikariální vaskulitidou) spojenou s bolestí kostí, svalů, kloubů. Následující vyšetření těchto pacientů odhalilo kostní změny (hyperostózu), lymfadenopatii a přítomnost monoklonálního imunoglobulinu třídy IgM [1–3]. V té době se jednalo o novou klinickou jednotku neznámé etiologie, která byla pojmenována po autorce prvního popisu jako Schnitzlerové syndrom.
Zásadním laboratorním znakem této nemoci je přítomnost monoklonálního imunoglobulinu typu IgM, výjimečně ale také typu IgG. Dominantní úlohu v rozvoji základních zánětlivých rysů této nemoci má však interleukin 1β. Zatím zůstává záhadou, jaký je vztah zvýšené koncentrace interleukinu 1β a monoklonálního imunoglobulinu [4–6].
Pro nemocné s touto chorobou nebyl dlouho dostupný žádný účinný lék, a tak velmi trpěli. Antihistaminika a imunosupresivní léky nebyly u těchto pacientů účinné, protizánětlivé léky jen tlumily bolest a přechodně snižovaly horečku. Pouze vysoké dávky glukokortikoidů mírnily příznaky nemoci, ale na druhé straně vyvolávaly známé nepříjemné projevy dlouhodobého podávání vysokých dávek glukokortikoidů. Teprve až se do klinické praxe dostaly léky blokující cíleně účinek interleukinu 1 (anakinra), bylo možné ve většině případů zcela odstranit příznaky této nemoci, či příznaky alespoň zmírnit [5,6].
Monoklonální gamapatie typu IgM provázející Schnitzlerové syndrom, se asi u 15–20 % pacientů při dlouhodobém sledování transformuje v maligní lymfoproliferativní onemocnění [7]. Pak by tito nemocní měli být léčeni postupy běžnými pro uvedené lymfoproliferativní onemocnění. Proto jsou pacienti se Schnitzlerové syndromem většinou léčeni a sledování hematoonkology.
Dříve byli tito pacienti při nedostupnosti účinné léčby při dlouhodobém průběhu nemoci poškozováni depozity AA-amyloidu, protože chronický zánětlivý proces vedl k dlouhodobě zvýšené koncentraci amylopeptidu A, který se ukládal ve formě AA-amyloidu [8,9].
V roce 2007 jsme popsali první případ v České republice, který byl úspěšně léčen preparátem anakinra. Postupně jsme tuto nemoc diagnostikovali a začali léčit u dalších 4 nemocných.
V době, v níž jsme tuto léčbu zahajovali, jsme si kladli otázku, jak dlouho bude tato léčba úspěšná a zda se v jejím průběhu neobjeví neočekávané nežádoucí účinky. V následujícím textu referujeme o dlouholeté léčbě celkem 5 pacientů. Prvnímu pacientovi je anakinra aplikována již 9. rokem. Léčba v průběhu let nic neztratila na své účinnosti. V diskusi přinášíme informace o diferenciální diagnostice této jednotky, která se řadí nejen do kategorie monoklonálních gamapatií, ale také do kategorie získaných autoinflamatorních chorob, a proto také ve velmi stručné podobně informujeme o této kategorii nemocí.
Popis souboru pacientů
První pacient
Muž (*1944) je na našem pracovišti sledován od roku 2002 (od svých 58 let) s monoklonální gamapatií typu IgM. Původní histologie kostní dřeně neprokázala maligní lymfoproliferaci. Trepanobiopsie byla obtížnější, kost kladla podstatně větší odpor bioptické jehle, než jsme zvyklí u pacientů s mnohočetným myelomem.
Pacient míval již před rokem 2002 ataky výsevů kopřivkových pupenů na velkých plochách. Kožní urtika byla v péči kožních specialistů a byl dlouho a marně hledán vyvolávající podnět. Žádná lokální léčba a perorálně podávaná antihistaminika nepomáhala. Urtika byla v tomto případně spojena s velmi silným svěděním, zatímco další čtyři pacienti svědění neudávali (obr. 1 a 2). Dalšími problémy byly bolesti v kostech dolních končetin a bolesti v oblasti pánve a kyčelních kloubů. Bolesti postihovaly dominantně ty části skeletu, v nichž byly zřetelné sklerotické (hyperostotické) změny při CT vyšetření a zvýšená aktivita na scintigrafii skeletu.

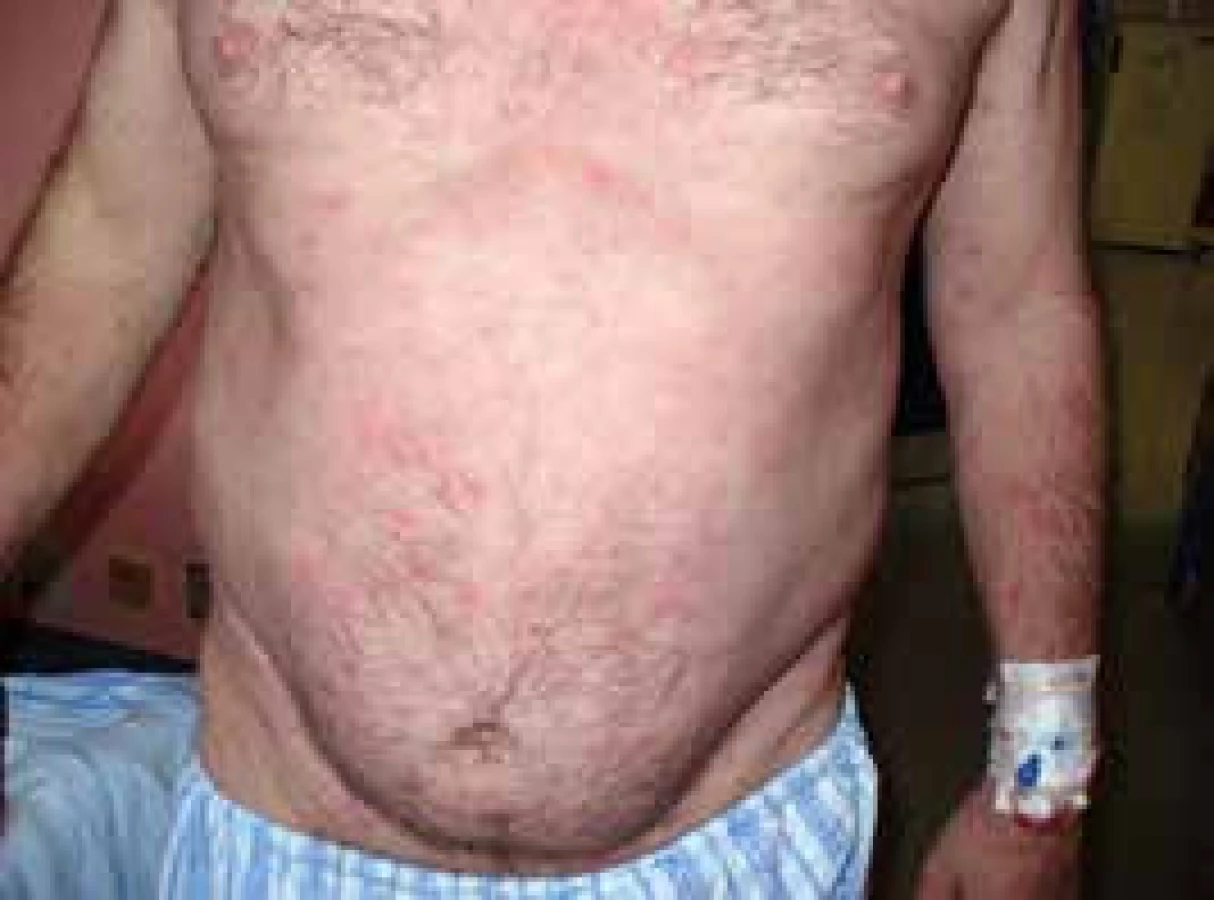
Ze všech léčebných alternativ, které jsme testovali od roku 2002 až do roku 2007, měl efekt pouze prednison, ale až v poměrně vysokých dávkách, 20–40 mg denně. Tyto dávky prednisonu indukovaly steroidní diabetes mellitus, kterým pacient trpí dodnes. Bolesti v kostech v místech osteosklerotických změn se zmenšovaly po aplikaci bisfosfonátů.
Podrobný popis celého případu až do roku 2007 jsme již publikovali [10].
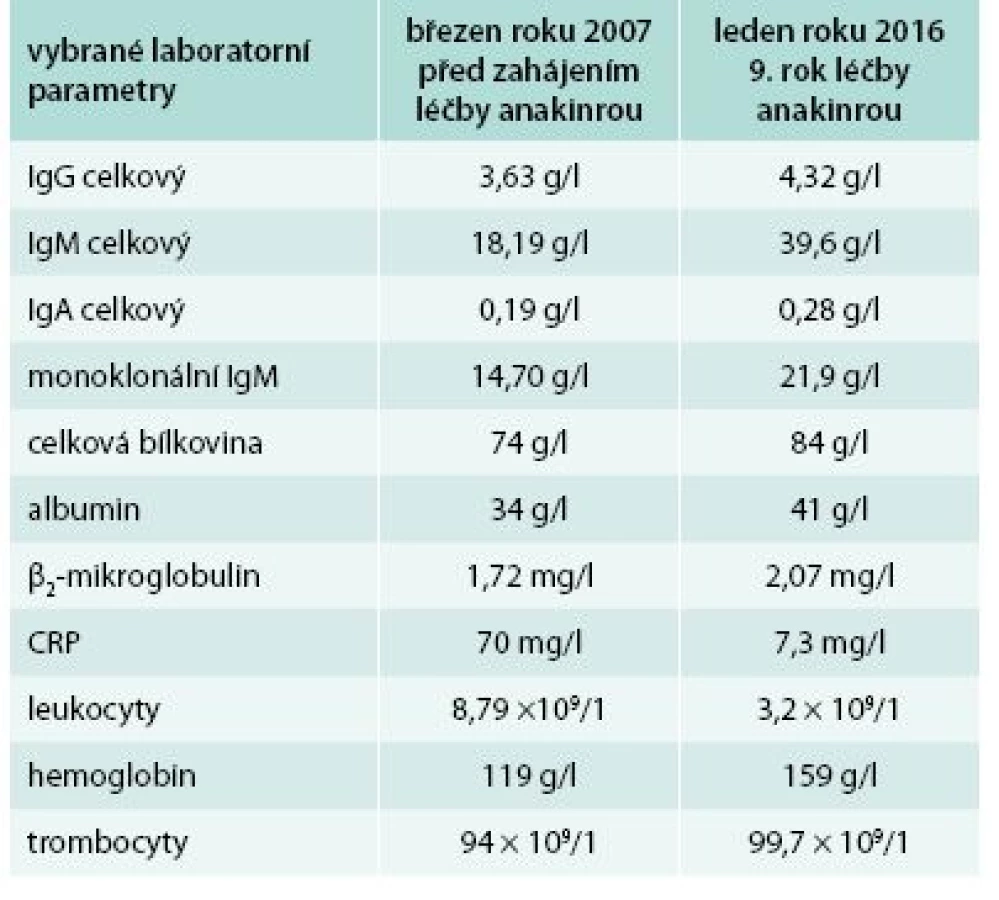
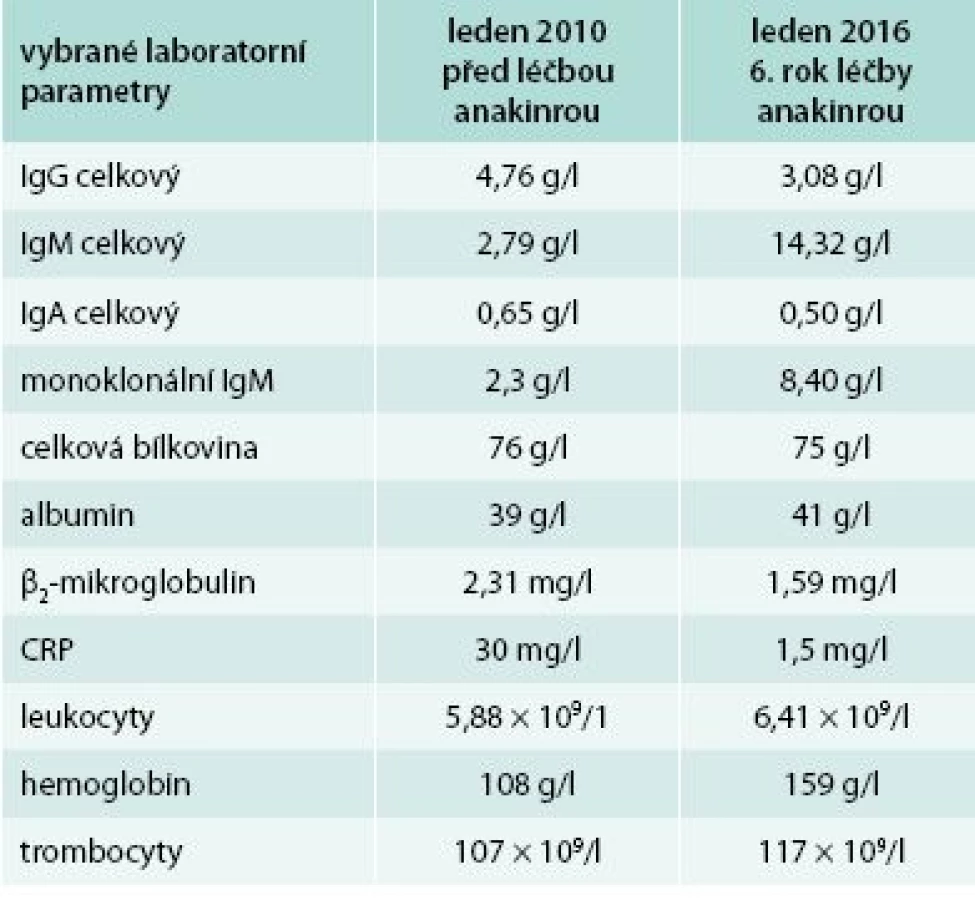
Od roku 2007 byla zahájena léčba preparátem anakinra v dávce 1 ampulka (100 mg) s.c. denně. Byl to první pacient s touto chorobou v ČR, u něhož byla v říjnu roku 2007 zahájena léčba anakinrou. Již po první aplikaci došlo k vymizení exantému. Svědící kopřivkový exantém se objevil pouze ve dnech, v nichž si pacient zapomněl aplikovat injekci anakinry. Zároveň s kožními změnami vymizely všechny ostatní příznaky – teploty, bolesti v oblasti skeletu a celková únava. Pacientův stav se natolik zlepšil, že je schopen v létě na kole zvládat trasy do 50 km. Vývoj laboratorních parametrů ukazuje tab. 1. Je vidět, že v průběhu sledování se postupně zvyšuje koncentrace celkového i monoklonálního IgM, a proto je v plánu kontrolní trepanobiopsie a kontrolní histologie kostní dřeně, abychom ověřili, zda nedošlo k transformaci v lymfoproliferativní onemocnění. Před zahájením léčby byla vysoká hodnota CRP provázena nižší hladinou hemoglobinu i albuminu jako důsledek chronického zánětu (anémie chronických chorob).
Druhý pacient
Muž (*1963) byl poprvé na našem pracovišti vyšetřen v roce 2005 ve svých 42 letech. Přišel k diferenciální diagnostice v té době asymptomatické krční lymfadenopatie. Histologické vyšetření uzliny neprokázalo žádnou malignitu, pouze nespecifické zánětlivé změny.
V roce 2006 si poprvé všiml kopřivkové vyrážky na celém těle. Kožní projevy nesvědily, nebolely. Na kůži byl zřetelný rudý drobný raš, difuzně postihující celé tělo a později se sléval do větších morf, které byly přítomné jak na trupu, tak na končetinách, jak ilustrují obr. 3 a 4.
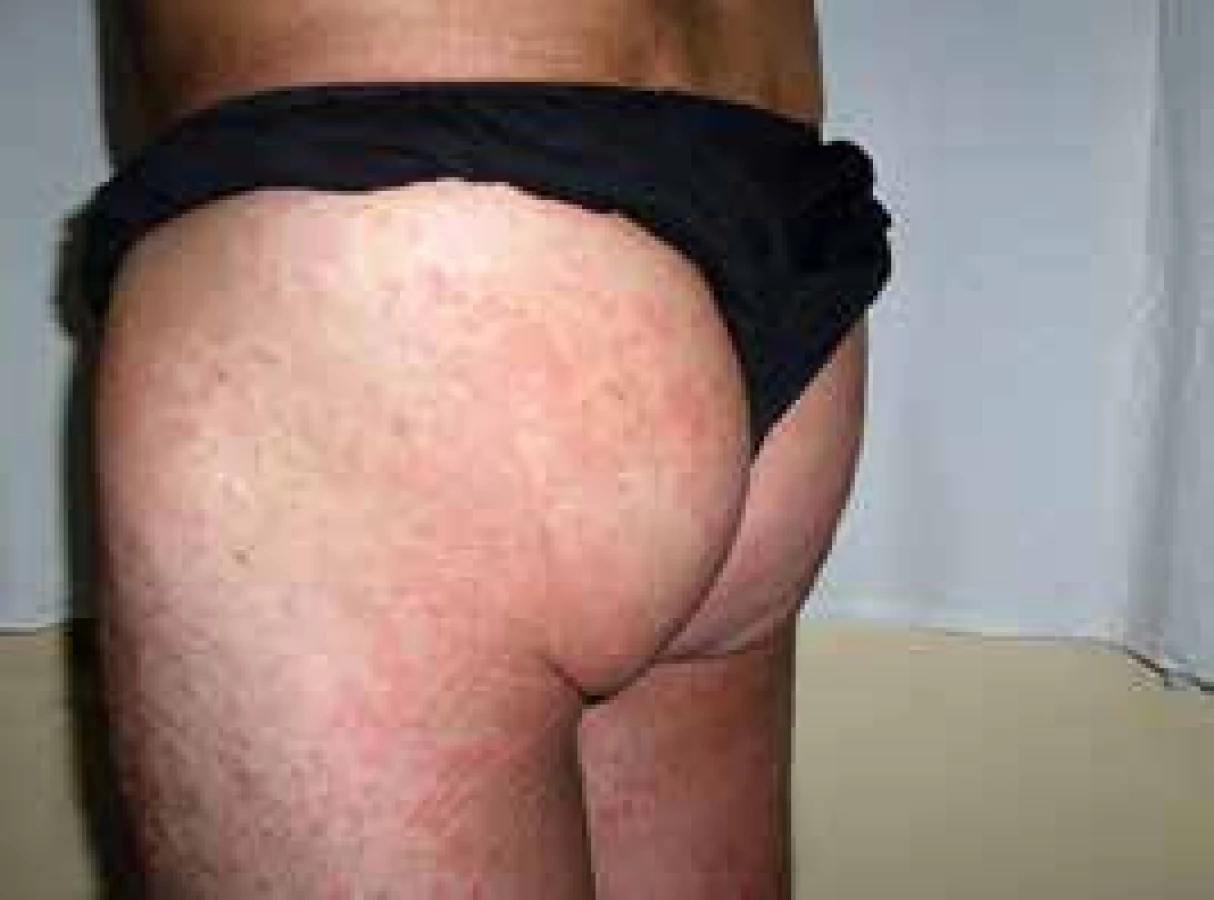
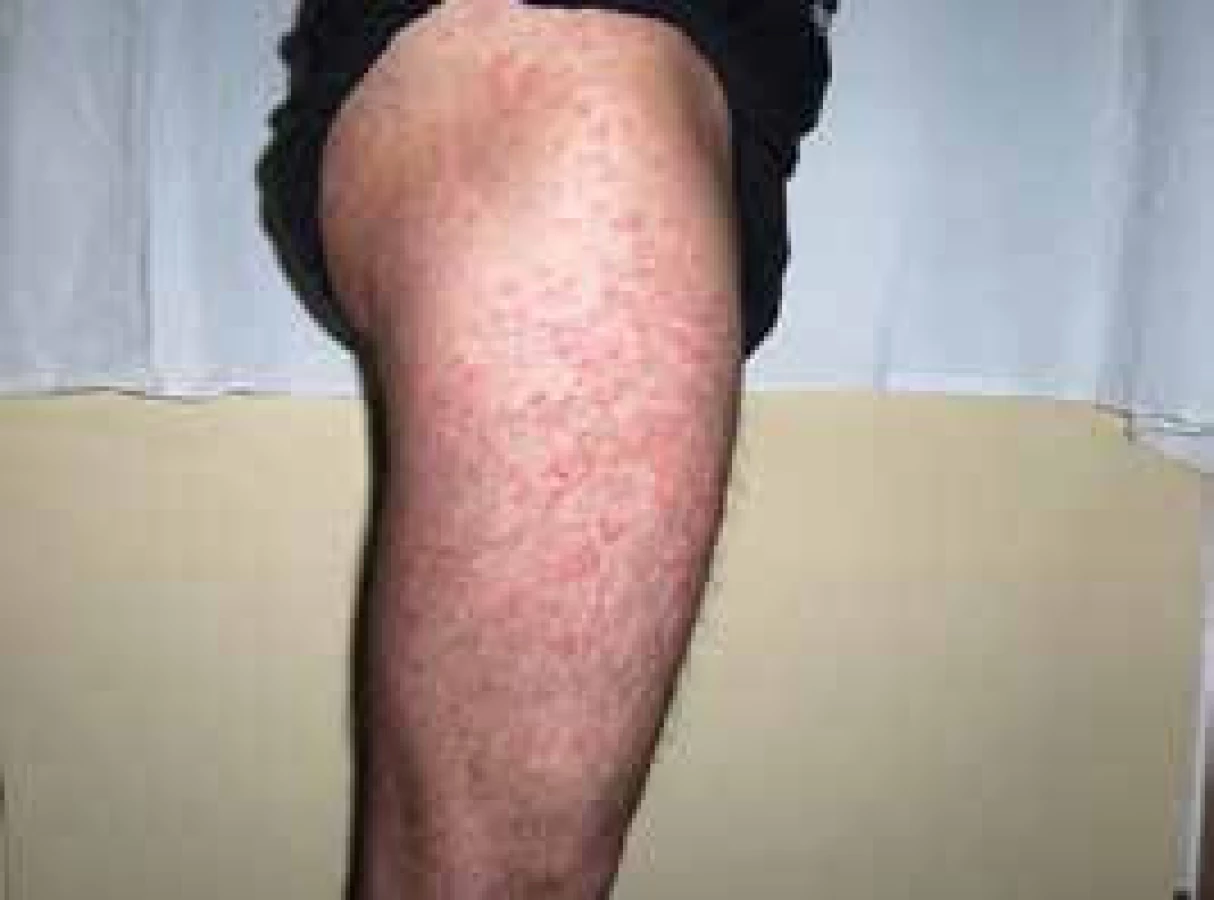
Při prvním vyšetření dostal lokálně kortikoidy a nemoc byla ošetřujícím kožním lékařem diagnostikována jako pityriasis (rosea) Gilbert. Léčba lokálně aplikovanými kortikoidy neměla léčebný efekt.
Po necelém roce od první erupce kopřivkového raše se objevily bolesti v kostech bérce. Intenzita bolestí v průběhu času sílila. A postupně se přidávaly i febrilie.
Byla hledána příčina horeček. V rámci hledání ložiska infekce přišel pacient o mnoho zubů zcela zbytečně, protože jejich odstranění nic neovlivnilo. V roce 2008 již bolesti nepostihovaly pouze dolní končetiny, ale také končetiny horní, klouby i kosti. Potíže někdy kulminovaly v záchvatech, během nichž se zvýšila teplota, zvýšila se intenzita bolestí a objevil se další výsev kopřivky a současně i pocit zimnice. Po těchto atakách se horšily i otoky kloubů.
Kožní lékaři, kteří o nemocného do roku 2009 pečovali, zjistili, že lokální kortikoidy a antihistaminika nemají vůbec žádný efekt. Potíže mírnil pouze prednison a později metylprednisolon. V době, kdy byl odeslán na naše pracoviště, užíval 54 mg metylprednisolonu (Medrol) denně, což způsobilo Cushingův syndrom a funkční útlum nadledvinek.
V roce 2009 se dostavil na naši ambulanci pro monoklonální gamapatie, protože měl prokázaný monoklonální imunoglobulin IgM (M-IgM) v séru. V té době pacienta pořád trápila urtika, bolesti kloubů, ale i kostí všech končetin, otoky kloubů a kolísavé febrilie. Vzestup teploty vždy signalizoval další výsev kopřivky a zhoršení bolestí.
Laboratorní vyšetření prokázalo monoklonální imunoglobulin typu IgM a zvýšené hodnoty CRP. Krevní obraz byl v mezích fyziologických hodnot.
Klasické snímky kostí neprokázaly nic patologického, zatímco MR skeletu prokázalo abnormální strukturu skeletu ve smyslu osteosklerotických změn. PET/CT vyšetření prokázalo sklerotizaci obou klíčních kostí a zesílení kortikalis v některých částech skeletu. Klasická scintigrafie skeletu prokázala abnormálně vyšší aktivitu v oblasti sterna, v obou humerech, v distální části levého femoru, v proximální polovině obou tibií a taktéž vysokou aktivitu v oblasti sakra. Tyto změny odpovídaly Schnitzlerové syndromu.
Léčba preparátem anakinra byla zahájena v únoru roku 2010. Již za 6 hod po první aplikaci vybledla vyrážka, do 24 hod zcela vymizela a v průběhu léčby anakinrou se již neobjevila. Po zahájení léčby vymizely zcela febrilie. Bolesti kloubů a otoky kloubů se zmenšily asi o 75 %, ale zcela při léčbě nevymizely.
Od února roku 2010 pokračuje léčba anakinrou. V únoru roku 2016 měl pacient za sebou 6 let léčby anakinrou. V průběhu léčby bylo vyzkoušeno přidání kolchicinu v dávce 0,5–1,0 mg denně s cílem dosáhnout úplného vymizení příznaků nemoci. Přidání kolchicinu v období od února roku 2014 do prosince roku 2014 do chronické medikace nepřineslo další zlepšení, navíc bylo ke konci podávání doprovázeno poklesem počtu krevních destiček na 50 × 109/l v terénu subklinické formy idiopatické trombocytopenické purpury s běžnými hodnotami trombocytů kolem 100 × 109/l. V průběhu léčby ale nebyly zjištěny žádné nežádoucí účinky anakinry.
Kontrolní NaF-PET/CT v roce 2015 neprokázalo žádný patologický nález mimo změny skeletu, které stále přetrvávají, protože aktivita nemoci u tohoto pacienta nevymizela zcela (obr. 5).
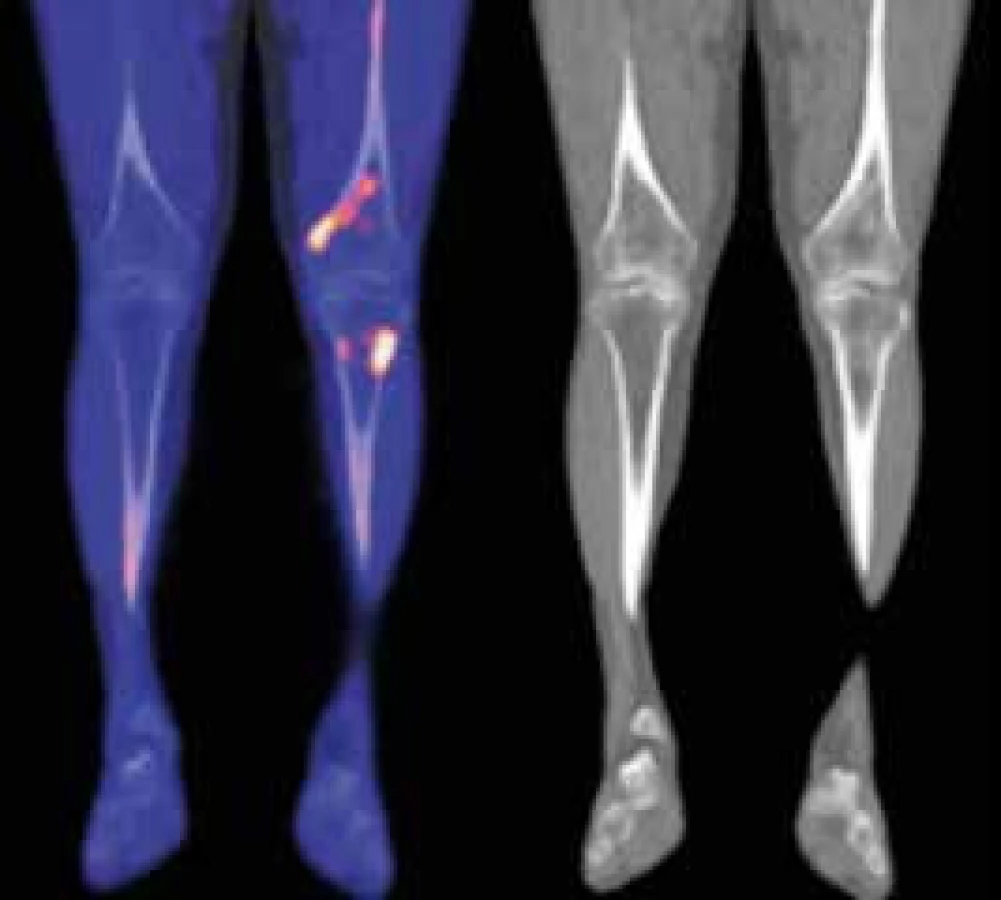
Změny ve vybraných laboratorních parametrech ukazuje tab. 2. Z tabulky je zřetelné, že před léčbou byla vyšší hodnota CRP, nižší koncentrace hemoglobinu, což se při zahájení léčby upravilo a nyní jsou hodnoty CRP i hemoglobinu normální, ale je vidět pomalé zvyšování koncentrace celkového i M-IgM
Třetí pacient
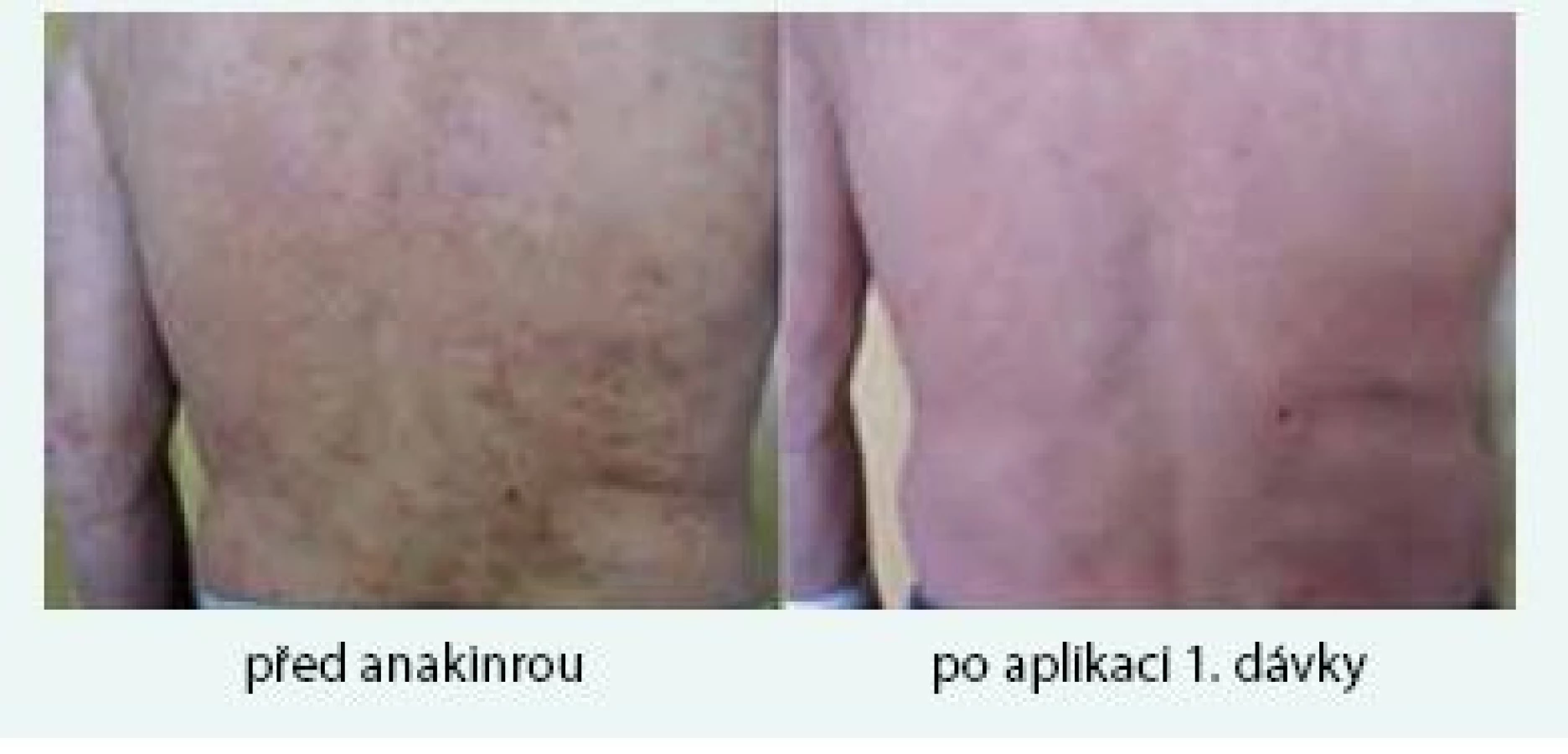
Muž (*1949) zpozoroval první příznaky nemoci, tedy patologickou únavu, poprvé v červnu roku 2009 ve svých 60 letech. Začátkem listopadu roku 2009 zjistil, že mívá večer vždy zvýšené teploty, kolem 37,5 °C, ne však vyšší než 38 °C. Počátkem prosince roku 2009 se objevily bolesti bércových kostí, nejintenzivněji je vnímal večer. Kosti byly bolestivé i na dotek. V prosinci roku 2009 také postřehl první výsevy kopřivkových pupenů. Kopřivkové morfy ale nikdy nesvědily. Kožní změny před a po podání přípravku anakinra dokumentuje obr. 6.
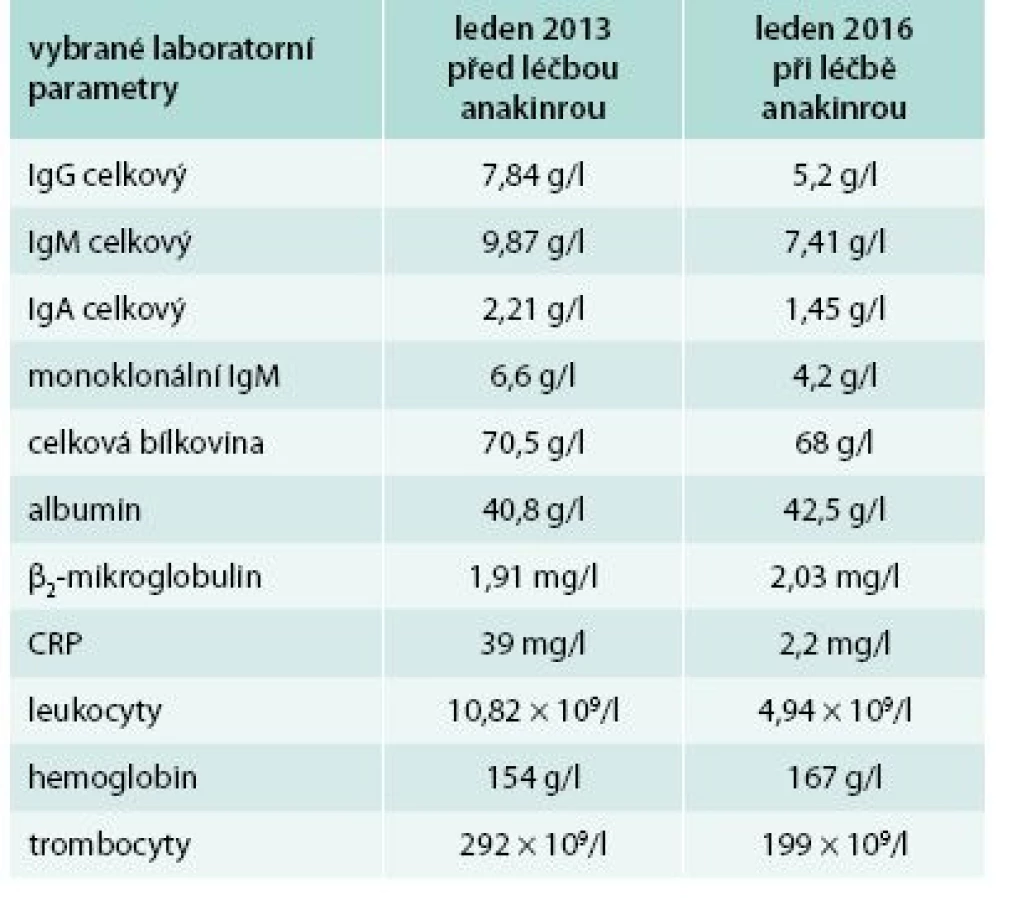
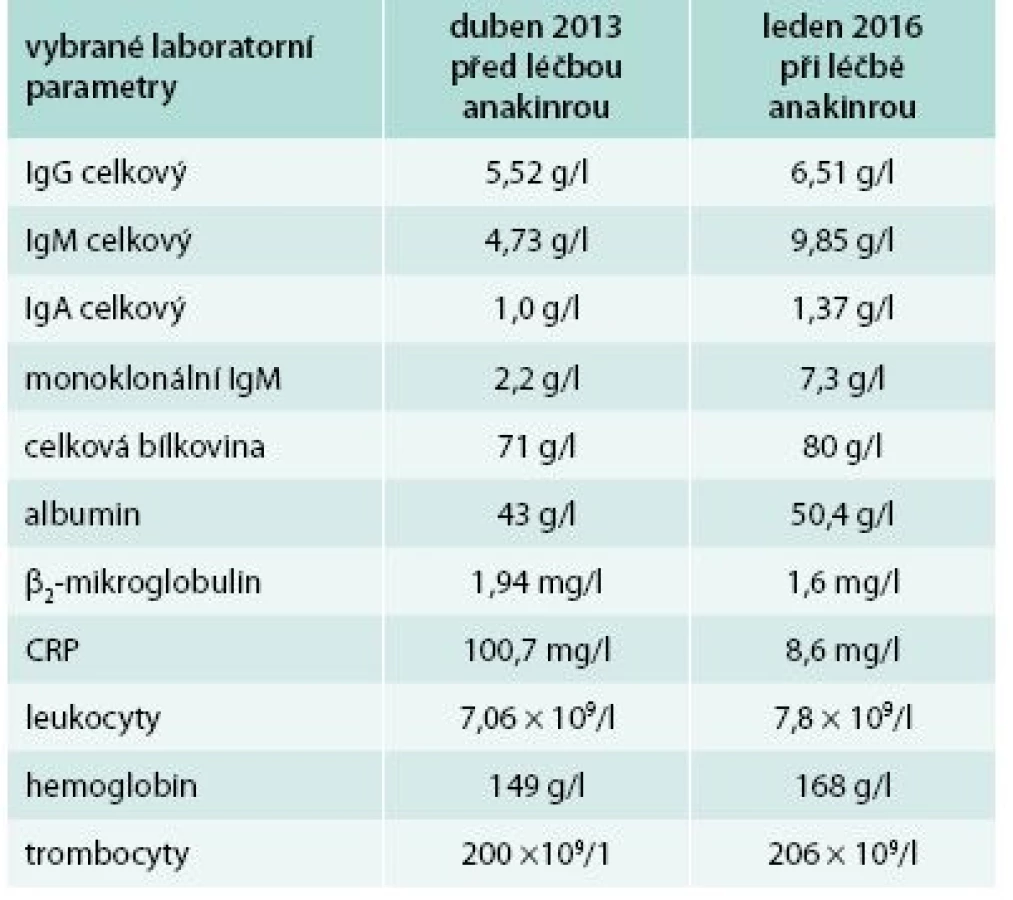
Bolesti v dlouhých kostech a horečky jej nakonec v květnu roku 2010 přivedly na interní oddělení regionální nemocnice. Hmotnost pacienta se snížila z 86 kg na 77 kg. Až při dalších vyšetřeních byl zjištěn M-IgM a tento nález vedl k odeslání nemocného na naše pracoviště. První vyšetření na naší klinice proběhlo v lednu roku 2013. Potvrdili jsme klasické příznaky Schnitzlerové syndromu: kopřivkový exantém, subfebrilie, bolesti kostí, patologický nález na scintigrafii skeletu a vypsali jsme žádost pro revizní lékaře o schválení úhrady léčby anakinrou. Léčbu jsme zahájili v dubnu roku 2013. Od té doby si jej pacient aplikoval v prvním roce 1krát denně. Veškeré příznaky nemoci při této léčbě vymizely. Po roce jsme otestovali prodloužení intervalů mezi aplikacemi anakinry a zjistili jsme, že symptomy nemoci se nevracejí, pokud se anakinra aplikuje v intervalu 48 hod, ne však v intervalech delších. Od té doby si je pacient aplikuje ob den sám. I při tomto dávkování se již nikdy nevrátily bolesti kostí a neinfekční teploty a celkový stav se upravil natolik, že pacient zvládá všechny aktivity podobně jako jeho vrstevníci. Jeho hmotnost se vrátila na hodnotu před začátkem nemoci. Laboratorní parametry před léčbou a při léčbě anakinrou v lednu roku 2016 uvádí tab. 3.
Čtvrtý pacient
Muž (*1955) si v roce 2008 ve svých 53 letech poprvé všiml nesvědících kopřivkových morf. Od té doby začal navštěvovat kožní lékaře. Erupce kopřivky byly spojené s difuzními bolestmi kostí, hlavně v oblasti dolních končetin. Po 3 letech ošetřování kožními lékaři byla odhalena přítomnost M-IgM, a proto byl v roce 2011 odeslán na odborné hematologické pracoviště v Praze s podezřením na Schnitzlerové syndrom. Klasická scintigrafie skeletu zobrazila ložiska zvýšeného vychytávání technecia v dlouhých kostech horních i dolních končetin.
Vzhledem k tomu, že se jednalo o IgM monoklonální gamapatii, byl v roce 2011 testován účinek chlorambucilu (Leukeran). Tato léčba probíhala dlouhodobě, ale s minimálním či žádným efektem. V lednu roku 2012 byl k chlorambucilu přidán prednison. Prednison se v této indikaci ukázal jako účinnější než chlorambucil a poněkud více tlumil potíže. Ale k žádné zásadní změně nedošlo. A tak byl pacient po 2 letech sledování předán k další léčbě na naše pracoviště. Po ověření diagnózy Schnitzlerové syndrom jsme vypsali žádost pro revizního lékaře o schválení úhrady léčby preparátem anakinra a léčba byla zahájena v červnu roku 2013. Od té doby léčba kontinuálně pokračuje. Při poslední kontrole na našem pracovišti (leden 2016), tedy po 2,5 roce léčby, pacient udává, že nemá žádné teploty, žádné bolesti kostí, žádnou kopřivku, prostě vůbec žádné symptomy nemoci při aplikaci anakinry 1krát denně. Jediný zdravotní problém je patologická únava. Ráno se probudí, aplikuje si anakinru, a pak ještě setrvá asi další 3 hod v posteli, než v sobě najde tolik energie, aby vstal a byl schopen úkonů běžného života. Po obědě je únava menší a je schopen se věnovat i svému koníčku – rybaření. Únava souvisí s kalendářním obdobím, v zimě je výraznější než v letních měsících.
Přetrvávající únavu jsme u tohoto pacienta zpočátku také hodnotili jako možný příznak nemoci a léčbu jsme doplnili přechodně od července roku 2014 do července roku 2015 o kolchicin v dávce 0,5 mg s postupným zvyšováním dávky až na 1,5 mg. Přidání kolchicinu a ani jeho opětovné vysazení však nijak neovlivnilo intenzitu patologické únavy. Odborné psychiatrické vyšetření tento příznak nyní uzavřelo jako chronickou depresivní poruchu. Ale ani antidepresiva, která mu psychiatři předepisují, tuto patologickou únavu zásadně nepotlačila.
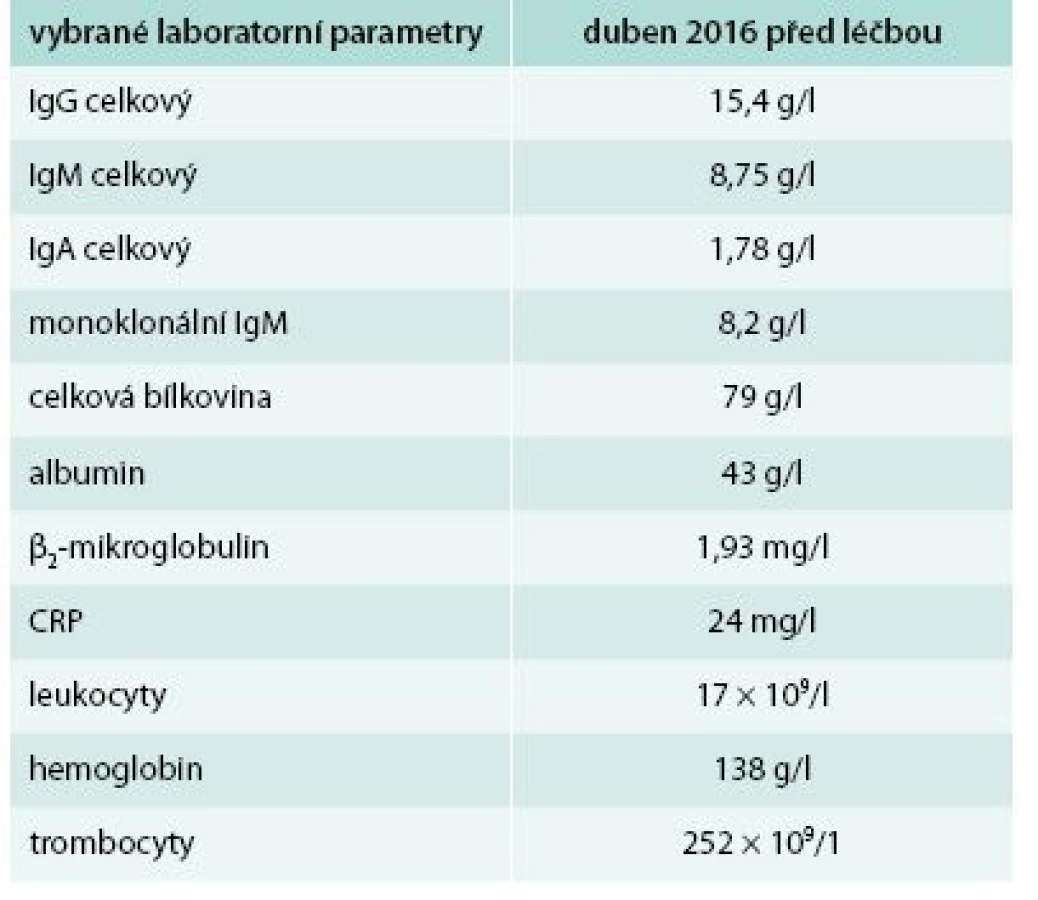
U tohoto pacienta lze léčbu trvající více než 2 roky zhodnotit jako dosažení kompletní klinické remise, ale stav komplikuje chronická depresivní porucha. Laboratorní parametry před zahájením léčby anakinrou a při poslední kontrole ukazuje tab. 4. Opět je zřetelný pokles CRP, vzestup koncentrace hemoglobinu a koncentrace M–IgM se v průběhu sledování jen nepatrně navýšil.
Pátý pacient
Pátý pacient je opět muž (*1971). První kopřivkové potíže se u něj objevily před 3 lety, tedy v roce 2013, ve 42 letech. Opět následovalo intenzivní testování a hledání možných alergenů, které by se daly označit za příčinu všech těchto problémů. Nebyl však nalezen vyvolávající alergen, ani se nepodařilo najít antihistaminikum, které by tlumilo potíže. Kopřivkový exantém pokrývá s různou intenzitou celé tělo vyjma hlavy. Pacient svými laboratorními parametry, opakovaně prokázanou leukocytózou se zvýšením počtu neutrofilních segmentů, opakovaně prokázanou zvýšenou hodnotou CRP, přítomným M-IgM a histologií kůže splnil Štrasburská kritéria Schnitzlerové syndromu. Bolesti kostí pacient sice neudával, ale popisuje bolesti velkých kloubů a bederní páteře. Teploty neměl, hmotnost zůstává stejná. V posledních letech však udává narůstající nevýkonnost a patologickou únavu. Všechny tyto symptomy se v posledních 3 letech postupně zintenzivňovaly a zvyšovala se leukocytóza a hodnota CRP. Oproti předchozím pacientům však jde o relativně časně stanovenou diagnózu. Vstupní laboratorní hodnoty ukazuje tab. 5. Pacient po schválení léčby revizním lékařem zahájil v květnu roku 2016 aplikaci anakinry. Po první injekci vymizel do večera exantém a v dalších dnech vymizela patologická únava a pacient se cítil tak skvěle jako před touto nemocí.
Diskuse
Kam zařadit Schnitzlerové syndrom? Na hematoonkologická pracoviště tyto pacienty přivádí přítomnost monoklonálního imunoglobulinu (M-Ig) a případně průkaz amyloidu. Přítomnost M-Ig je důvodem, proč je Schnitzlerové syndrom řazen do velké skupiny nemocí, nazvané „monoklonální gamapatie“.
Zásadní role interleukinu 1 v etiopatogenezi příznaků této nemoci zase vede k řazení této choroby do skupiny autoinflamatorních chorob. Tato skupina chorob představuje nejmladší skupinu nemocí, a proto v diskusi připomeneme velmi stručně a orientačně i základní choroby z této skupiny. Cílem tohoto stručného přehledu je dosáhnout lepšího pochopení variability klinických projevů Schnitzlerové syndromu i variabilitu léčebných odezev a pomoci při diferenciální diagnóze, protože některé ze stručně zmíněných autoinflamatorních chorob je nutno mít na mysli i při diferenciální diagnostice Schnitzlerové syndromu. Potvrzení diagnózy níže uvedených autoinflamatorních chorob je v ČR možné na Ústavu imunologie, ev. na revmatologickém oddělení FN Motol Praha a také ve VFN v Praze, na revmatologické ambulanci Kliniky dětského a dorostového lékařství LF UK.
Obecná charakteristika autoinflamatorních chorob
Je již tomu mnoho desetiletí, co byly definovány autoimunitní choroby, neboli choroby způsobené tvorbou autoprotilátek poškozujících organizmus. Tyto autoprotilátky pak slouží jako základ diagnostiky autoimunitních chorob.
Ale teprve relativně nedávno, koncem 90. let minulého stolení, byla definována nová kategorie patologických stavů, autoinflamatorní choroby. Jsou způsobeny změnami vrozených imunitních reakcí. Zatímco autoimunitní nemoci jsou způsobeny poruchou tolerance a jsou zprostředkovány protilátkami, ev. buněčnými reakcemi namířenými proti vlastním tkáním, u autoinflamatorních chorob jsou základním mechanizmem opakovaná vzplanutí zánětu. Tento zánět není spouštěn infekcí, ale vychází z porušených mechanizmů regulace zánětu, většinou spočívá v poruchách reakcí vrozené imunity.
Jedna skupina autoinflamatorních chorob je již molekulárně definována. Jednotky, které jsou řazeny do této skupiny, jsou důsledkem mutace jednoho genu. Druhá skupina autoinflamatorních chorob zatím jednotně definované molekulárně-biologické znaky nemá, ale dle všech ostatních znaků do kategorie autoinflamatorních chorob jednoznačně patří.
Vrozené monogenní autoinflamatorní choroby se začínají projevovat v dětství, mnohdy časně po porodu. Stejné či podobné mutace mohou vzniknout de novo v průběhu embryonálního života. Autoinflamatorní choroby mohou mít podle své povahy a dalších doplňujících vlivů časnou postnatální manifestaci, ale i opožděné klinické projevy objevující se až v dospělosti.
Porucha mechanizmů vrozené imunity, většinou doprovázená zvýšenou produkcí a špatnou regulací prozánětlivých cytokinů, způsobuje podobné klinické příznaky: ataky horeček, často periodických, bolesti kloubů, strukturální změny kostí (hyperostózy, osteosklerotická ložiska) spojené s bolestmi kostí, a změny na kůži. Autoinflamatorní choroby jsou provázeny zvýšenými humorálními (biochemickými i hematologickými) známkami zánětu [11–22]. Při přehlédnutí charakteristiky těchto nemocí je zřetelné, že příznaky se mnohdy prolínají, opakují a je pravděpodobné, že poznání této kategorie nemocí se bude postupně rozvíjet a budou přibývat molekulárně definované nemoci, podobně jak je tomu v případě myeloproliferativních nemocí. Schnitzlerové syndrom je řazen do skupiny získaných autoinflamatorních chorob, zatím bez jasně definované molekulární podstaty této, a proto pro stanovení diagnózy používáme Štrasburská kritéria. V průběhu následujících let bude jistě odhaleno více z molekulární biologie této získané nemoci, a to jistě povede i ke změně terminologie.
Geneticky definované autoinflamatorní choroby
Familiární středomořská horečka
Familiární středomořská horečka (familiar mediterranean fever – FMF) je způsobená mutací genu mediterrání horečky (mediterranean fever – MEVF). Nemoc se vyskytuje ve východním středomoří, v Turecku s incidencí až 1 : 1 000 obyvatel. Popsáno je více mutací a bylo prokázáno, že typ mutace souvisí s mírou agresivity nemoci. Příznaky začínají v dětství, vyvolává je námaha či stres, ale často není vyvolávající příčina jasná. Nemoc způsobuje horečku, serositidu, synovitidu a tendosynovitidu jako hlavní typické příznaky. Familiární středomořská horečka má i kožní příznaky, erytém podobný erysipelu. Serositida může imitovat „akutní břicho“ a vést k operaci. Teploty trvají krátce, kolem 3 dnů. V léčbě familiární středozemní horečky se osvědčil kolchicin, pomáhá zvládat akutní záchvaty a dlouhodobé užívání snižuje frekvenci atak. Kortikoidy také pomáhají zvládnout akutní ataku. V refrakterních případech se osvědčila anakinra [11,14,17,23].
Tumor necrosis factor receptor associated periodic sydrome – TRAPS
Pro nemoc není ustálený český překlad, takže ponecháváme anglický termín. Choroba se vyskytuje v Irsku, Skotsku a severní Evropě (hibernian fever), ale s lepšími možnostmi diagnostiky byla tato nemoc prokázána i v jiných zemích, včetně České republiky. Manifestuje se v dětství horečkou, bolestmi kloubů, svalů a často i břicha, migrujícím kožním rašem, konjuktivitidou, periorbitálním edémem, ulceracemi v ústech. Tyto febrilní periody trvají podstatně déle než u familiární středomořské horečky, 1–3 týdny. Laboratorně provázejí ataky vždy vzestup všech zánětlivých faktorů. V etiopatogenezi TRAPS syndromu se uplatňuje mutace genu receptoru pro TNF. V léčbě se dominantně osvědčil etanercept a někdy také anakinra. Infliximab vedl naopak ke zhoršení. Incidence ve Velké Británii se odhaduje na 1–2/1 milion obyvatel. Stanovení diagnózy je dnes založeno na prokázání patologických genetických změn [11–23].
Mevalonate kinase deficiency – MKD – hyper-IgD syndrom
Syndrom „mevalonate kinase deficiency – MKD“ je zvaný též jako hyper-IgD syndrom. Dříve byl výskyt popisován v severozápadní Evropě, ale s rozšiřující se dostupností genetické diagnostiky byl prokázán prakticky všude, včetně České republiky. Manifestuje se brzy v prvních letech života horečkami trvajícími 3–7 dní. Dalšími příznaky jsou průjem, artralgie, artritida, aftózní ulcerace, bolesti břicha. V etiopatogenezi hraje hlavní roli mutace mevalonátové kinázy, byť nejsou jasné patofyziologické důsledky této mutace způsobující projevy nemoci. U většiny pacientů je koncentrace imunoglobulinu IgD > 100 IU/dl, ale etiopatogenetická souvislost není jasná. Pacienti nemívají jen zvýšené IgD, ale také v 80 % zvýšenou koncentraci IgA. Jako lék volby se u této nemoci uvádí anakinra. Kolchicin a steroidy jsou také někdy účinné. Souvislost molekulárního defektu s příznaky není zatím zcela objasněna [11–23].
Kryopyrin asociovaný periodický syndrom – CAPS
Kryopyrin asociovaný periodický syndrom (neboli kryopyrinopatie) je autoinflamatorní porucha spojená s mutací genu NLRP3, která způsobuje zvýšenou aktivaci inflamasomu, a to pak ve svém důsledku vede ke zvýšené aktivitě interleukinu 1. Podobné změny byly nyní nově zachyceny i u syndromu Schnitzlerové. Porucha může být jak hereditární, tak mutací de novo. Incidence se odhaduje na 1–2/1 milion obyvatel. Do skupiny kryopyrin asociovaného syndromu se řadí 3 klinicky definované poruchy, projevující se u dětí. Tyto 3 formy jsou dnes považovány za různé závažnosti jedné nemoci. Všechny 3 poruchy se manifestují brzy po narození a trvají po zbytek života, tedy i v dospělosti, kdy mohou být odhaleny při hledání příčiny AA-amyloidózy.
Nejméně závažnou poruchou z této skupiny je familiar cold autoinflamatory syndrome – FCAS. Familiární chladová urtika je vyvoláván chladem a manifestuje se neutrofilní nesvědící urtikarií, febriliemi, konjunktivitidou, artralgií a artritidou.
Mucleův Wellsův syndrom představuje závažnější formu této nemoci s trvalými projevy zánětlivé odezvy s intermitentními exacerbacemi. V 2. či 3. dekádě se rozvíjí porucha sluchu.
Nejzávažnější forma nemoci se nazývá neonatal-onset multisystem inflamatory disorder –NOMID.
Dnes jsou všechny tyto 3 choroby řazeny do jedné skupiny zvané cryopyrin-associated periodic syndrom – CAPS. Lékem volby pro tyto choroby je anakinra. Dalším účinným lékem je kanakinumab a rilonacept [15–25]. Právě tato skupina nemocí nese hodně podobností se syndromem Schnitzlerové, a proto se i Mucleův Wellsův syndrom u dospělého uvádí v diferenciální diagnostice Schnitzlerové syndromu.
Disfunction interleukin 1 antagonist – DIRA
CAPS syndromu podobná porucha vzniká mutací v genu pro interleukin 1 receptor antagonist, porucha se označuje akronymem DIRA (disfunction interleukin 1 antagonist). DIRA se opět manifestuje v 1. roce života teplotami a systémovým zánětem, otoky kloubů a bolestmi kostí a charakteristickými kostními změnami s hyperostózami. Kožní změny na rozdíl od CAPS syndromu mají charakter pustul. Lékem volby je opět anakinra [15–26].
Geneticky dosud nedefinované autoinflamatorní choroby
Mimo tyto genetické definované autoinflamatorní choroby existují nemoci, u nichž dosud není molekulární podstata odhalena.
Periodic fever, aphthous stomatitis, pharyngitis, and adenitis – PFAPA
PFAPA je nemoc, jejíž příznaky jsou obsaženy v názvu nemoci. Postihuje více děti než dospělé. Projevuje se febriliemi trvajícími 4–5 dní a angínou, aftózní stomatitidou a faryngitidou bez přítomnosti jakékoliv prokazatelné infekce horních dýchacích cest. Pacienti s touto nemocí mohou mít i bolesti břicha, nevolnost a zvracení. Tato nemoc velmi dobře a rychle reaguje na glukokortikoidy. Etiologie není jasná, ale předpokládá se nadprodukce interleukinu 1 monocyty. Léčba není jasně definována díky vzácnosti případů, ale i u těchto případů byla s úspěchem použita anakinra [23,24]. Někteří autoři doporučují tonsilektomii [27].
Pyogenic arthritis – pyoderma gangresnosum – PAPA
Je to opět dominantně dědičné onemocnění, vedoucí k nadprodukci interleukinu 1. Příznaky jsou uvedeny v názvu nemoci, neprojevuje se však teplotami. Kožní změny dobře reagovaly na inhibitory TNF, ale také na inhibitory interleukinu 1 [15–26]. Nově i PAPA má svůj patognomický gen – PSTPIP1.
SAPHO syndrom
SAPHO syndrom (synovitis, acne, pustulosis, hyperostosis, osteitis) je onemocnění charakterizované pustulózní psoriázou, palmoplantární pustulózou, hnisavou hidraadenititou a závažným akné. Tato nemoc je ovlivnitelná jak anakinrou, tak také léky blokujícími TNF [28].
Behçetova choroba
Je to multisystémová zánětlivá porucha, která způsobuje ulcerace v ústech a na genitálu, oční záněty, intermitentní kožní raše, gastrointestinální ulcerace, neurologické postižení, horečky a artritidy, aniž by byly prokázány jakékoliv protilátky podílející se na etiopatogenezi této nemoci. Léčebná odpověď byla prokázána po protilátce proti interleukinu l (kanakinumab, gevokizumab) [22–28]. Asociace s HLA B 51 je silná a opakovaně dokázána.
Stillova nemoc dospělých
Z autoinflamatorních chorob, které se manifestují až v dospělém věku, je důležité připomenout Stillovu chorobu dospělých. V dětství se tato nemoc manifestuje jako idiopatická artritida, také zvaná SOJIA (systemic onset jevenile idiopatic arthritis). Dnes je řazena mezi multigenní autoinflamatorní choroby [29].
Je to opět onemocnění nejasné etiologie, bez jakýchkoliv pro tuto nemoc typických autoprotilátek, které se diagnostikuje pouze a jen na základě klinických příznaků, podobně jako Schnitzlerové syndrom. Pro tuto nemoc je typická vysoká horečka, artralgie či artritida, nesvědící makulární a makulopapulární kožní exantém lososovité barvy, neutrofilní leukocytóza. Dále bývá bolest v hrdle, lymfadenopatie, hepatosplenomegalie, abnormální hodnoty jaterních testů, negativní výsledky vyšetření antinukleárních protilátek a revmatoidního faktoru. U 30–40 % pacientů jsou přítomny serozitidy. Z laboratorních parametrů je vyjma leukocytózy přítomna zvýšená hladina feritinu (až 5krát nad normu) a zvýšená aktivita jaterních enzymů. Pro Stillovu chorobu bylo publikováno více kritérií, jejich přehled uvádí Malegová et al. Snad největší akceptovanost mají stále původní Yamaguchiho kritéria, která uvádíme v tab. 6, abychom demonstrovali odlišnosti od Schnitzlerové syndromu. Všechna kritéria Stillovy horečky dospělých se shodují ve velkém kritériu – febrilie nad 39 °C. U Stillovy nemoci dospělých je popisována extrémně zvýšená koncentrace feritinu pouze u 10–20 % nemocných, takže extrémně vysoká hladina feritinu není podmínkou stanovení diagnózy Stillovy nemoci dospělých [29–33].

Léčbou prvé volby zde bývají kortikoidy v dávce 1 mg/kg s postupným snižováním. Dávky kortikoidů může snížit použití perorálního metotrexátu. Nejúčinnějším lékem pro tyto nemocné je anakinra [34–39].
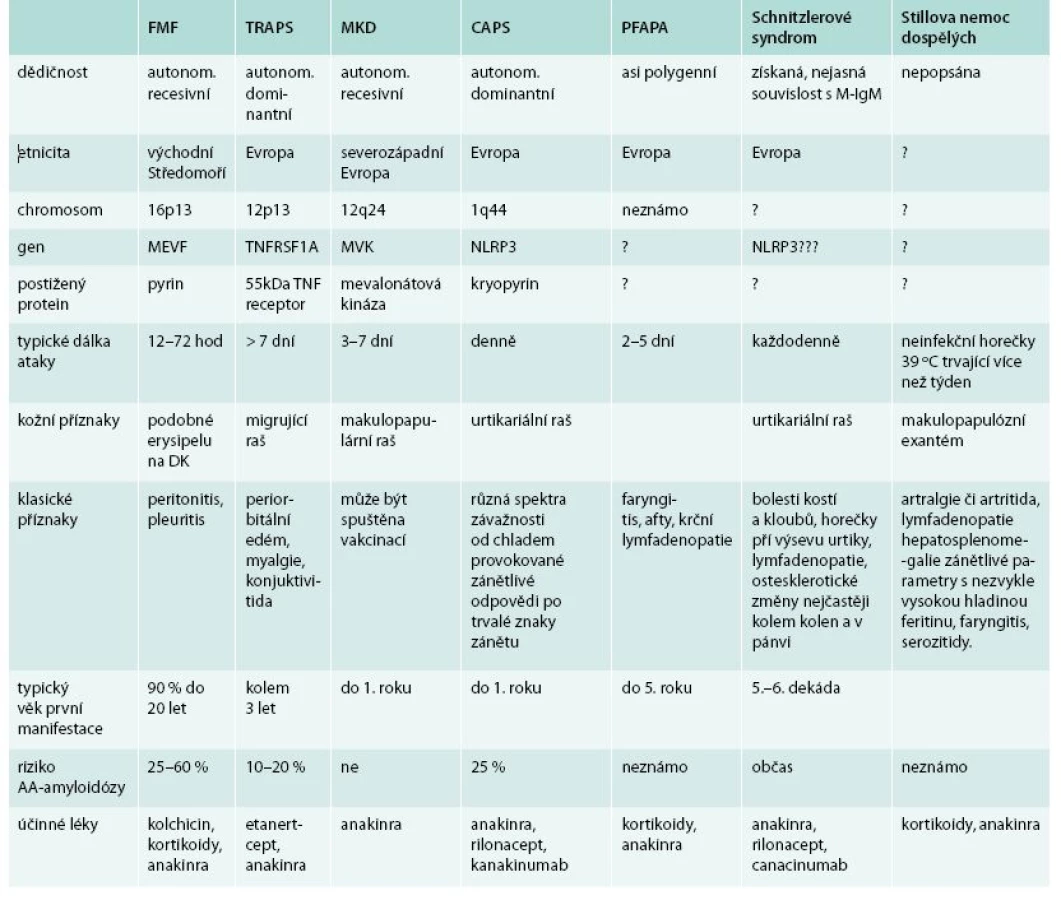
Přehledem nejznámějších hereditárních a 2 získaných autoinflamatorních chorob v tab. 7 chceme ilustrovat podobné rysy těchto nemocí, byť se některé nemoci vyskytují ve velmi mladém věku, zatímco Schnitzlerové syndrom a Stillova nemoc jsou choroby dospělého věku. A všechny tyto choroby mají podobnou léčbu – blokádu aktivity interleukinů, nejčastěji interleukinu 1.
Schnitzlerové syndrom
Etiologie Schnitzlerové syndromu
Schnitzlerové syndrom je choroba, která v mnohém připomíná geneticky definované autoinflamatorní nemoci, ale manifestuje se až ve věku dospělém. Má hodně podobných příznaků se skupinou nemocí zvanou cryopyrin associated periodic syndrome – CAPS. Kostní změny u syndromu Schnitzlerové se podobají kostním změnám popisovaným u dětí při deficiency interleukin 1 antagonist (DIRA). U některých případů Schnitzlerové syndromu byla analyzována mutace genu NLRP3 a byl prokázán somatický mozaicizmus genu NLRP3 v linii myeloidních buněk [40–42]. Také u jednoho z našich pacientů byl prokázán polymorfizmus v genu NLRP3 [43].
Tyto první výsledky signalizují, že Schnitzlerové syndrom bude v průběhu let molekulárně-biologicky definován.
Je zřejmé, že podobnou variabilitu závažnosti průběhu, jakou vidíme u CAPS syndromu v dětství, můžeme vidět i u Schnitzlerové syndromu v dospělosti. Vysvětlujeme si tím rozdílnou intenzitu léčebné odezvy na anakinru u našich pacientů [44]. U 4 z 5 demonstrovaných pacientů anakinra příznaky nemoci úplně odstranila, zatímco u 1 pacienta s polymorfizmem v genu NLRP3 došlo jen k výraznému ústupu potíží, ale ne k jejich úplnému vymizení.
Klinické příznaky Schnitzlerové syndromu
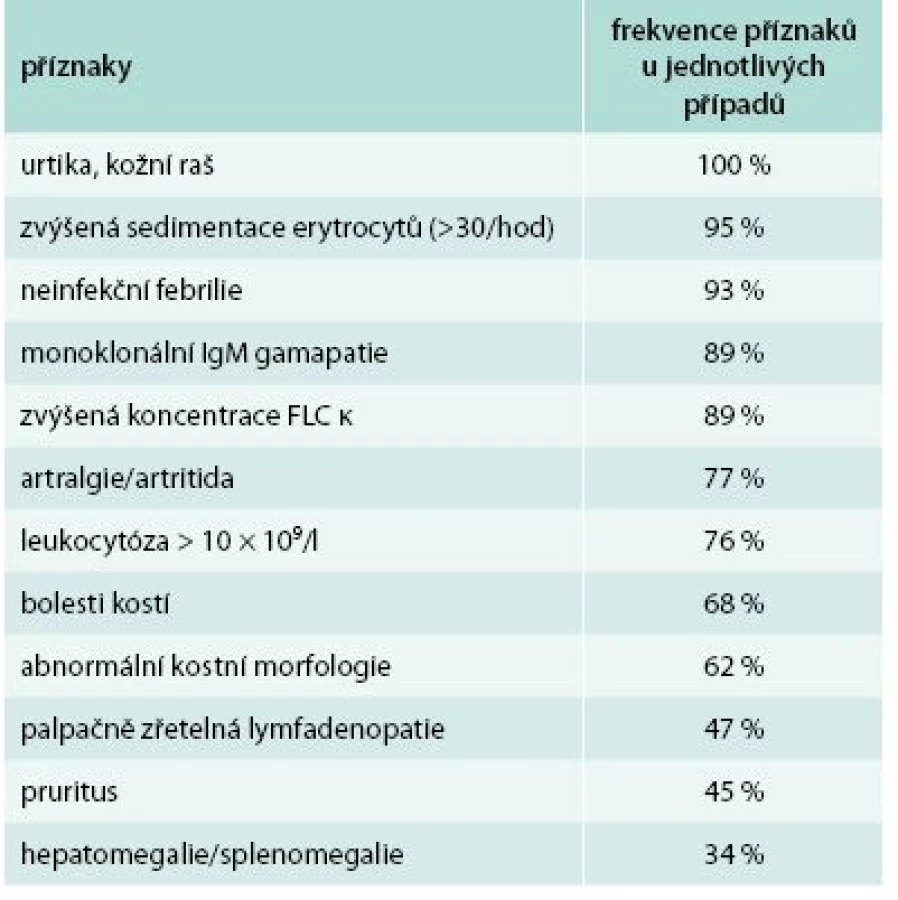
Na první pohled je nejzřetelnějším znakem kopřivkový raš, který obvykle nesvědí. V našem souboru pacientů měli 4 nemocní nesvědící kopřivku a 1 měl kopřivku provázenou značným svěděním. Někdy může mít kopřivka podobu velkých kopřivkových makul, jindy jde o difuzní krupičkový kožní raš. Kopřivka je tedy zásadním znakem Schnitzlerové syndromu, podobně jako přítomnost M-Ig. Pokud by pacient měl přítomny všechny ostatní příznaky a neměl kopřivkový raš, doporučuje se tento stav nazývat Schnitzler-like syndrome [44–47].
Pokud je přítomna kopřivka a jeden z dalších typických příznaků uvedených v tab. 8, doporučuje se vždy zapátrat po ostatních možných příznacích a pak prozkoumat, zda pacient nesplňuje Štrasburksá kritéria Schnitzlerové syndromu [9] uvedená v tab. 9.

Muskuloskeletární postižení je dalším důležitým znakem této nemoci, vyskytuje se asi u 80 % nemocných. Schnitzlerové syndrom totiž způsobuje i změny kostního metabolizmu. Zvýšení kostní denzity je nejčastější radiologický nález. V oblastech zvýšené kostní denzity nemocný často pociťuje bolesti.
Bolesti kostí jsou popisovány u 59 % nemocných a často jsou ještě doplňovány bolestmi kloubů (59 %), které však nevedou k jejich deformacím ani k destrukci. Kostní bolesti jsou nejčastěji popisovány v oblasti pánve a v kosti holenní. Stehenní kosti, paže, předloktí a klíční kosti jsou postihovány méně často. Bolesti kloubů bývají v kyčli, koleni, zápěstí či lokti, méně často v ostatních kloubech těla. V některých případech byly uvedeny i myalgie. Změny na kostech charakteru osteosklerózy lze detekovat pomocí klasických kostních CT skenů, ale jsou viditelné i při MR zobrazení skeletu [48–52]. Ložiska zvýšeného kostního metabolizmu lze zobrazit klasickou scintigrafií kostí radioaktivním techneciem nebo metodou PET/CT s použitím radioaktivního natriumfluoridu.
Diagnóza Schnitzlerové syndromu
V květnu roku 2012 se konala ve Štrasburku (ve Francii) konference expertů, jejímž cílem bylo definovat kritéria této nemoci, její optimální léčbu a sledování pacientů. Závěrem této konference byla dohodnuta nová definice (kritéria) této nemoci, která uvádí tab. 9. Dále bylo vytvořeno doporučení pro diagnostiku, léčbu a sledování pacientů s touto nemocí [9].
Diferenciální diagnóza Schnitzlerové syndromu
Na diagnózu syndromu Schnitzlerové se obvykle dochází v rámci širší diagnostiky horečky nejasného půdu – fever of unknown origin – FUO. V rámci diferenciální diagnostiky febrilního stavu je vždy třeba také vyloučit infekční, nádorová a autoimunitní – protilátkami zprostředkovaná onemocnění.
Z infekčních nemocí připadá v úvahu jako příčina více než 3 týdny probíhajících horeček infekce HIV, infekční endokarditida, borelióza, yersinióza, mykoplazmová infekce, syfilis, tuberkulóza, osteomyelitis, případně jiné skryté dlouhodobě probíhající infekce. Musí se provést odpovídající sérologická a kultivační vyšetření.
Je třeba vyloučit neoplazie, které mohou způsobovat horečku, tedy světlobuněčný Grawitzův karcinom ledviny, lymfomy, leukemie, ale také uzlinová onemocnění nejasné etiologie, jako je Castlemanova choroba či Rosaiova-Dorfmanova choroba. Tyto choroby lze odhalit s pomocí FDG-PET/CT vyšetření a odběrem histologie v případě nálezu patologického ložiska.
Dále je třeba vyloučit autoimunitní onemocnění, polymyalgia revmatica, obrovskobuněčnou arteriitidu a další systémové nekrotizující vaskulitidy. To je možné pomocí sérologického vyšetření a opět pomůže PET/CT vyšetření [53–55], které prokáže i přítomnost vaskulitidy.
Je třeba provést sérologická vyšetření k vyloučení lupusu, revmatoidní artritidy (antinukleární protilátky, revmatoidní faktor, anti-citrulin-peptidy, ANCA protilátky) a také vyloučit Bechtěrevovu chorobu. Také je nutno vyloučit střevní specifické záněty, pokud by byly odpovídající příznaky. Pokud se nic z uvedeného neprokáže, je možné vznést podezření na autoinflamatorní chorobu [56].
V diferenciální diagnostice autoinflamatorních chorob dospělých musíme myslet na Schnitzlerové syndrom a Stillovu nemoc dospělých.
Podobné kožní projevy může mít ale také urtikariální vaskulitida a kryoglobulinemická vaskulitida, systémový lupus erythematodes a chronická idiopatická kopřivka. Stillova choroba dospělých může v mnohém napodobovat Schnitzlerové syndrom, protože i pro Stillovu chorobu jsou typické kožní projevy, febrilie, bolesti kostí a kloubů, leukocytóza a nepřítomnost specifických diagnostických markerů. U Stillovy nemoci se často popisuje počáteční faryngitida, zatímco pro Schnitzlerové syndrom není typická. U Schnitzlerové syndromu nebývá také zvýšení transamináz, to je častěji pozorované u Stillovy nemoci. Také velmi vysoké hodnoty feritinu jsou typické pro Stillovu chorobu, nikoli však pro Schnitzlerové syndrom [56]. Urtikariální vaskulitida, zvláště její varianta se sníženou hladinou komplementu, může také svými příznaky (raš, teploty, kloubní bolesti) Schnitzlerové syndrom připomínat. V těchto případech pomůže ke stanovení diagnózy kožní biopsie, která v případě urtikariální vaskulitidy prokáže opravdovou vaskulitidu s fibrinoidní nekrózou malých cévek, což u Schnitzlerové syndromu nebývá. V případě urtikariální vaskulitidy je přítomná konsumpce komplementu a jsou přítomny anti-C1q protilátky, které u Schnitzlerové syndromu přítomny nebývají.
A také je třeba si uvědomit, že může jít o současnou manifestaci 2 nezávislých odchylek, monoklonální gamapatie a idiopatické kopřivky, protože incidence obou těchto odchylek narůstá po 50. roku života [57].
Léčba Schnitzlerové syndromu
Anakinra
Léčba je indikována v případě závažných klinických potíží. Lékem volby je nyní stále anakinra, což je antagonista receptoru pro interleukin 1. Biologický poločas anakinry je ale jen 4–6 hod. Standardní doporučená dávka je 100 mg denně. Z nežádoucích účinků se zmiňuje o něco vyšší frekvence závažných infekcí ve srovnání s placebem a občas možný pokles absolutního počtu neutrofilů. U pacientů s renální insuficiencí není předepsána žádná redukce, pouze v dokumentaci léku se vybízí ke zvýšené opatrnosti.
U Schnitzlerové syndromu se anakinra podává zpočátku v dávce 100 mg denně. V případě nedostatečného účinku anakinry se doporučuje navýšit denní dávku anakinry až na 3krát 100 mg denně [58–60].
Efekt anakinry nastupuje za několik hodin po aplikaci. Dle publikovaných i našich zkušeností léčebný účinek anakinry nemizí ani při dlouhodobé léčbě. Autoři multicentrické studie s mediánem sledování 36 měsíců (2–79) uvádějí, že v 83 % případů bylo dosaženo kompletní remise a jen u 17 % parciální remise [61].
V případně úplného vymizení příznaků je možné zkoušet najít nejnižší ještě účinnou dávku, tedy podávat tento lék s nižší frekvencí než 1krát denně. Pokud se léčbou navodí kompletní remise, je možné po 2 letech otestovat přerušení léčby: v případě recidivy nemoci se v přerušené léčbě ihned pokračuje. Efekt preparátu anakinra je potvrzen velkým počtem publikací, anakinra tedy představuje pro tuto nemoc standardní lék prvé volby [62–90].
Kanakinumab
V roce 2015 se v zahraničí dostávají do klinické praxe dlouhodobě účinné inhibitory interleukinu 1, které není nutné podávat denně a které pro tyto nemocné představují výhodnou alternativu. Jedná se o kanakinumab a rilonacept.
Kanakinumab je humánní monoklonální protilátka proti lidskému interleukinu 1β. O jeho přínosu pro nemocné trpící Schnitzlerové syndromem již referovalo více autorů [91–94]. Zatím jsme jej u našich u našich pacientů nepoužili.
Rilonacept
Rilonacept je další lék s podstatně delším účinkem, než má anakinra. Rilonacept je dimerický fúzní protein, skládající se z extracelulární částí receptoru pro interleukin 1 a z jeho akcesorního proteinu, které jsou oba navázány na Fc fragment protilátky typu IgG. Tato látka je obecně charakterizována jako „IL1 trap“ čili „past na IL1“.
Zatím jsou pouze ojedinělé zprávy o léčbě Schnitzlerové syndromu lékem zvaným rilonacept, který jiným mechanizmem inhibuje aktivitu interleukinu 1, a to na podstatně delší dobu než 1 aplikace anakinry [95]. Rilonacept je USA registrován agenturou FDA pro léčbu nemocí ze skupiny CAPS.
Protilátky proti interleukinu 6
Léčba pomocí protilátky proti interleukinu 6 zvaná tocilizumab byla popsána taktéž jako účinná [96].
Kolchicin
Další alternativou, kterou uvádí literatura, je přidání kolchicinu. Kolchicin v monoterapii vykazoval totiž u této nemoci slabou aktivitu. Lipsker uvádí, že kolchicin navodil alespoň mírné zlepšení u 25 % pacientů se Schnitzlerové syndromem léčených kolchicinem v monoterapii. Proto byla naděje, že jeho přidání k léčbě anakinrou zvýší efekt léčby [4,6]. U našich pacientů léčených anakinrou však kolchicin již nevedl k dalšímu zlepšení, a tak jej nikomu ze 4 popsaných pacientů léčených anakinrou již nepodáváme.
Pefloxacin
V rámci úplnosti přehledu literatury je nutno zmínit ojedinělé publikace popisující efekt pefloxacinu. Pefloxacin je chinolonové antibiotikum, které bylo používáno na střevní infekce. Příznivý efekt pefloxacinu byl popsán pouze ve 2 publikacích [97,98]. Z nízkého počtu pozitivních hodnocení usuzujeme, že účinek byl minimální. Mimoto tento lék u nás nyní není dostupný. Jeho použití je spojeno s nežádoucími účinky v podobě tendinopatií.
Rituximab s chemoterapií
Zatím jen jedna práce popisuje použití chemoterapie obsahující antiCD20 protilátku rituximab u pacienta se Schnitzlerové syndromem, jehož IgM gamapatie již měla charakter Waldenströmovy makroglobulinemie. Léčba, která dosáhla kompletní hematologické remise, vedla i k remisi Schnitzlerové syndromu [99].
Sledování při léčbě
V průběhu léčby se doporučuje sledovat klasické parametry zánětu, počet leukocytů a hodnotu CRP. Někteří autoři zvažují i monitorování sérového amylopeptidu A, protože při jeho vysoké koncentraci hrozí rozvoj AA-amyloidózy. Dále je třeba sledovat koncentraci M-Ig a při výrazném vzestupu provést přešetření, zda již nedošlo k transformaci v maligní lymfoproliferaci, což je výjimečně také možné.
Závěr pro praxi
- Horečka nejasné etiologie (fever of unknown origin) je vždy velkým stresem pro pacienty i jejich lékaře. Vzhledem k velkému počtu možných příčin je vždy vhodné diferenciální diagnostiku rozplánovat dle doporučení pro diagnostiku [53–56,100–103].
- FDG-PET/CT vyšetření je velkou oporou pro diagnostiku těchto nejasných stavů, protože umí rozpoznat ložiska zánětu, vaskulitidy, ale i lymfomy a jiné neoplazie, ale i Schnitzlerové syndrom má své znaky rozpoznatelné na PET/CT zobrazení.
- Schnitzlerové syndrom a Stillova nemoc představují dvě autoinflamatorní choroby, které se mohou v dospělosti manifestovat horečkami a kožními změnami. Na autoinflamatorní nemoci je také nutno myslet v rámci diferenciální diagnostiky neinfekčních febrilních stavů, zvláště pokud jsou provázeny kožními projevy [104–106]. Schnitzlerové syndrom a Stillova nemoc dospělých nemají etiologickou definici, a proto jsou definovány pomocí mezinárodních kritérií pro tyto nemoci.
- Pro všechny případy Schnitzlerové syndromu vyžadující léčbu je lékem volby antagonista receptoru interleukinu 1 – anakinra, v roce 2015 se objevují první zprávy o dalších účinných lécích blokujících účinky interleukinu 1, kanakinumab a rilonacept, které mají delší účinek než anakinra a v následujících letech zřejmě se dostanu do léčby těchto nemocí.
- Léčba pomocí preparátu anakinra ani po mnohaleté aplikaci neztrácí nic ze své účinnosti, jak dokumentujeme na našich pacientech a jak uvádí literatura.
Děkujeme kolektivu pracovníků OKRZP Ústředí VZP ČR a MUDr. Haně Burkoňové, revizní lékařce VZP ČR, která posuzovala celou problematiku, za povolení úhrady léčby velmi vzácné autoinflamatorní nemoci nazvané dle francouzské kožní lékařky Liliane Schnitzler, pro kterou neexistují v ČR oficiální léčebná doporučení ani pro tuto nemoc registrované léky.
Publikace byla sepsána v na podporu grantů: MUNI/A/1180/ 2015 a dále MZ ČR – RVO MOÚ, 00209805 a projektem LO 1413, MZ ČR – RVO FNBr, 65269705.
prof. MUDr. Zdeněk Adam, CSc.
z.adam@fnbrno.cz
Interní hematologická a onkologická klinika LF MU a FN Brno,
pracoviště Bohunice
www.fnbrno.cz
prim. prof. MUDr. Anna Šedivá, DrSc.
Anna.Sediva@fnmotol.cz
Ústav imunologie 2. LF UK a FN v Motole,
Praha
Doručeno do redakce 10. 2. 2016
Přijato po recenzi 4. 5. 2016
Sources
1. Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde ?) Cas cliniques, n° 46 B. Journée Dermatologique d‘Angers, 28 octobre 1972.
2. Schnitzler L, Schubert B, Boasson M et al. Urticaire chronique, lésions osseuses, macroglobulinémie IgM: maladie de Waldenström? 2ème présentation. Bull Soc Fr Dermatol Syphil 1974; 81 : 363–366.
3. Schnitzler L, Hurez D, Verret JL. Urticaire chronique, ostéocondensation, macroglobulinémie. Cas princeps. Etude sur 20 ans. Ann Dermatol Venereol 1989; 116(8): 547–550.
4. Lipsker D, Veran Y, Grunenberger F et al. The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore) 2001; 80(1): 37–44.
5. de Koning HD, Bodar EJ, van der Meer JW. Schnitzler Syndrome Study Group. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum 2007; 37(3): 137–148.
6. Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis 2010; 8 : 38. Dostupné z DOI: <http://dx.doi.org/10.1186/1750–1172–8-38>.
7. Kyle RA, Therneau TM, Rajkumar SV et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003; 102(10): 3759–3764.
8. Claes K, Bammens B, Delforge M et al. Another devastating complication of the Schnitzler syndrome: AA amyloidosis. Br J Dermatol 2008; 158(1): 182–184.
9. Simon A, Asli B, Braun-Falco M et al. Schnitzler syndrome: diagnosis, treatment and follow up. Alergy 2013; 68(5): 562–568. Dostupné z DOI: <http://dx.doi.org/10.1111/all.12129>.
10. Adam Z, Krejčí M, Pour L et al. Schnitzlerův syndrom – popis čtrnáctiletého průběhu nemoci a přehled informací o této nemoci. Vnitř Lék 2008; 54(12): 1140–1153.
11. Cetkovská P, Benáková N. Autoinflamatorní syndromy s kožními projevy. Čes Dermat 2015; 90(4): 144–155.
12. Tichá M. Terapie autoinflamatorních onemocnění a periodických horeček. Alergie 2015; 17(2): 128–135.
13. Šedivá A. Nové poznatky a pokroky v oblasti primárních imunodeficiencí. Postgraduální medicína 2015; 17(3): 294–303.
14. Blažová K, Horáčková M, Horváth R et al. Kardiorenální syndrom při AA amyloidóze v důsledku kombinace dvou mutací genů pro periodické horečky. Aktuality v nefrologii 2013; 19(4): 127–130.
15. Doležalová P. Syndrom CINCA – nejzávažnější onemocnění ze skupiny kryopyrinopatií. Farmakoterapie 2012; 8(Suppl 1): 8–9.
16. Šedivá A. Autoinflamatorní onemocnění v dermatologii. Čes Dermat 2012; 2(4): 239–243.
17. Šedivá A. Autoinflamatorní onemocnění a familiární středozemní horečka. Alergie 2012; 14(Suppl 2): 26.
18. Šedivá A. Periodické horečky a autoinflamatorní onemocnění – klinický přístup. Postgraduální medicína 2012; 14(2): 174–179.
19. Šedivá A. Periodické horečky AU. Alergie 2009; 11: P1-P16.
20. Šedivá A. Periodické horečky a další syndromy s poruchou regulace zánětlivé odpovědi. Českoslov Ped 2011; 66(3): 187–191.
21. Němcová D, Doležalová D, Brejchová I. Autoinflamatorní onemocnění kostí. Čes Revmat 2010; 18(3): 10–15.
22. Doležalová P, Król P, Němcová D. Autoinflamatorní onemocnění – přehled. Čes Revmat 2010; 18(3): 144–145.
23. Cush JJ. Autoinflamatory syndromes. Dermatol Clin 2013; 31(3): 471–480. Dostupné z DOI: <http://dx.doi.org/10.1016/j.det.2013.05.001>.
24. Yu JR, Leslie KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep 2011; 11(1): 12–20. Dostupné z DOI: <http://dx.doi.org/10.1007/s11882–010–0160–9>.
25. Kötter I, Schedel J, Kümmerle-Deschner JB. Periodic fever syndrome/autoinflammatory syndrome. Z Rheumatol 2009; 68(2): 137–148.
26. Jesus AA, Goldbach-Mansky R. Il-1 blokade in autoinflamatory syndromes. Annu Rev Med 2014; 65 : 233–244. Dostupné z DOI: <http://dx.doi.org/10.1146/annurev-med-061512–150641>.
27. Katra R, Dytrych P, Groh D et al. PFAPA syndrom v ORL oblasti a jeho indikace k tonzilektomii. Otorinolaryngologie a foniatrie 2010; 59(4): 197–201.
28. Beer HD, Contassot E, Frech LE. The inflammasome in autoinflamatory disease with skin involvement. J Investigative Dermatol 2014; 134(7): 1805–1810. Dostupné z DOI: <http://dx.doi.org/10.1038/jid.2014.76>.
29. Valentin MG, Jamilloux Y, Iwaz Y et al. Adult onset Sill´s disease. Autoimmun Rev 2014; 13(7): 708–722. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2014.01.058>.
30. Malegová J, Koen L, Horák P. Stillova nemoc dospělých, obtížná cesta k diagnóze přes horečku a výpotky nejasné etiologie. Vnitřní Lék 2014; 60(5–6): 520–526.
31. Skácelová, M, Horák P. Stillova nemoc dospělých. Čes Revmat 2012; 20(4): 198–202.
32. Skácelová M, Horák P. Stillova choroba dospělých. Postgraduální medicína 2015; 17(4): 391–394.
33. Houzarová A. Stillova nemoc. Referátový výběr z revmatologie 2007; 47(3): 1214–1216.
34. Fischer-Betz R, Specker C, Schneider M. Successful outcome of two pregnancies in patients with adult-onset Still‘s disease treated with IL-1 receptor antagonist (anakinra). Clin Exp Rheumatol 2011; 29(6): 1021–1023.
35. Jamilloux Y, Gerfaund-Valentin M, Henry T et al. Treatment of adult-onset Still´s disease: a review. Ther Clin Risk Manag 2015; 11 : 33–43. Dostupné z DOI: <http://dx.doi.org/10.2147/TCRM.S64951>.
36. Ortiz-Sanjuán F, Blanco R, Riancho-Zarrabeitia L et al. Efficacy of Anakinra in Refractory Adult-Onset Still‘s Disease: Multicenter Study of 41 Patients and Literature Review. Medicine (Baltimore) 2015; 94(39): e1554. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000001554>.
37. Jamilloux Y, Gerfaud-Valentin M, Henry T et al. Treatment of adult-onset Still‘s disease: a review. Ther Clin Risk Manag 2014; 11 : 33–43.
38. Hong D, Yang Z, Han S et al. Interleukin 1 inhibition with anakinra in adult-onset Still disease: a meta-analysis of its efficacy and safety. Drug Des Devel Ther 2014; 8 : 2345–2357. Dostupné z DOI: <http://dx.doi.org/10.2147/DDDT.S73428>.
39. Gerfaud-Valentin M, Jamilloux Y, Iwaz J at al. Adult-onset Still‘s disease. Autoimmun Rev 2014; 13(7): 708–722.
40. de Koning HD, van Gijn ME, Stoffels M et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol 2015; 135(2): 561–564.
41. Pizzirani C, Falzoni S, Govoni M et al. Dysfunctional inflammasome in Schnitzler‘s syndrome. Rheumatology (Oxford) 2009; 48(10): 1304–1308.
42. Loock J, Lamprecht P, Timmann C et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol 2010; 125(2): 500–502.
43. Szturz P, Šedivá A, Žurek M et al. Léčba anakinrou u Schnitzler-syndromu – výsledky první retrospektivní multicentrické studie šesti pacientů z České republiky. Klin Onkol 2014; 27(2): 111–126.
44. Lamprecht P. Adult-onset Still‘s disease, Schnitzler syndrome, and autoinflammatory syndromes in adulthood. Z Rheumatol 2009; 68(9): 740–746. Dostupné z DOI: <http://dx.doi.org/10.1007/s00393–009–0490-y>.
45. Simon A, Asli B, Braun-Falco M et al. Schnitzler‘s syndrome: diagnosis, treatment, and follow-up. Allergy 2013; 68(5): 562–568. Dostupné z DOI: <http://dx.doi.org/10.1111/all.12129>.
46. Patel S, Sindher S, Jariwala S et al. Chronic urticaria with monoclonal IgG gammopathy: a clinical variant of Schnitzler syndrome? Ann Allergy Asthma Immunol 2012; 109(2): 147–148.
47. Sokumbi O, Drage LA, Peters MS. Clinical and histopathologic review of Schnitzler syndrome: the Mayo Clinic experience (1972–2011). J Am Acad Dermatol 2012; 67(6): 1289–1295.
48. Flórez AF, Gallardo Agromayor E, García-Barredo R et al. Radiological aid to clinical diagnosis of Schnitzler‘s syndrome: multimodality imaging approach. Clin Rheumatol 2008; 27(1): 107–110.
49. Lee KY, Grattan CE. Intracostal neuralgia as a previously undescribed symptom of Schnitzler‘s syndrome. Br J Dermatol 2012; 167(6): 1392–1393. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2133.2012.11057.x>.
50. Terpos E, Asli B, Chistoulas D et al. Increased Angiogenesis and Enhanced Bone Formation in Patients with IgM Monoclonal Gammopathy and Urticarial Skin Rash: New Insight into the Biology of the Schnitzler Syndrome. Haematologica 2012; 97(11): 1699–1703. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2012.067306>.
51. Niederhauser BD, Dingli D, Kyle RA et al. Imaging findings in 22 cases of Schnitzler syndrome: characteristic paraarticular osteosclerosis and the hot knees sign differential diagnosis. Skeletal Radiol 2014; 43(7): 905–915. Dostupné z DOI: <http://dx.doi.org/10.1007/s00256–014–1857-y>.
52. Willekens I, Walgraeve N, Goethals L et al. Correlative bone imaging in a case of Schnitzler´s syndrome a briew review of the literatura. Hell J Nucl Med 2015; 18(1): 71–73.
53. Jarůšková M, Bělohlávek O. Role of FDG-PET and PET/CT in the diagnosis of prolonged febrile states. Eur Journal Nucl Med Mol Imaging 2006; 33(8): 913–918.
54. Kotík L. Teploty nejasného původu. Interní med pro praxi 2006; 8(11): 493–495.
55. Ferdová E, Záhlava J, Ferda J. Horečky nejasného původu, význam hybridního zobrazení 18F-FDG-PET/CT. Čes Radiol 2008; 62(1): 23–33.
56. Lachmann HJ. Autoinflammatory syndromes as causes of fever of unknown origin. Clin Med (Lond) 2015; 15(3): 295–298. Dostupné z DOI: <http://dx.doi.org/10.7861/clinmedicine.15–3-295>.
57. Barbosa NS, Schoch JJ, Ringler MD et al. Chronic urticarial eruption associated with monoclonal gammopathy. Am J Hematol 2015; 90(4): 365–366. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.23873>.
58. Šedivá A, Slíva J, Doležalová P et al. Anakinra. Farmakoterapie 2011; 7(6): 621–629.
59. Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med 2014; 65 : 223–44. Dostupné z DOI: <http://dx.doi.org/10.1146/annurev-med-061512–150641>.
60. Pazyar N, Feily A, Yaghoobi R. An overview of interleukin-1 receptor antagonist, anakinra, in the treatment of cutaneous diseases. Curr Clin Pharmacol 2012; 7(4): 271–275.
61. Neel A, Henry B, Barbarot S et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler´s syndrome. A French multicenter study. Autoimmune Rev 2014; 13(10): 1035–1041. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2014.08.031>.
62. Martinez-Taboada VM, Fontalba A et al. Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum 2005; 52(7): 2226–2227.
63. de Koning HD, van der Meer JW, Simon A Comment on: Schnitzlers syndrome-exacerbation after anti-TNF treatment. Rheumatology (Oxford) 2007; 46(11): 1741.
64. Besada E, Nossent H. Dramatic response to IL-1-RA treatment in longstanding multidrug resistant Schnitzler‘s syndrome: a case report and literature review. Clin Rheumatol 2010; 29(5): 567–571. Dostupné z DOI: <http://dx.doi.org/10.1007/s10067–010–1375–9>.
65. Billey T, Beldjerd M, Popa L et al. Schnitzler syndrome: a dramatic improvement with anakinra. Presse Med 2010; 39(12): 1338–1339. Dostupné z DOI: <http://dx.doi.org/10.1016/j.lpm.2010.07.009>.
66. Cascavilla N, D‘Arena G, Dell‘OLio M. Schnitzler syndrome. Br J Haematol 2008; 143(2): 152. 10.1111/j.1365–2141.2008.07287.x
67. Cascavilla N, Bisceglia M, D‘Arena G. Successful treatment of Schnitzler‘s syndrome with anakinra after failure of rituximab trial. Int J Immunopathol Pharmacol 2010; 23(2): 633–636.
68. Crouch R, Akhras V, Sarkany R. Schnitzler‘s syndrome: successful treatment with anakinra. Australas J Dermatol 2007; 48(3): 178–181.
69. Devlin LA, Wright G, Edgar JD. A rare cause of a common symptom Anakinra is effective in the urticaria of Schnitzler Syndrome: a case report. Cases J 2008; 1(1): 348. Dostupné z DOI: <http://dx.doi.org/10.1186/1757–1626–1-348>.
70. Dybowski F, Sepp N, Bergerhausen HJ et al. Successful use of anakinra to treat refractory Schnitzler‘s syndrome. Clin Exp Rheumatol 2008; 26(2): 354–357.
71. Eiling E, Möller M, Kreiselmaier I et al. Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol 2007; 57(2): 361–364.
72. Eiling E, Schröder JO, Gross WL et al. The Schnitzler syndrome: chronic urticaria and monoclonal gammopathy – an autoinflammatory syndrome? J Dtsch Dermatol Ges 2008; 6(8): 626–631. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1610–0387.2008.06627.x>.
73. Frischmeyer-Guerrerio PA, Rachamalla R, Saini SS. Remission of Schnitzler syndrome after treatment with anakinra. Ann Allergy Asthma Immunol 2008; 100(6): 617–619. Dostupné z DOI: <http://dx.doi.org/10.1016/S1081–1206(10)60064–6>.
74. Gilson M, Abad S, Larroche C et al. Treatment of Schnitzler syndrome with anakinra. Clin Exp Rheumatol 2007; 25(6): 931.
75. Klemmer N, Lenain P, Balguerie X et al. Effectiveness of anti-IL1 in Schnitzler‘s syndrome. Joint Bone Spine 2007; 74(5): 509–510.
76. Kluger N, Rivière S, Guillot B et al. Efficacy of interleukin 1 receptor antagonist (anakinra) on a refractory case of Schnitzler‘s syndrome. Acta Derm Venereol 2008; 88(3): 287–288. Dostupné z DOI: <http://dx.doi.org/10.2340/00015555–0404>.
77. Larocca CA, McEvoy JW, Ellis CL et al.. Schnitzler‘s syndrome associated with pancreatitis: a disease of IL-1 dysregulation. Clin Rheumatol 2012; 31(1): 169–174. Dostupné z DOI: <http://dx.doi.org/10.1007/s10067–011–1804–4>.
78. Loock J, Lamprecht P, Timmann C et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol 2010; 125(2): 500–502. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2009.10.066>.
79. Ryan JG, de Koning HD, Beck LA et al. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol 2008; 121(1): 260–262.
80. Saiz E, Gálvez J, Mora A et al. Anakinra and Schnitzler‘s syndrome. Med Clin (Barc) 2008; 130(9): 358–359.
81. Schneider SW, Gaubitz M, Luger TA et al. Prompt response of refractory Schnitzler syndrome to treatment with anakinra. J Am Acad Dermatol 2007; 56(5 Suppl): S120-S122.
82. Schuster C, Kränke B, Aberer E et al. Schnitzler syndrome: response to anakinra in two cases and review of the literature. Int J Dermatol 2009; 48(11): 1190–1194. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–4632.2009.04151.x>.
83. Sokumbi O, Drage LA, Peters MS. Clinical and histopathologic review of Schnitzler syndrome: the Mayo Clinic experience (1972–2011). J Am Acad Dermatol 2012; 67(6): 1289–1295. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaad.2012.04.027>.
84. Thonhofer R, Uitz E, Graninger W. Schnitzler‘s syndrome–exacerbation after anti-TNF treatment. Rheumatology (Oxford) 2007; 46(6): 1041–1042.
85. Tinazzi E, Puccetti A, Patuzzo G et al. Schnitzler syndrome, an autoimmune-autoinflammatory syndrome: report of two new cases and review of the literature. Autoimmun Rev 2011; 10(7): 404–409. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2011.01.003>.
86. Treudler R, Kauer F, Simon JC. Striking effect of the IL-1 receptor antagonist Anakinra in chronic urticarial rash with polyclonal increase in IgA and IgG. Acta Derm Venereol (Stockh) 2007; 87(3): 280–281.
87. van Deuren M, Kroot JJ, Swinkels DW. Time-course analysis of serum hepcidin, iron and cytokines in a C282Y homozygous patient with Schnitzler‘s syndrome treated with IL-1 receptor antagonist. Haematologica 2009; 94(9): 1297–1300. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2009.005975>.
88. Vandenhende MA, Bentaberry F, Morlat P et al. Anakinra: an effective treatment in the Schnitzler syndrome. Joint Bone Spine 2011; 78(6): 636–637. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jbspin.2011.03.021>.
89. Volz T, Wölbing F, Fischer J et al. Dermal Interleukin-1 Expression and Effective and Long-lasting Therapy with Interleukin-1 Receptor Antagonist Anakinra in Schnitzler Syndrome. Acta Derm Venereol 2012; 92(4): 393–394. Dostupné z DOI: <http://dx.doi.org/10.2340/00015555–1307>.
90. Wastiaux H, Barbarot S, Gagey-Caron V et al. Schnitzler syndrome: a dramatic improvement with anakinra. J Eur Acad Dermatol Venereol 2009; 23(1): 85–87. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1468–3083.2008.02708.x>.
91. Pesek R, Fox R. Successful treatment of Schnitzler syndrome with canakinumab. Cutis 2014; 94(3): E11-E12.
92. de Koning HD, Schalkwijk J, van der Ven-Jongekrijg J et al. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler‘s syndrome. Ann Rheum Dis 2013; 72(10): 1634–1638. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2012–202192>.
93. Vanderschueren S, Knockaert D. Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum 2013; 42(4): 413–416. Dostupné z DOI: <http://dx.doi.org/10.1016/j.semarthrit.2012.06.003>.
94. de Koning HD, Schalkwijk J, van der Meer JW et al. Successful canakinumab treatment identifies IL-1β as a pivotal mediator in Schnitzler syndrome. J Allergy Clin Immunol 2011; 128(6): 1352–1354. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2011.05.023>.
95. Krause K, Weller K, Stefaniak R et al. Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy 2012; 67(7): 943–950. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1398–9995.2012.02843.x>.
96. Krause K, Feist E, Fiene M et al. Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol 2012; 129(3): 848–850. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2011.10.031>.
97. Asli B, Bienvenu B, Cordoliani F et al. Chronic urticaria and monoclonal IgM gammopathy (Schnitzler syndrome): report of 11 cases treated with pefloxacin. Arch Dermatol 2007; 143(8): 1046–1050.
98. Kastritis E, Katoulis A, Terpos E et al. Schnitzler‘s syndrome: increased levels of bone formation and angiogenesis factors are reduced after successful pefloxacin treatment. Clin Lymphoma Myeloma 2008; 8(6): 359–362. Dostupné z DOI: <http://dx.doi.org/10.3816/CLM.2008.n.053>.
99. Aouba A, Pressiat C, Pricopi M et al. Complete remission of Schnitzler syndrome and Waldenström macroglobulinemia under rituximab-cyclophosphamide-dexamethasone. Dermatology 2015; 230(1): 18–22. Dostupné z DOI: <http://dx.doi.org/10.1159/000368349>.
100. Król P, Doležalová P. Horečka jako hlavní projev nemoci. Pediatrie pro praxi 2011; 12(2): 111–114.
101. Doležalová P, Król P. Recidivující horečky u dětí. Vox pediatriae 2010; 10(3): 16–20.
102. Tomíčková D. Horečka neznámého původu. Lékařské listy 2009; 58(8): 9–11.
103. Křivanová A, Adam Z, Mayer J. Teplota nejasné etiologie: příčiny a diagnostický postup. Vnitř Lék 2007; 53(2): 169–178.
104. Toplak N, Dolezalova P, Constantin T et al. [Eastern/Central European autoinflammatory collaborating group for the Paediatric Rheumatology International Trials Organization (PRINTO) and Eurofever Project]. Periodic fever syndromes in Eastern and Central European countries: results of a pediatric multinational survey. Pediatr Rheumatol Online J 2010; 8 : 29. Dostupné z DOI: <http://dx.doi.org/10.1186/1546–0096–8-29>.
105. Król P, Böhm M, Sula V et al. PFAPA syndrome: clinical characteristics and treatment outcomes in a large single-centre cohort. Clin Exp Rheumatol 2013; 31(6): 980–9877.
106. Toplak N, Frenkel J, Ozen S et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis 2012; 71(7): 1177–1182. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2011–200549>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 9
Most read in this issue
- Arytmogenní kardiomyopatie levé komory
-
Schnitzlerové syndrom
Diferenciální diagnostika, přehled léčebných možností a popis 5 případů léčených anakinrou - Perorálne antikoagulanciá v primárnej a sekundárnej prevencii vénovej tromboembólie
- Klinicky relevantné možnosti a limity diferenciálnej diagnostiky megaloblastovej anémie a myelodysplastického syndrómu typu refraktérnej anémie v trepanobioptických vzorkách kostnej drene