
Hemoglobinopatie
Hemoglobinopathies
This article summarize molecular-genetic basis of hemoglobinopathies, their classification and phenotypic manifestations. The description of individual subgroups is supplemented with a case reports of patients diagnosed in the Czech population. This paper provides an overview of 14 types of α-thalassemic mutations, 34 β-thalassemic alleles, 4 δβ-thalassemic alleles and 22 hemoglobin variants identified in the Czech population in 876 persons from 579 families. In more detail are described hemoglobinopathies, that have been diagnosed and described as novel: β-thalassemic mutation CD 38/39 (-C); Hb Olomouc; Hb Hana; Hb Hradec Kralove and 18.3 kb deletion downstream of α-globin cluster leading to a new mechanism of α-thalassemia-2. The fact that until the end of 2017 hemoglobinopathies were diagnosed in nearly 900 patients shows that they are not rare in the Czech Republic. This brings increased demands for their diagnostics, including prenatal diagnosis.
Key words:
hemoglobinopathies – hemoglobinopathy with high affinity to oxygen – sickle cell anemia – thalassemia – thalassemic hemoglobinopathy – unstable hemoglobins
:
Karel Indrák 1; Martina Divoká 1; Dagmar Pospíšilová 2; Jaroslav Čermák 3; Monika Beličková 3; Monika Horváthová 4; Vladimír Divoký 1,4
:
Hemato-onkologická klinika LF UP a FN Olomouc
1; Dětská klinika LF UP a FN Olomouc
2; Ústav hematologie a krevní transfuze, Praha
3; Ústav biologie LF UP Olomouc
4
:
Vnitř Lék 2018; 64(5): 476-487
:
Přehledný článek přibližuje molekulárně-genetickou podstatu hemoglobinopatií, jejich klasifikaci a fenotypické projevy. Popis jednotlivých podskupin je doplněn o kazuistické příklady nemocných z české populace. Je uveden přehled 14 typů α-talasemických mutací, 34 β-talasemických alel, 4 δβ-talasemické alely a 22 hemoglobinových variant identifikovaných v české populaci u 876 osob z 579 rodin. Podrobněji jsou popsány hemoglobinopatie, které zde byly diagnostikovány prioritně: β-talasemická alela CD 38/39 (-C); Hb Olomouc; Hb Haná; Hb Hradec Králové a α-tal.-2 delece DNA o délce 18,3 kb, jejíž identifikace umožnila odhalit nový mechanizmus vzniku α-talasemie-2. Skutečnost, že hemoglobinopatie byly v ČR do konce roku 2017 diagnostikovány u téměř 900 nemocných, ukazuje, že u nás nejsou vzácné. To klade zvýšené nároky na jejich diagnostiku, včetně diagnostiky prenatální.
Klíčová slova:
hemoglobinopatie – hemoglobinopatie s vysokou afinitou ke kyslíku – nestabilní hemoglobiny – srpkovitá anémie – talasemické hemoglobinopatie – talasemie
Definice
Hemoglobinopatie jsou vrozená onemocnění způsobená poruchou tvorby jednoho nebo více globinových řetězců. Příčinou jsou mutace nebo delece v genech kódujících globinové řetězce. Patogeneticky hemoglobinopatie dělíme na (1) hemoglobinopatie způsobené kvantitativním snížením syntézy globinových řetězců hemoglobinu – talasemie a (2) hemoglobinopatie způsobené poruchou struktury molekuly hemoglobinu – strukturální hemoglobinopatie.
Úvod
Lidský hemoglobin je tetramer tvořený 2 páry polypeptidových (globinových) řetězců – 2 dimery. Tyto dimery jsou v definitivní erytropoéze tvořeny kombinací jednoho α a jednoho non-α (β, γ nebo δ) řetězce. Každý řetězec obepíná hem umístěný v hemové kapse. Hem je protoporfyrin tvořený čtveřicí pyrolových jader – tetrapyrol. Ve středu tetrapyrolového jádra je šestivazný a dvojmocný iont železa (obr. 1), který je schopen reverzibilně vázat molekulu kyslíku. Tím umožňuje hemoglobinu plnit jeho hlavní funkci – přenos kyslíku z plic do tkání.

V letech 1977 a 1978 zjistili Deisseroth et al [1,2], že α globinové geny řídící syntézu α globinových řetězců jsou lokalizovány na chromosomu 16 v tzv. α-globinovém lokusu (HBA lokus) a β-globinové geny řídící syntézu β-globinových řetězců jsou lokalizovány na krátkém rameni chromosomu 11 v tzv. β-globinovém lokusu (HBB lokus). Jednotlivé geny jsou na těchto lokusech uspořádány v pořadí jejich postupné exprese od embryonálního období do dospělosti. Na HBA lokusu jsou geny uloženy v pořadí od 5´ k 3´konci: ζ2, ψζ1, 2 pseudogeny α a 2 α globinové geny pojmenované jako α2 a α1. Tzv. „pseudogeny“ jsou nefunkčními homology globinových genů a značí se písmenem ψ. Na konci lokusu se nachází gen θ s neznámou funkcí (schéma 1). Na HBB lokusu (schéma 2) jsou geny uspořádány v pořadí od 5´ k 3´konci: ε, dva γ globinové geny (Gγ a Aγ), ψβ, δ a β gen. Transkripce globinových genů se spouští po interakci transkripčních faktorů s promotory a vzdálenými tkáňově specifickými zesilovači genové exprese: „hypersensitive site 40“ (HS-40) na HBA lokusu a „β-locus control region“ (β-LCR) na HBB lokusu. Jednotlivé globinové geny jsou tvořeny třemi do mRNA se přepisujícími exony, které jsou od sebe odděleny dvěma introny. V nepřepisované 5´ oblasti (5´UTR) mRNA globinového genu leží konzervované sekvence odpovědné za transkripci genu [3]. Nepřepisovaná oblast 3´UTR je kódovaná sekvencí zodpovědnou za signál pro sestřih 3´ konce primárního transkriptu a přidání poly (A) konce slouží k udržení stability výsledné RNA molekuly (schéma 3). U zdravých jedinců je exprese α a β-globinových genů balancovaná. V dospělosti tvoří většinu hemoglobinu krve (asi 97 %) hemoglobin A (α2β2). Hemoglobin A2 (α2δ2) tvoří asi 1,5–3 %. Zvýšená hladina HbA2 je typická (diagnostická) pro velkou většinu nosičů β-talasemické alely. Snížená hladina HbA2 se vyskytuje u nosičů α-, δ - nebo δβ-talasemické alely. Hemoglobin F (α2γ2) tvoří v dospělosti 1 % hemoglobinu. Množství HbF se může u novorozence pohybovat v rozmezí 60–95 %. V dospělosti je jeho množství ovlivněno věkem a pohlavím (mírně vyšší bývá u žen). U některých jedinců přetrvává zvýšená hladina HbF i v dospělosti. Důvodem může být hereditární perzistence fetálního hemoglobinu (HPFH).



Kvantitativní hemoglobinopatie – talasemie
Talasemie se vyskytují především v malarických oblastech, v nichž je jejich zvýšená incidence a prevalence důsledkem selekčního tlaku malarického parazita – talasemická alela částečně chrání svého nosiče před malárií. Talasemie se většinou dědí autosomálně recesivně. V zemích západní, severní a střední Evropy, v nichž byly talasemie vzácné, se v důsledku populační migrace obyvatel z malarických oblastí jejich výskyt zvyšuje. Molekulárně-genetickou podstatou talasemií jsou bodové mutace, delece či inzerce různě dlouhých úseků DNA v oblasti globinových genů. Talasemie se projevují sníženou (x+) nebo chybějící (x0) expresí některého z globinových genů. Globinový řetězec, jehož syntéza je mutací alterována, dává talasemii jméno – tedy α-talasemie mají sníženou produkci α-globinových řetězců, β-talasemie mají sníženou produkci β-globinových řetězců atd. Vzácné jsou δβ - a εγδβ-talasemie. Snížená produkce některého globinového řetězce nastoluje nerovnováhu v poměru α a non-α-globinových řetězců v erytrocytech. Velikost této nerovnováhy se přímo úměrně projevuje tíží anémie. Molekulárně-geneticky i klinicky se α-talasemie a β-talasemie liší. β-talasemie jsou nejčastěji způsobeny bodovými mutacemi, delecemi či inzercemi několika nukleotidů v β-globinovém genu. α-talasemie vznikají nejčastěji v důsledku dlouhých delecí DNA, a protože volné β-globinové a γ-globinové řetězce jsou schopné tvořit nestabilní solubilní tetrametry (např. β4-Hb-H), projevují se α-talasemie klinicky méně závažně.
A-talasemie
A-talasemie jsou ve více než 80 % způsobeny jednoduchou delecí funkčních α-globinových genů. Protože, na rozdíl od pouze jednoho β-globinového genu, má haploidní lidský genom dva α-globinové geny, značíme normální genotyp schematicky αα/αα. Podle rozsahu DNA postižení a klinických příznaků můžeme α talasemie rozdělit do 4 skupin (schéma 4):
- a) u heterozygotů pro α+-talasemii (-α/αα) mluvíme o němém (tichém) nosičství
- b) u homozygotů pro α+-talasemii (-α/-α) nebo u heterozygotů pro α0–talasemii ( - -/αα) hovoříme o nosičství α-talasemie, heterozygoti mívají mírnou mikrocytární hypochromní anémii (MCV 60–70 fl)
- c) ztráta tří α-globinových genů ( - -/-α) vede k chorobě HbH s produkcí HbH (β4) – pacienti jsou bledí a ikteričtí (mají hemolytickou anémii), mívají hepatosplenomegalii, masivní otoky a hydrops podobný jako při inkompatibilitě Rh faktoru
- d) chybění všech čtyř α-globinových genů ( - -/ - -) není slučitelné se životem a vede ke vzniku „hydrops fetalis“ nebo syndromu Hb Bart‘s (γ4) – většinou se rodí hydropicky změněné dítě

Delece postihující α-globinové geny a způsobující α-talasemii mohou být různě dlouhé. Nedeleční formy α-talasemií jsou vzácnější. První přehledné sdělení o výskytu α-talasemií v ČR publikovali Divoká et al v roce 2016 [4]. V ČR jsme do konce roku 2017 molekulárně-geneticky diagnostikovali 14 typů delečních i nedelečních α-talasemických mutací u 147 osob ze 130 nepříbuzných rodin (tab. 1). Jedinci postižení α-talasemií jsou v ČR vesměs heterozygoti. Nejčastější u nás identifikovanou α-talasemickou mutací je delece DNA o délce 3,7 kb zahrnující části α2 - i α1-globinových genů (-α3,7), která vytváří jeden HBA2/HBA1 hybridní gen (schéma 5). Druhá, v ČR nejčastěji detekovaná mutace, je delece DNA označovaná jako --SEA zahrnující nejen celé geny α2 i α1, ale i pseudogeny ψα2 a ψα1. Deleci celého HBA lokusu jsme identifikovali u 12 osob ze 7 rodin. Delece regulační oblasti HS-40 byla v ČR detekována u 6 osob ve 4 α-talasemických rodinách, z nichž 3 byly českého původu. Dále jsme v souboru nemocných odhalili 2 unikátní delece zahrnující α2 - i α1-globinové geny (regulační oblast HS-40 byla zachována), všichni postižení byli českého původu. U 37letého muže polského původu s hraniční mikrocytární hypochromní anémií (Hb 126 g/l, Ery 5,84 × 1012/l, MCV 67,8 fl, MCH 21,6 pg, MCHC 31,6 g/dcl, HbA2 2,3 %) byla diagnostikována α-tal.-2 delece DNA o délce 18,3 kb (schéma 5), která vede díky negativnímu pozičnímu chromosomálnímu efektu k utlumení exprese obou α-globinových genů a projevuje se mikrocytózu a hypochromazií, tedy fenotypem α-tal.-1, fenotypem typickým pro genotyp (--/αα), byť se jedná o deleci α-tal.-2 s genotypem (-α/αα). Jedná se o nový, do té doby neznámý mechanizmus vzniku α-tal.-2 [5,6]. HbH [β4, ztráta tří α-globinových genů ( - -/α)] jsme diagnostikovali u 3 nemocných. Jedna z nich byla Češka, jejíž předkové pocházeli z Itálie, další dvě nemocné pocházely z Vietnamu a z Afriky. Česká nemocná byla vyšetřována pro těžkou mikrocytární hypochromní anémii: Hb 87 g/l; Ery 4,96 × 1012/l; Hkt 0,29; MCV 59,3 fl, MCH 17,5 pg. Hladiny železa a feritinu měla v normě, hladina HbA2 byla na spodní hranici normy (1,3 %) a hladina HbF byla normální (0,5 %). Elektroforéza Hb ukazovala standardní hemoglobinové spektrum (HbH lze detekovat elektroforeticky jen u zcela čerstvě připraveného hemolyzátu). V nátěru periferní krve obarveném brilliant-kresylovou modří jsme v erytrocytech nalezli H-inkluze typické pro HbH onemocnění (volné β-globinové řetězce vytvářejí tetrametry, které precipitují na membránách erytrocytů), obr. 2. Molekulárně-geneticky jsme identifikovali deleci regulační oblasti HS-40 na jednom chromosomu a deleci -α3.7 na druhém chromosomu, a tedy inaktivaci exprese 3 α-globinových genů. Nemocné z Afriky a z Asie měly podobné hodnoty krevního obrazu s odpovídající klinikou a při obarvení nátěru periferní krve briliant-kresylovou modří jsme u nich identifikovali typické H-inkluze. Na molekulární úrovni jsme zjistili genotyp HbH (ztrátu 3 α-globinových genů) u nemocné původem z Afriky způsobený -α4.2/--MED, resp. u nemocné z rodiny pocházející z Vietnamu, způsobený delecí --SEA/-α3.7. Výskyt α-talasemií je v ČR zřejmě daleko větší, než se dosud soudilo. Dá se předpokládat, že α-talasemické alely zde budou se stupňující se migrací přibývat.


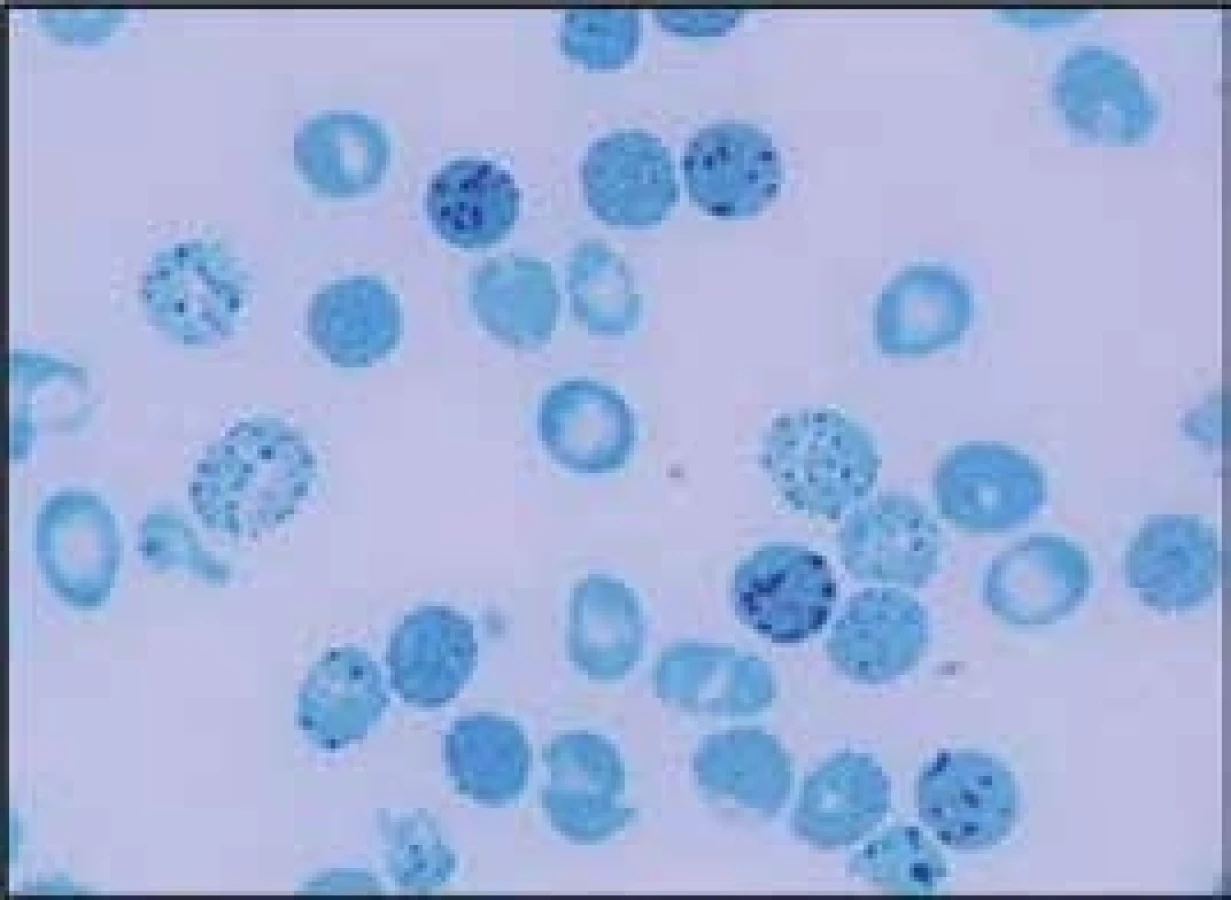
B-talasemie
Molekulárně-genetickou podstatou β-talasemií jsou většinou bodové mutace. Jak již bylo řečeno, rozhodujícím faktorem ovlivňujícím tíži klinických projevů talasemií je velikost nerovnováhy v syntéze α a β-globinových řetězců. Nerovnováha nemusí být podmíněna jen redukcí tvorby β-globinových řetězců, ale může být významně ovlivněna i navýšením (např. triplikací α-globinového genu) nebo redukcí produkce α-globinových řetězců (α-talasemie). Podle typu genetického postižení a klinických příznaků se β-talasemie projevuje jako:
B-talasemie major (Cooley’s anemia): geneticky se jedná o homozygoty nebo složené heterozygoty, s postižením obou β-globinových genů β0 mutací nebo s postižením jednoho genu β0 a druhého β+ mutací. Nemocní mají hepatosplenomegalii, deformity kostí a často trpí i retardací růstu, v krevním obraze nacházíme těžkou mikrocytární a hypochromní anémii, pro kterou jsou postižení jedinci celoživotně transfuzně závislí. V dospělosti u nich bývá detekován pouze HbF, minoritní frakce HbA2 a výrazně snížený HbA (označovaný také jako zbytkový HbA) u β0/β+ složených heterozygotů nebo žádný HbA u β0/β0 homozygotů. Důsledkem četných transfuzí a nízké hladiny hepcidinu [7], která vede ke zvýšení absorpce železa ve střevě, jsou projevy přetížení organizmu železem.
B-talasemie intermedia: geneticky se jedná o homozygoty nebo složené heterozygoty, z nichž jeden gen nese β0 a druhý mírnou β+ mutaci nebo oba β-globinové geny nesou β+ mutaci. Postižení jedinci mají středně těžkou mikrocytární hypochromní anémii. Transfuzní substituci vyžadují sporadicky. Příčinou přetížení organizmu železem je především nízká hladina hepcidinu [7].
B-talasemie minor: geneticky se jedná o heterozygoty, nosiče β-talasemické alely s β0 nebo β+ mutací na jednom β-globinovém genu. Druhý β-globinový gen je intaktní. V naší populaci je to nejčastější forma, při níž mají postižení jedinci lehkou anémii s mikrocytózou (MCV 60–70 fl) a hypochromazii (MCH < 27 pg) a s erytrocytózou (> 5 × 1012/l). Nemocní jsou klinicky asymptomatičtí a nevyžadují transfuzní léčbu. Diagnóza bývá většinou stanovena náhodně, při vyšetření krevního obrazu z jiného důvodu nebo při vyšetření talasemického rodu. V rámci diferenciální diagnostiky náhodně zjištěné mikrocytární hypochromní anémie vylučujeme v první řadě nedostatek železa. Pro diagnózu β-talasemie minor svědčí mírně zvýšená hladina HbA2 (3,5–6 %) a někdy i zvýšená hladina HbF (většinou 2–5 %), erytrocytóza a samozřejmě většinou také pozitivní rodinná anamnéza. Pomocí molekulárně-genetických vyšetření určujeme typ mutace.
Autosomálně dominantní β-talasemie, resp. talasemické hemoglobinopatie: jako dominantní β-talasemie označujeme velmi vzácné β-globinové mutace, které produkují hypernestabilní β-globinové řetězce a vytvářejí extrémně nestabilní hemoglobinové varianty. Nestabilní β-globinové řetězce precipitují ještě před spojením s α globinovými řetězci. Volné α-globinové řetězce se v krvi hromadí a díky inefektivní erytropoéze dávají vzniknout fenotypu β-talasemie intermedia. Jak říká název, tyto mutace se dědí autosomálně dominantně. Jsou-li rodiče postiženého jedince hematologicky normální, může se jednat o spontánní mutaci. Diagnózu potvrzuje sekvenační analýza HBB genu. Do této skupiny tzv. talasemických hemoglobinopatií patří i Hb Hradec Králové popsaný u matky a dcery z Čech [8]. Nemoc byla identifikována u 32leté matky a její 6leté dcery. Obě měly středně těžkou hemolytickou anémii s Heinzovými tělísky (Hb 96, resp. 98 g/l, Ery 3,37, resp. 3,84 × 1012/l, bilirubin 71, resp. 40 μmol/l). Zvýšené hladiny HbA2 4,2 %, resp. 3,2 % svědčily pro talasemii. Zvýšená hladina HbF 4,0 %, resp. 13,4 % ukazovala na možnost přetrvávání vysoké produkce tohoto hemoglobinu. Matce byla v 5 letech provedena splenektomie, která ale klinický obraz choroby nijak nezměnila. Na možnost hemoglobinopatie ukazovala při normálním elektroforetickém nálezu pozitivita izopropanolového testu. Diagnóza nestabilního hemoglobinu byla stanovena až pomocí analýzy β-globinového genu, která odhalila mutaci GCC-GAC v kodonu 115, která vede k záměně alaninu za aspartát (Ala-Asp). Tato mutace mění aminokyseliny v místě β-globinového řetězce, které je zodpovědné za vazbu α1β2 řetězců. Tato mutace byla doc. Divokým a spol. popsána prioritně a podle místa bydliště obou nemocných dostala jméno Hemoglobin Hradec Králové [8].
První souborný přehled o výskytu 8 β-talasemických alel u 97 osob z 36 rodin v ČR publikovali v roce 1992 Indrák et al [9]. V ČR se většinou vyskytují nosiči β-talasemické alely, kteří v 80 % nesou mediteránní mutace. Jen výjimečně se zde vyskytuje β-talasemie intermedia. Někdy může být v rodině kombinován výskyt β-talasemické alely s jinou poruchou produkce globinových řetězců. Při analýze souboru nemocných Indrák zjistil u 17leté dívky s vrozenou mikrocytární a hypochromní hemolytickou anémií nevyžadující transfuzní léčbu (Hb 95 g/l, Hkt 0,35, Ery 5,24 × 1012/l, MCV 66 fl, MCH 18,1 pg, bilirubin 60 mg/ml), u jejího bratra z Olomouce a při následném vyšetření i u dalších 5 členů rodiny s mírnější mikrocytární a hypochromní anémií talasemický fenotyp s vysokou hladinou HbA2 (5,1–6,0 %) a nízkou hladinu HbF (cca 0,3 %). Molekulárně-genetická analýza ukázala, že se jedná o β0 mutaci v kodonech 38/39 (-C) [10]. Tato mutace byla popsána prioritně, a jak se během let ukazuje, je specifická pro ČR (nikde jinde zatím nebyla popsána), do dnešního dne byla zjištěna ve 12 nepříbuzných rodinách u 34 osob. Sestra matky propozity (teta propozity) je také heterozygot pro tuto β0-talasemii, ale byla u ní zjištěna 10krát vyšší hladina HbF (3,1 %) než u ostatních nosičů této β-talasemické mutace. Analýza HbF ukázala, že je tvořen z 95 % Gγ řetězcem a jen stopami Aγ a AγT řetězců. Dva synové tety nezdědili β-talasemickou alelu (v krevním obraze neměli mikrocytózu, hypochromazii ani zvýšený HbA2), a hladinu HbF měli cca 0,8 %, ale ten byl tvořen z 94–99 % Gγ řetězcem, jen stopami Aγ a nebyl identifikován žádný AγT řetězec. S ohledem na normální hodnoty KO obou bratranců propozity je tento nález hodnocen jako tzv. švýcarský typ přetrvávání hladiny hemoglobinu F (HPFH – Hereditary Persistence of Fetal Hemoglobin). Molekulárně-genetická analýza ukázala, že příčinou je mutace C-T v pozici -110 Gγ [10,11]. Další zajímavé β-talasemické mutace popsali Divoký et al [12,13] a Divoká et al [14]. K dnešnímu dni bylo v ČR popsáno 34 β-talasemických alel v 363 rodinách u 610 osob. Specifická pro naši populaci je již zmíněná alela 38/39 (-C). Největší incidenci/prevalenci mají mediteránní alely IVS-I-1 (G-A) s 25,6 %, IVS-I-1110 (G-A) s 19,3 %, IVS-I-6 (T-C) 10,7 % a alela CD 39 (C-T) 10,2 %. Tyto 4 alely reprezentují 69 % všech mutací identifikovaných v naší populaci. U imigrantů nebo jejich potomků byly diagnostikovány 2 případy β-talasemie major a 2 případy dvojitě heterozygotního, resp. homozygotního postižení (HbS). Zajímavé je srovnání incidence β-talasemických alel v ČR a SR. V SR se nejčastěji vyskytuje alela IVS-II-1 (G-A) v 30,3 % (v ČR jen 6,7 %), IVS-I-110 (G-A) v 29,9 % (v ČR 19,3 %) a IVS-I-6 (T-C) 12,9 % (v ČR 10,7 %). Tyto 3 mutace reprezentují 73,1 % všech β-talasemických alel identifikovaných na Slovensku. V SR se nevyskytuje pro ČR specifická mutace v kodonech 38/39 (-C). (Data o slovenských β-talasemiích byla získána s laskavostí paní doc. V. Fábryové, primářky v Nemocnici sv. Michala v Bratislavě a docentky na Slovenskej zdravotníckej univerzite a Vysokej škole sv. Alžbety). Přehled 34 β-talasemických alel identifikovaných v naší populaci a incidenci jejich záchytu ukazuje tab. 2
ΔΒ-talasemie
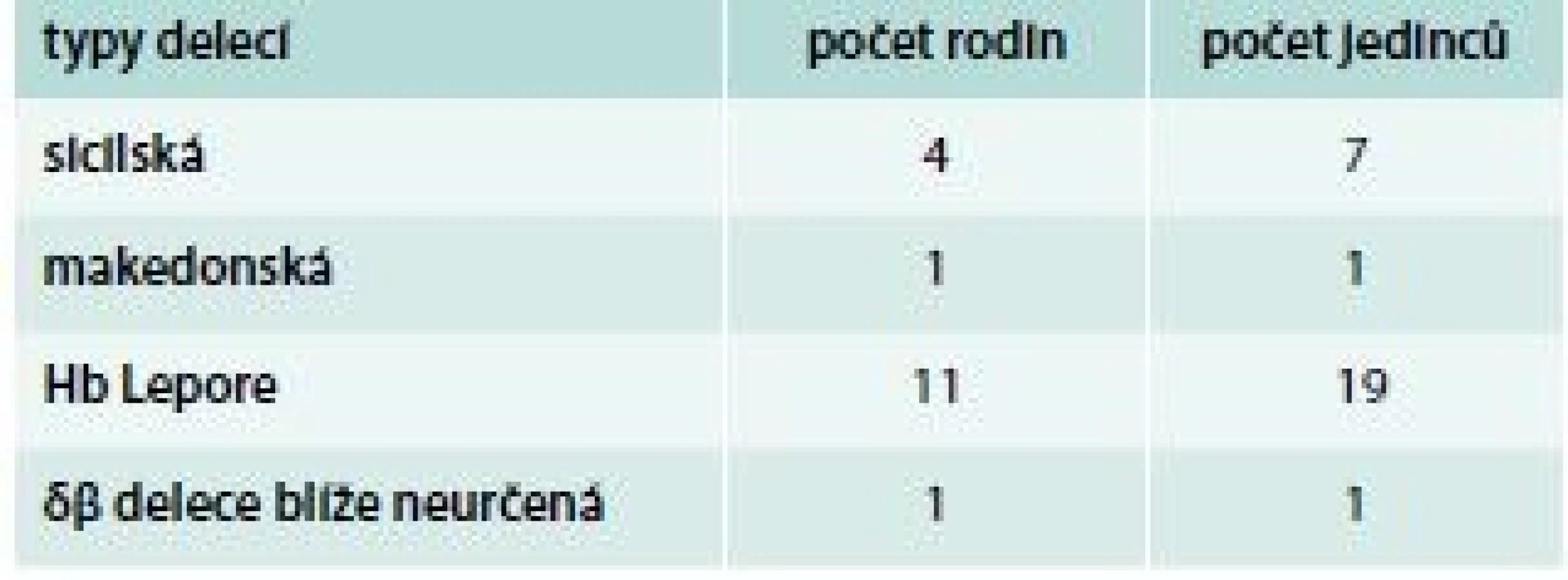
Jsou relativně vzácné, příčinou je nejčastěji delece δ - a β-globinových genů. Syntéza δ - a β-globinových řetězců je v těchto případech nahrazena syntézou γ-globinových řetězců. Typy některých delecí v HBB lokusu vedoucí k δβ-talasemii, které byly diagnostikovány v naší populaci, ukazuje schéma 6. Klinický obraz heterozygotů pro δβ-talasemii připomíná β-talasemii minor s lehkou mikrocytární anémií; produkce HbF je u nich však zvýšena až na 5–20 %. Homozygoti pro δβ-talasemii tvoří jen HbF a hematologicky připomínají β-talasemii intermedia s lehkou anémií a mírnou splenomegalií. V diferenciální diagnostice pomáhá určení hladiny HbA2, která je u heterozygotů s δβ-talasemií normální. V ČR byla v jedné rodině diagnostikována makedonská, ve čtyřech rodinách sicilská delece, v 1 rodině se rozsah δβ-delece zatím nepodařilo blíže určit (tab. 3). Vznik hybridních δ/β globinových genů vede ke vzniku talasemických hemoglobinových variant, např. Hb Lepore, kdy dochází k fúzi N terminální aminokyselinové sekvence normálního δ-globinového řetězce s C terminální částí normální sekvence β-globinového řetězce (HBD-HBB fúze) [15]. Hb Lepore se dědí autosomálně recesivně. Heterozygoti mají KO podobný jako nosiči β-talasemií – lehkou anémii s výraznou hypochromazií a mikrocytózou. Homozygoti mají během prvních dvou let života fenotyp podobný β-talasemia major. V ČR byl Hb Lepore s heterozygotním postižením diagnostikován v 11 rodinách.

Hemoglobinopatie způsobené poruchou struktury molekuly hemoglobinu (strukturální)
Jedná se o onemocnění způsobená záměnou aminokyselin v globinovém řetězci, jejímž výsledkem je strukturní změna v molekule hemoglobinu. Nejčastější příčinou na genové úrovni jsou bodové mutace v kódujících sekvencích globinových genů. Každá mutace však ještě nemusí být provázena změnou funkčních vlastností hemoglobinu. Fenotyp postižených jedinců se může projevovat jako těžká anémie s hemolýzou nebo může být téměř bezpříznakový. Fenotyp se odvíjí od stability a ev. změny afinity hemoglobinu ke kyslíku. Obecně můžeme strukturální hemoglobinopatie rozdělit do 4 skupin:
- abnormální varianty (nestabilní hemoglobiny) s hemolýzou a tendencí precipitovat
- hemoglobinové varianty tvořící srpkovité krvinky
- hemoglobinové varianty s abnormální vazbou ke kyslíku (např. Hb Olomouc) a varianty s kongenitální cyanózou (abnormální methemoglobiny)
- hemoglobinové varianty s abnormální syntézou hemoglobinu – tzv. talasemické hemoglobiny (např. HbE, Hb Lepore)
Nestabilní hemoglobiny
Nestabilní hemoglobiny jsou způsobeny mutacemi, které mění sekvenci aminokyselin jednoho z globinových řetězců a způsobují jeho nestabilitu, jejímž fenotypickým důsledkem je anémie nemocného. Výskyt nestabilních hemoglobinů je v ČR vzácný. Více než 80 % těchto mutací postihuje β-globinový gen. Jedná se o autosomálně dominantně dědičná onemocnění, ale někdy může nestabilní hemoglobinopatie vzniknout jako „de novo“ mutace. U heterozygotů nacházíme vedle hemoglobinu A i nestabilní hemoglobin. Příčinou nestability hemoglobinu může být:
- záměna nepolární, hydrofobní aminokyseliny hemové kapsy za hydrofilní aminokyselinu (Hb St. Louis, Hb Santa Ana) nebo zeslabení vazby hemu na globin, které snižuje stabilitu celé podjednotky (Hb Sydney, Hb Köln). Hemolytická anémie je v těchto případech důsledkem uvolnění hemu a vzniku vysoce nestabilních tetramerů s následnou tvorbou hemichromů a Heinzových tělísek, Hb St. Louis a Hb Köln byly diagnostikovány i v české populaci [16,17].
- mutace histidinu (proximálního nebo distálního), tj. hem-kontaktní aminokyseliny, za aminokyselinu, která nemůže vázat hem (Hb Haná). Hemoglobin Haná byl diagnostikován u 26leté matky a jejích 2 dcer ve věku 6 a 0,5 roku [18,19]. Šestiletá dcera měla mírnou hemolytickou anémii s Heinzovými tělísky (Hb 110 g/l, Ery 4,0 × 1012/l, MCV 85 fl, retikulocyty 0,063, bilirubin 56 µmol/l), hladiny HbA2 (2,7 %) a HbF (0,53 %) byly v normě a zvýšená byla hodnota methemoglobinu (5,5 %). Pozitivita testů tepelné stability a izopropanolového testu ukazovala na nestabilní hemoglobin. Podobné hodnoty krevního obrazu měla v té době půlroční sestra nemocné, zatímco matka měla normální hodnoty (Hb 148 g/l, Ery 5,0 × 1012/l, MCV 87 fl, retikulocyty 0,014), hraniční hodnoty HbA2 (3 %) a HbF (1,02 %) a mírně zvýšenou hodnotu methemoglobinu (2,3 %). I u matky svědčily pozitivní testy tepelné stability a izopropanolový test na nestabilní hemoglobin. Molekulárně-genetická analýza odhalila u všech 3 jedinců v β-globinovém genu v pozici 63 (E7) záměnu histidinu za asparagin. Tento mírně nestabilní hemoglobin dostal název hemoglobin Haná podle regionu, ve kterém rodina nemocné žila. Tento nález ale nevysvětloval rozdíly v hodnotách krevního obrazu mezi matkou a jejími dcerami se stejnou genetickou abnormitou. Ani podrobné vyšetření β - a α-globinových genů a poměru α - a β-globinových řetězců nepřineslo vysvětlení. To přineslo až vyšetření erytrocytárních enzymů, které ukázalo, že příčinou rozdílů mezi dcerami a matkou je částečný deficit glutationreduktázy (GR) v erytrocytech obou dcer, zatímco u matky je hladina tohoto enzymu v erytrocytech v normě [19].
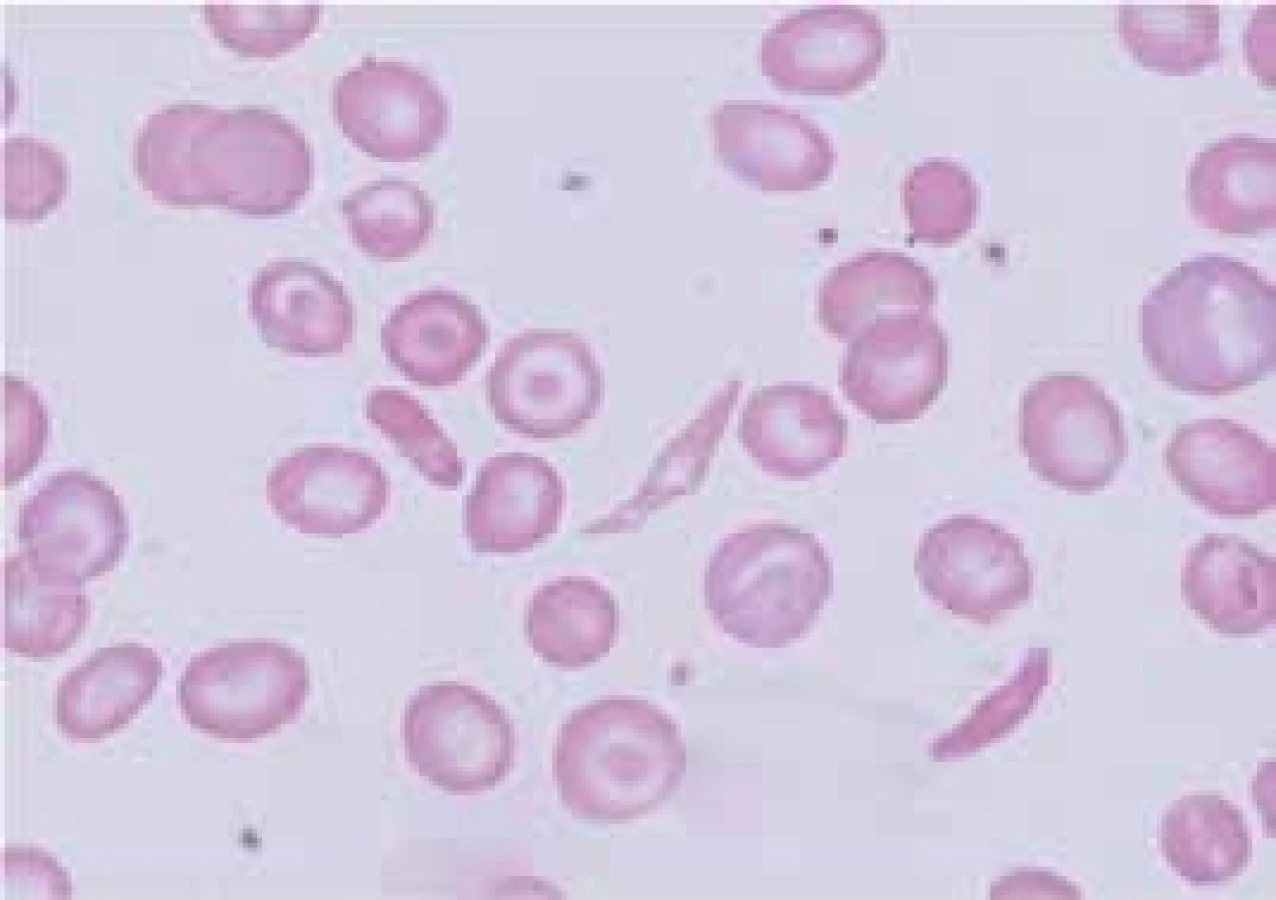
- mutace narušující α1β1 nebo α1β2 vazby (Hb Hradec Králové, [8]) způsobují nestabilitu hemoglobinu. Dochází k narušení vazby α1β1 nebo α1β2 dimerů a k jejich precipitaci. Vzniklé precipitáty tvoří Heinzova tělíska, a ta narušují plasticitu erytrocytární membrány a díky zhoršené deformabilitě erytrocytu zhoršují průchodnost erytrocytů mikrocirkulací. To vede k deformaci a rozpadu červené krvinky a k anemizaci postiženého jedince. Homozygoti a složení heterozygoti pro mutace vedoucí k nestabilitě hemoglobinu dosud nebyli identifikováni, protože pravděpodobně nejsou schopni života. Nemocní s hemoglobinopatií s nestabilním hemoglobinem mají většinou fenotyp hereditární nesférocytární hemolytické anémie s Heinzovými tělísky. Fenotypický projev těchto nestabilních hemoglobinů je dán stupněm jejich nestability. V důsledku kompenzační nadprodukce erytropoézy v kostní dřeni nacházíme v periferní krvi zvýšené hodnoty retikulocytů. Hemolytickou krizi mohou u nemocných s nestabilním hemoglobinem vyvolat infekce nebo oxidativně působící léky. Ve fyzikálním nálezu dominuje bledost, ikterus a splenomegalie, častá je cholelitiáza. Pacienti mívají v období krize tmavou moč. V krevním obraze nacházíme snížené hodnoty hemoglobinu, makrocytózu (především v důsledku retikulocytózy), hypochromazii, poikilocytózu, polychromazii, anizocytózu a bazofilní tečkování erytrocytů. Běžně se v nátěru periferní krve při barvení brilantní kresylovou modří objevují i Heinzova tělíska. Pro diagnostiku nestabilních hemoglobinů je důležitý průkaz Heinzových tělísek v nátěru periferní krve obarveném brilantní kresylovou modří (obr. 3). Elektroforéza Hb může, ale nemusí odhalit abnormální frakci Hb. Spolehlivý screening poskytují testy tepelné denaturace (stability) a izopropanolový test. Stanovení definitivní diagnózy nestabilního Hb umožňuje analýza globinových řetězců pomocí vysokotlaké kapalinové chromatografie. Nejnestabilnější řetězce však precipitují již v erytroblastech kostní dřeně a nevytvářejí tetramery (vedou k inefektivní erytropoéze). Nejdou proto pomocí těchto metod identifikovat – diagnózu umožní stanovit až analýza DNA na úrovni sekvenace globinových genů. Do této skupiny patří i Hb Hradec Králové [8]. Tyto hemoglobinopatie je proto možné detekovat jen molekulárně-genetickými metodami. Léčebně může být u nemocných s nestabilním hemoglobinem přínosná splenektomie, ale při rozhodování o její indikaci je vhodné vycházet z empirických zkušeností dostupných v odborné literatuře.

Hemoglobinové varianty tvořící srpkovité krvinky
HbS vzniká záměnou kyseliny glutamové za valin v místě 6. aminokyseliny β-globinového řetězce (β6 Glu-Val). Příčinou je tedy bodová mutace v β-globinovém genu. HbS je charakterizován polymerizací a agregací neokysličeného HbS a formací nitrobuněčných polymerů, které deformují červené krvinky do typické podoby srpkovitého erytrocytu (obr. 4). Polymery mění biofyzikální vlastnosti erytrocytů, které jsou hůře deformovatelné a adherují k endotelu. Srpkovatění je zpočátku reverzibilní, ale opakované srpkovatění erytrocytu poškozuje jeho membránu a srpkovitý tvar erytrocytu se stane ireverzibilní. Vznik srpkovatění závisí na řadě faktorů, jako jsou intracelulární koncentrace hemoglobinu, přítomnost jiné hemoglobinopatie, nedostatečné okysličení krve a snížení hladiny 2,3-bisfosfoglycerátu. Srpkovité erytrocyty zvýšeně adherují k endotelu cév. Jejich shluky způsobují stázu krve, zvýšení mikrovaskulární viskozity a v důsledku trombóz vedou k infarzaci a poškození tkání. Gen pro srpkovitou anémii je nalézán především v Africe, kde je až 40 % heterozygotů pro HbS, ale vyskytuje se i v celé řadě dalších oblastí (Střední Východ, Řecko, Indie). Geografická souvislost výskytu HbS v oblastech s vysokým výskytem malárie je i zde důsledkem genetického tlaku zvýhodňujícího nosiče HbS proti vzniku malarické infekce. Srpkovitá anémie se klinicky projevuje jen u homozygotů (HbSS); heterozygoti (HbA/HbS) jsou hematologicky normální. Onemocnění má výrazně rozdílný interindividuální klinický průběh, jehož tíže závisí na řadě faktorů – např. jsou-li nemocní homozygoti (HbSS) nebo dvojití heterozygoti pro HbS a jinou běžnou hemoglobinopatii (např. HbS/HbC), na míře přetrvávající exprese fetálního hemoglobinu (HbF) v dospělosti apod. U heterozygotů pro HbS tvoří méně než 50 % hemoglobinu v erytrocytu hemoglobin S (přibližně 40 %) a zbytek tvoří normální hemoglobin, hlavně HbA. Ten postiženého jedince účinně chrání proti srpkovatění, s výjimkou zvláštních situací, jakými je hypoxie a hyperosmolarita. Morbidita a mortalita nosičů srpkovité anémie je mimořádně nízká. U homozygotních dětí (HbSS) patří k převažujícím klinickým projevům bolesti, infekce a různé záněty. Hyperplazie erytropoézy způsobuje rozšíření medulárních prostor v kostní dřeni a ztenčení kortexu. Obratle mohou být bikonkávní a mohou připomínat rybí páteř. Nekrózy kostí mohou být provázeny periostální reakcí a oblastmi osteosklerózy. U dospělých homozygotů mají klinické projevy ráz chronického onemocnění s postižením různých orgánů. Řada nemocných ale po většinu života nemá žádné zdravotní problémy. Vazookluzivní krize nebo ataky bolesti patří k nejčastějším manifestacím srpkovité anémie a u jednotlivých nemocných se jejich incidence výrazně liší – od denně se vyskytujících potíží až k potížím objevujícím se jednou za rok a někteří jedinci nemají potíže po celou dobu života. Generalizované okluze mohou způsobit syndrom akutní srpkovité krize. Ta je charakterizovaná nesnesitelnými bolestmi v kostech a v kloubech. Tkáňová hypoxie a infarkty se mohou vyskytnout kdekoliv. Mezi časté lokalizace patří cévní mozkové příhody (především u dětí), nekrózy kostí, priapizmus, „acute chest syndrome“ charakterizovaný teplotou, leukocytózou, plicními infiltráty a akutním plicním edémem. V dětství je častá splenomegalie, ale v důsledku opakovaných infarktů nacházíme v dospělosti malou, fibroticky změněnou slezinu (autosplenektomie). Chronická vazookluze vede k papilární nekróze ledvin s hematurií, mikroalbuminurií a proteinurií zhoršující se s věkem a končící často uremií. Častou komplikací u dospělých je plicní hypertenze, srdeční selhání a bércové vředy. Sekvestrační krize se vyskytují u kojenců nebo vzácně u starších dětí a příležitostně u dospělých se zvětšenou slezinou. Při nich dochází k náhlému masivnímu shluknutí erytrocytů ve slezině, které může způsobit hypotenzní šok a případně i smrt. Srpkovité anémie jsou charakterizovány výrazným zkrácením přežívání erytrocytů. Infekce, především parvovirem B19, mohou vést k přechodné aplastické krizi s dramatickým poklesem hodnot hemoglobinu. Důsledkem chronické hemolýzy je výskyt cholelitiázy u 50–75 % dospělých jedinců. V graviditě se incidence bolestivých krizí zvyšuje a může dojít k preeklampsii. Hladina Hb se u homozygotů většinou pohybuje mezi 50–110 g/l. Anémie je normochromní a normocytární s větší distribuční šíří velikosti erytrocytů (RDW). V nátěru periferní krve nacházíme retikulocytózu, a srpkovité krvinky a terčovité buňky (obr. 4). Častá je i leukocytóza a trombocytemie, především u nemocných s akutními problémy, a mírné zvýšení hladiny železa, ale onemocnění z přetížení železem je vzácné. HbS můžeme identifikovat pomocí elektroforézy a potvrdit molekulárně-geneticky. Prenatální diagnostika se provádí vyšetřením DNA z vilů choria, které se získávají amniocentézou. Nesmírně důležitá je pečlivá diferenciální diagnostika k odlišení bolestí v důsledku srpkovité krize od bolestí, které byly vyvolány z jiného důvodu. Pacienti se srpkovitou anémií by se měli vyhýbat vysokým nadmořským výškám, prochlazení a infekcím, které v důsledku hypoxemie vedou k srpkovatění erytrocytů. Přínosné může být podávání kyseliny listové. Děti by měly být vakcinovány pneumokokovou vakcínou. Co nejdříve by měla být zahájena léčba každé infekce. Omezení srpkovatění přináší zvýšení hladiny HbF pomocí hydroxyurey, která stimuluje tvorbu γ-globinových řetězců a tvorbu HbF. Chronická léčba hydroxyureou by měla být volbou léčby u nemocných s častými a těžkými srpkovitými krizemi. Alogenní transplantace krvetvorných buněk je jedinou kurativní léčbou srpkovité anémie. Transfuze zvyšují hladinu hemoglobinu a snižují podíl srpkovitých erytrocytů. Chronické podávání transfuzí proto působí jako prevence mozkových příhod. Nemocní s cévní komplikací by měli být souběžně s transfuzní léčbou i dostatečně hydratováni a měli by dostávat adekvátní analgetickou léčbu. Pečlivé sledování vyžadují zvláště gravidní ženy s HbS, ale přínos pravidelné transfuzní léčby je u nich sporný. Nízká porodní váha a zvýšené potraty žen s HbS jsou pravděpodobně způsobeny cévními okluzemi v placentě. Cholestáza způsobená srpkovitou anémií může být velmi vážnou a dokonce fatální komplikací a i v tomto případě může mít léčebný efekt výměnná transfuze krve. V oblastech s vysokým výskytem HbS existují specializované klinicko-diagnostické jednotky pro tyto nemocné (Sickle cell centres).
Hemoglobinopatie s vysokou afinitou ke kyslíku
První hemoglobinopatii s vysokou afinitou ke kyslíku u starého muže s familiárním výskytem erytrocytózy popsali v roce 1966 Charache et al [20]. Hemoglobin byl označen jménem nemocnice, ve které byl diagnostikován – Hb Chesapeake. Většina dosud známých Hb variant s vysokou afinitou ke kyslíku má aminokyselinovou substituci v jedné ze tří oblastí, které hrají zásadní roli pro fungování hemoglobinu:
- styčná plocha α1β1 spojení
- C-terminální konec β-globinového řetězce
- místo vazby 2,3-bisfosfoglycerátu (BPG)
Projevem zvýšené vazby Hb ke kyslíku je posun disociační křivky kyslíku doleva. Musíme si ale uvědomit, že k posunu disociační křivky mohou vést i intracelulární faktory – např. změna pH, změna hladiny 2,3-bisfosfoglycerátu (2,3-BPG), modifikace hemové skupiny u methemoglobinemie nebo karboxyhemoglobinemie. Všichni nemocní s hemoglobinopatií se zvýšenou afinitou ke kyslíku jsou heterozygoti – kauzální mutace jsou autosomálně dominantní. Množství hemoglobinu A je v jejich erytrocytech přinejmenším stejné jako množství hemoglobinové varianty. Homozygoti nejsou schopni života. Většina nemocných nemá žádné klinické projevy. Při fyzikálním vyšetření dominuje červené zbarvení celého těla. Zcela výjimečně bývá nevelká splenomegalie. Diagnóza bývá stanovena na základě vyšetření krevního obrazu a náhodně zjištěné erytrocytózy bez známek hyperviskozity či projevů tkáňové hypoxie. Hodnoty Hb se pohybují kolem 190 g/l, zvýšení erytrocytární masy bývá menší než u pravé polycytemie. Hodnoty leukocytů a trombocytů bývají v normě a nenacházíme JAK2 V617F mutaci. Podezření na hemoglobinopatii se zvýšenou afinitou ke kyslíku podporuje nález erytrocytózy u více členů rodu. Při elektroforetickém vyšetření hemoglobinu na acetátu celulózy se daří odseparovat od HbA jen asi polovinu hemoglobinových variant. U ostatních variant jsou k separaci nutné změněné podmínky elektroforézy. Podezření na tuto hemoglobinopatii podporuje snížená hodnota pO2(50) nebo vyšetření disociační křivky hemoglobinu ke kyslíku s nálezem posunu doleva. Většina nemocných nevyžaduje žádnou léčbu, v případě vzácného vzestupu hematokritu > 0,55 s projevy hyperviskozity je vhodné snížení hodnot hematokritu < 0,50 erytrocytaferézou nebo venepunkcí. Záměna této erytrocytózy s pravou polycytemií by mohla vést k nevhodnému zahájení cytoredukční léčby se zvýšením kancerogenního rizika nebo k nadměrné venepunkční léčbě s následnou hypoxemií. Od roku 2005, kdy byla u nemocných s pravou polycytemií identifikována mutace JAK2 V617F [21–24], kterou nese více než 95 % nemocných, je ale riziko této záměny malé. Vysoká afinita Hb ke kyslíku byla prokázána také u asi třetiny nestabilních hemoglobinových variant. Dominujícím klinickým projevem zde však je akcelerovaná destrukce erytrocytů (hemolýza). Všechny uvedené znaky hemoglobinů s vysokou afinitou ke kyslíku splňovaly i nálezy u 45letého muže sledovaného 8 let pro nejasnou absolutní erytrocytózu s normální saturací arteriální krve kyslíkem (Hb 193 g/l, Ery 6,0 × 1012/l, leukocyty a trombocyty v normě, objem erymasy 37 ml/kg, saturace arteriální krve kyslíkem 98 %). Až vyšetření pO2(50) arteriální krve ukázalo sníženou hodnotou 2,77 kPa (N = 3,2–4,2) na možnost hemoglobinopatie s vysokou afinitou ke kyslíku. Podezření ještě zvýšil nález podobných laboratorních hodnot u syna nemocného a potvrdila jej elektroforéza hemoglobinu na acetátu celulózy. Separace a izolace HbX s následnou izolací β-globinových řetězců a jejich analýza ukázala, že se jedná o dosud neznámou variantu hemoglobinu β86 Ala-Asp, která dostala podle místa bydliště nemocného název Hb Olomouc [25].
Další formou hemoglobinopatií se zvýšenou afinitou ke kyslíku jsou methemoglobinemie způsobené hemoglobinovými variantami označovanými souborně jako HbM. Methemoglobin (Met-Hb) je heterodimer, ve kterém jsou jeden nebo více fero-iontů (Fe2+) hemu oxidovány na feri-ionty (Fe3+). Vzhledem ke známé dynamice oxygenace deoxyhemoglobinu na oxyhemoglobin vede přítomnost feri-iontu (Fe3+), byť jen v jednom hemu, ke zvýšené afinitě hemoglobinu ke kyslíku u zbývajících 3 hemových skupin hemoglobinové molekuly. Přítomnost Met-Hb tedy vede k posunu disociační křivky Hb doleva a ke snížení schopnosti postiženého hemoglobinového řetězce vázat a přenášet kyslík. Důsledkem je snížené uvolňování kyslíku do tkání. Přítomnost Met-Hb proto způsobuje lehkou polyglobulii s mírnou trvalou cyanózou. Vrozené methemoglobiny jsou velmi vzácné a mají autosomálně dominantní dědičnost. Vrozené methemoglobinemie se u heterozygotů manifestují nahnědlým nebo břidlicově šedým zbarvením kůže. Pro hemoglobinopatii s přítomností HbM je typická přítomnost cyanózy a mírné polycytemie i u dalších členů rodiny ve více generacích. Při HbM je břidlicově-šedé zbarvení dáno spíše molekulou Met-Hb než vzestupem deoxyhemoglobinu. Krev se zvýšenou hladinou methemoglobinu má typicky čokoládově hnědou barvu, která se nemění ani v přítomnosti vzdušného kyslíku. U některých forem HbM může být přítomná mírná hemolytická anémie. Postižení jedinci obvykle nejsou dyspnoičtí, nemají žádná omezení fyzické aktivity a mají předpoklad normální průměrné délky života. Při pátrání po vrozené methemoglobinemii je důležitá důkladná rodinná a osobní anamnéza s cílenými dotazy na výskyt cyanózy u ostatních členů rodiny a na použití léků nebo kontakt s chemikáliemi způsobujícími cyanózu. K přesnému stanovení diagnózy je nutné určení hladiny Met-Hb v krvi. Klasická pulzní oxymetrie v těchto případech není spolehlivá, protože odlišuje pouze oxyhemoglobin a redukovaný hemoglobin. Moderní analyzátory krevních plynů jsou vybaveny optickým systémem, který umožňuje stanovit koncentraci hemoglobinu a jeho derivátů přímou spektrofotometrií. K vyloučení vrozeného enzymového defektu je nutno spektrofotometricky stanovit hladinu methemoglobinreduktázy. Přítomnost patologického hemoglobinu M je možno prokázat pomocí elektroforézy hemoglobinu, případně pomocí sekvenačního vyšetření globinových genů. Léčba nemocných s HbM většinou není nutná. Některé přidružené stavy jako anémie, kardiopulmonální choroby nebo požití léků s oxidačním účinkem však mohou stav pacienta zhoršit a vyžadují léčbu.
Hemoglobinové varianty s abnormální syntézou hemoglobinu – tzv. talasemické Hb (např. HbE, Hb Lepore)
Hemoglobin E je abnormální hemoglobin vzniklý záměnou glutamové kyseliny v pozici 26 β-globinového genu. Mutace vytváří alternativní sestřihové místo mRNA v kodonech 25–27 β-globinového genu. Výsledkem je mírný nedostatek normální β mRNA a snížená tvorba β řetězců, která může vést ke vzniku β-talasemie. Tato hemoglobinopatie je charakterizována i slabým spojením mezi α-globinovými a β-globinovými řetězci, což vede k nestabilitě v případě oxidativního stresu nebo při virové infekci. Hemoglobin E je velmi častý v jihovýchodní Asii a v Indii. Můžeme jej detekovat pomocí elektroforézy. Někteří nemocní jsou asymptomatičtí (heterozygoti nebo homozygoti pro HbE) s mírnou mikrocytární hypochromní anémií, zatímco nemocní s častou kombinací HbE a β-talasemie (složení heterozygoti pro HbE a β-talasemii) mohou mít těžkou anémii a mohou být transfuzně závislí. V české populaci jsme u 13leté dívky s těžkou vrozenou hemolytickou anémií (Hb 55 g/l, Ery 1,99 × 1012/1, MCV 94 fl, MCH 27,6 pg, retikulocyty 0,28 ‰; erytroblasty 1 000/100 Le) s transfuzní závislostí a známkami přetížení železem identifikovali HbE β-talasemii. Otec a teta nemocné z otcovy strany měli jen mírnou mikrocytární hypochromní anémii v důsledku nosičství β-talasemie (Hb 11,5, resp. 10,9 g/l, Ery 5,96, resp. 4,89 × 1012/l, MCV 63,4, resp. 66,9 fl, MCH 19,3, resp. 22,3 pg, HbA2 5,2, resp. 4,7 % a HbF < 1 %), matka nemocné a bratr nemocné měli mírnou anémii (Hb 118 g/l), resp. normální hodnoty Hb (138 g/l) a byli nosiči HbE s 26 %, resp. 28,4 % HbE. Molekulárně-genetická analýza ukázala, že otec a jeho sestra jsou nosiči β-talasemické mutace IVS-I-1 (G-A), matka nemocné a bratr nemocné jsou nosiči HbE. Nemocná získala od otce β-talasemickou mutaci IVS-I-1 (G-A) a od matky mutaci pro HbE. Kombinace těchto mutací je vzácná a vede ke vzniku těžké chudokrevnosti, která vesměs, na rozdíl od naší nemocné, nevyžaduje transfuzní léčbu. Možné vysvětlení těžké anémie nemocné přinesla až analýza α-globinových genů, která ukázala, že má vedle dvou mutací v β-globinovém genu i třetí mutaci – triplikaci α-globinového genu (ααα/αα), která u ní zvyšuje nerovnováhu mezi α-globinovými a β-globinovými řetězci a dále prohlubuje chudokrevnost, makrocytózu a hypochromazii nemocné [26]. Mezi talasemické hemoglobinové varianty se řadí i některé varianty s hypernestabilními globiny vedoucí k autosomálně dominantnímu β-talasemickému fenotypu (např. Hb Hradec Králové, viz výše) [8,27,28].
Přehled 20 hemoglobinových variant s mutací v β-globinovém řetězci identifikovaných do konce roku 2017 v ČR přináší tab. 4. Další dvě varianty s mutací v α globinovém řetězci jsou uvedeny v tab. 1.
Závěr
Hemoglobinopatie nejsou v ČR tak vzácné, jak se dříve myslelo. Do roku 2017 byly diagnostikovány u 876 osob z 579 rodin. Jejich incidence/prevalence se v ČR v posledních letech stále zvyšuje, což souvisí s lepšící se informovaností lékařů o těchto chorobných jednotkách a s lepší dostupností specializované diagnostiky. Dá se však předpokládat, že se v naší populaci stále vyskytují jedinci s nerozpoznanou hemoglobinopatií. Proto zde chceme připomenout charakteristické znaky základních typů hemoglobinopatií a jejich diagnostiku i některé vzácné typy a klinické projevy, z nichž některé byly prioritně popsány právě v naší populaci. Poddiagnostikováni jsou především nosiči talasemických alel, kteří mají nenápadný fenotyp mikrocytární hypochromií anémie s mírnou erytrocytózou a normální hladinou feritinu i sérového železa. Klinicky jsou tito jedinci asymptomatičtí, ale v důsledku změn v krevním obraze jim někdy bývala mylně stanovena diagnóza sideropenické anémie. Následná „léčba“ železem, která nemá žádný vliv na změny v krevním obraze, může tyto jedince s charakteristicky nízkou hladinou hepcidinu naopak poškodit. Problematiku hemoglobinopatií v ČR připomínáme i proto, že jejich význam stoupá i se současnou vlnou migrace obyvatelstva z malarických oblastí do Evropy. V uzavřených a dnes již relativně početných komunitách mladých migrantů s vysokou prevalencí heterozygotního postižení hemoglobinopatií jsou často uzavírány sňatky uvnitř komunity (např. mezi Vietnamci). S ohledem na závažnost klinických projevů u homozygotů a složených heterozygotů proto nabývá na významu i prenatální diagnostika homozygotního postižení. Tu dnes již rozvinulo centrum vzácných chorob na Ústavu hematologie a krevní transfuze v Praze a na Klinice dětské hematologie a onkologie 2. LF UK a FN Motol Praha ve spolupráci s Oddělením lékařské genetiky FN Motol Praha a Centrem vzácných chorob na Hemato-onkologické klinice LF UP a FN v Olomouci.
prof. MUDr. Karel Indrák, DrSc.
Hemato-onkologická klinika LF UP a FN Olomouc
Doručeno do redakce 7. 12. 2017
Přijato po recenzi 15. 3. 2018
Sources
- Deisseroth A, Nienhuis A, Turner P et al. Localization of the human alpha-globin structural gene to chromosome 16 in somatic cell hybrids by molecular hybridization assay. Cell 1977; 12(1): 205–218.
- Deisseroth A, Nienhuis A, Lawrence J et al. Chromosomal localization of human beta globin gene on human chromosome 11 in somatic cell hybrids. Proc Natl Acad Sci USA 1978; 75(3): 1456–1460.
- Sgourou A, Routledge S, Antoniou M et al. Thalassaemia mutations within the 5‘UTR of the human beta-globin gene disrupt transcription. Br J Haematol 2004; 124(6): 828–835.
- Divoká M, Partschová M, Pospíšilová D et al. Alfa-talasemie u 45 českých rodin a 37 rodin cizinců žijících v České republice: přehled literatury a molekulárně-genetická diagnostika. Transfuze Hematolo Dnes 2016; 22(3): 201–210.
- Indrák K, Gu YC, Novotný J et al. A new alpha-thalassemia-2 deletion resulting in microcytosis and hypochromia and in vitro chain imbalance in the heterozygote. Am J Hematol 1993; 43(2): 144–145.
- Barbour VM, Tufarelli C, Indrak K et al. Alpha-thalassemia resulting from a negative chromosomal position effect. Blood 2000; 96(3): 800–807.
- Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med 2015; 21(3): 221–230. Dostupné z DOI: <http://dx.doi.org/10.1038/nm.3814>.
- Divoky V, Svobodova M, Indrak K et al. Hb Hradec Kralove (Hb HK) or alpha2beta2 115 (G17) Ala→Asp, a severely unstable hemoglobin variant resulting in a dominant beta-thalassemia trait in a Czech family. Hemoglobin 1993; 17(4): 319–328.
- Indrak K, Brabec V, Indrakova J et al. Molecular characterization of beta-thalassemia in Czechoslovakia. Hum Genet 1992; 88(4): 399–404.
- Indrák K, Indráková J, Kutlar F et al. Compound heterozygosity for a beta zero-thalassemia (frameshift codons 38/39; – C) and a nondeletional Swiss type of HPFH (A-C at NT -ll0; G) in a Czechoslovakian family. Ann Hematol 1991; 63(2): 111–115.
- Indrák K, Indráková J, Kutlarová F et al. Dvojnásobná heterozygozita pro beta thalassemii (posunutá mutace v kodonech 38/39:-C) a pro nedeleční – švýcarský – typ dědičné persistence hemoglobinu F (A-C v nukleotidu-110: ‚G‘gama) v olomoucké rodině. Hematológia a transfuziológia 1992; 2(1): 32–41.
- Divoký V, Gu LH, Indrák K et al. A new beta zero-thalassemia nonsense mutation (codon 112, T-A) not associated with a dominant type of thalassemia in the heterozygote. Br J Haematol 1993; 83(3): 523–524.
- Divoký V, Baysal E, Indrák K et al. The T-->C mutation at position +96 of the untranslated region 3‘ to the terminating codon of the beta-globin gene is a rare polymorphism that does not cause a beta-thalassemia as previously ascribed. Hum Genet 1994; 93(1): 77–78.
- Divoká M, Partschova M, Kucerova J et al. Molecular Characterization of beta-Thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutation. Hemoglobin 2016; 40(3): 156–162. Dostupné z DOI: <http://dx.doi.org/10.3109/03630269.2016.1152581>.
- Steinberg MH, Adams JG. Thalassemic hemoglobinopathies. Am J Pathol 1983; 113(3): 396–409.
- Wiedermann B, Indrák K, Wilson JB et al. Hb Saint Louis or alpha 2 beta 2(28)(B10)Leu----Gln in a Czechoslovakian male. Hemoglobin 1986; 10(6): 673–676.
- Indrák K, Brabec V, Wilson JB et al. Hb Köln or alpha 2 beta 2(98)(FG5)Val----Met in a Czechoslovakian family. Hemoglobin 1991; 15(1–2): 133–135.
- Divoký V, Indrák K, Divoká M et al. Hemoglobin Haná nebo alfa2 beta2 63 (E7) HIS-ASN: nová nestabilní varianta hemoglobinu identifikovaná v Moravské rodině z Hané. Vnitř Lék 1997; 43(5): 267–272.
- Mojzikova R, Dolezel P, Pavlicek J et al. Partial glutathione reductase deficiency as a cause of diverse clinical manifestations in a family with unstable hemoglobin (Hemoglobin Haná, beta63(E7) His-Asn). Blood Cells Mol Dis 2010; 45(3): 219–222.
- Charache S, Weatherall D, Clegg JB. Polycythemia associated with a hemoglobinopathy. J Clin Invest 1996; 45(6): 813–822. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI105397>.
- Baxter EJ, Scott LM, Campbell PJ et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365(9464): 1054–1061. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(05)71142–9>. Erratum in Lancet 2005; 366(9480): 122.
- James C, Ugo V, Le Couédic JP et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434(7037): 1144–1148. Dostupné z DOI: <http://dx.doi.org/10.1038/nature03546
- Levine RL, Wadleigh M, Cools J et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7(4): 387–397. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ccr.2005.03.023>.
- Kralovics R, Passamonti F, Buser AS et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med 2005; 352(17): 1779–1790. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa051113>.
- Indrák K, Wiedermann B, Bátěk F et al. Hb OLOMOUC or alpha2 beta2 86 /F2/ ALA-ASP, a new high oxygen affinity variant. Hemoglobin 1987; 11(2): 151–155.
- Indrák K, Fei YJ, Li HW et al. A Czechoslovakian teenager with Hb E-beta zero-thalassemia [IVS-I-1 (G----A)] complicated by the presence of an alpha-globin gene triplication. Ann Hematol 1991; 63(1): 42–44.
- Divoký V, Walczysková S, Pospíšilová D et al. Některé vzácnější formy hereditárních anémií vyskytující se v dospělé populaci v ČR – beta-talasemie a nestabilní hemoglobinové varianty. Vnitř Lék 2005; 91(7–8): 886–893.
- Divoký V, Indrák K, Mojzíková R. Hemoglobinopatie: talasémie a strukturní Hb varianty. In: Pospíšilová Š, Dvořáková D, Mayer J (eds). Molekulární hematologie. Galén: Praha 2013 : 270–283. ISBN 978–80–7262–942–8.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2018 Issue 5
Most read in this issue
- Autoimmune hemolytic anemia
- Differential diagnosis of anemia
- Congenital and acquired bleeding disorders
- Hemoglobinopathies