
Imunodeficience v rámci diferenciální diagnostiky intersticiálních plicních procesů
Immunodeficiency in the differential diagnosis of interstitial lung diseases
Interstitial lung processes (IPP), or diffuse parenchymal lung diseases, are a broad group of diseases characterized by varying degrees of pulmonary fibrosis and inflammation affecting predominantly, but not exclusively, pulmonary interstitium. IPP mostly occur in adulthood with maximum manifestation between 40 and 70 years of age. Although IPP mostly present as a primary diagnosis, they also belong to the portfolio of pulmonary disorders in patients with primary immunodeficiencies. The authors present case reports of patients with interstitial lung involvement and primary immunodeficiencies [particularly those manifesting also in adulthood, such as common variable immunodeficiency (CVID), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) deficiency]. In addition, we report the case of silicosis patient with severe lymphopenia. Therefore, in patients with newly diagnosed interstitial lung disease, congenital immune system disorder should be considered. Basic immunological laboratory examination of humoral and cellular immunity should be an essential part of the differential diagnosis algorithm for interstitial lung disease.
Keywords:
common variable immunodeficiency – cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) deficiency – immunology laboratory investigation – interstitial lung disease – primary immunodeficiencies
Authors:
Martina Doubková 1; Jana Špeldová 1; Zita Chovancová 2
Authors‘ workplace:
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno, pracoviště Bohunice
1; Ústav klinické imunologie a alergologie LF MU a FN u sv. Anny v Brně
2
Published in:
Vnitř Lék 2019; 65(11): 685-693
Category:
Overview
Intersticiální plicní procesy (IPP), neboli difuzní parenchymatózní onemocnění plic, jsou širokou skupinou onemocnění, která jsou charakterizována různým stupněm plicní fibrózy a zánětu, a to nejen na úrovni plicního intersticia. IPP se vyskytují zejména v dospělosti s maximem manifestace mezi 40.–70. rokem věku. Ačkoli se IPP vyskytují nejčastěji samostatně jako základní diagnóza, patří též do portfolia plicního postižení u pacientů s některými primárními imunodeficiencemi. Autoři prezentují kazuistiky pacientů s primárními imunodeficiencemi, u kterých se vyskytuje intersticiální plicní postižení a které se zároveň mohou manifestovat až v dospělém věku. Jedná se zejména o běžnou variabilní imunodeficienci (Common Variable ImmunoDeficiency – CVID) a deficienci cytotoxického T-lymfocytárního antigenu 4 (CTLA4). Navíc uvádí kazuistiku pacienta se silikózou, u kterého byla zjištěna významná lymfocytopenie. U pacientů s nově zjištěným intersticiálním plicním postižením tedy musíme myslet na rozvoj primární poruchy imunitního systému. Základní imunologické laboratorní vyšetření humorální a buněčné imunity by proto mělo tvořit nezbytnou součást diferenciálně diagnostického algoritmu intersticiálních plicních postižení.
Klíčová slova:
běžná variabilní imunodeficience – deficience cytotoxického T-lymfocytárního antigenu 4 – imunologické laboratorní vyšetření – intersticiální plicní procesy – primární imunodeficience
Úvod
Intersticiální plicní procesy (IPP), jinak zvané i difuzní parenchymatózní nemoci plic, jsou širokou skupinou onemocnění. IPP jsou charakterizovány různým stupněm zánětu a plicní fibrózy nejen na úrovni plicního intersticia, ale i plicních kapilár, endotelu a bronchiolů (dle Americké hrudní společnosti/American Thoracic Society – ATS) [1]. Tato skupina chorob má řadu společných rysů, včetně podobných klinických příznaků, radiologického nálezu a obdobné patofyziologie. Z těchto důvodů není často diferenciální diagnostika snadná. Uplatňuje se multidisciplinární přístup vyžadující spolupráci mezi kliniky a radiology, chirurgy a patology. IPP se třídí do skupin podle různých hledisek: na IPP ze známých příčin, idiopatické intersticiální pneumonie, granulomatózní procesy a jiné [1]. IPP mohou být primární (sarkoidóza) nebo sekundární spojené s jinými onemocněními a setkáváme se s nimi také u jedinců s primárními či sekundárními poruchami imunitního systému.
Etiopatogeneze IPP není sice dosud objasněna, nicméně se předpokládá, že se na jejich rozvoji významným způsobem podílejí mechanizmy imunitního systému [1]. V roce 1973 byla poprvé popsána přítomnost folikulárních agregátů v bronchiální stěně králíků, která byla nazvána „s bronchy asociovaná lymfatická tkáň“ (Bronchus Associated Lymphoid Tissue – BALT), která byla řazena původně mezi sekundární lymfatické orgány (SLO) podobně jako lymfatické uzliny, slezina nebo s gastrointestinálním traktem asociovaná lymfatická tkáň (GALT) včetně Peyerových plátů ve střevě [2,3]. SLO vytvářejí optimální prostředí pro rozvoj imunitní odpovědi a dochází v nich ke kontaktu mezi T-lymfocyty a B-lymfocyty a antigen prezentujícími dendritickými buňkami. Později bylo zjištěno, že se BALT značně odlišuje od SLO charakteru Peyerových plátů ve střevě [4], a proto byla následně zařazena mezi tzv. terciární lymfatické orgány (TLO). Ty představují vysoce organizované imunologické struktury, které vznikají v místě infekce, autoimunitních procesů, chronické imunitní stimulace nebo nádorového bujení [5]. Podílejí se na rozvoji a udržování imunitní odpovědi zejména v případě přetrvávající extravazace leukocytů a přítomnosti dostatečného množství antigenů, tak jak je tomu např. v místě chronické infekce, autoimunitních procesů nebo rejekce transplantátu [5]. BALT byla nalezena u IPP v rámci CVID, systémových onemocnění pojiva, idiopatické plicní fibrózy, hypersenzitivní pneumonie, a podílí se tak pravděpodobně na rozvoji zánětu a plicní fibrózy [6,7].
Většina imunodeficitních onemocnění není dnes chápána pouze jako porucha funkce imunitního systému ve smyslu prostého snížení jeho funkce, ale komplexněji též v rámci přítomné dysregulace imunitního systému. S tím souvisí také to, že s celou řadou imunodeficitních onemocnění se pojí vyšší výskyt autoimunitních procesů. Prakticky všechny imunodeficientní stavy mohou mít i respirační symptomatologii. Navíc u některých z nich může dojít přímo k rozvoji intersticiálního plicního procesu tak jako v následujících kazuistikách.
Kazuistiky
Kazuistika 1: Běžná variabilní imunodeficience (CVID)
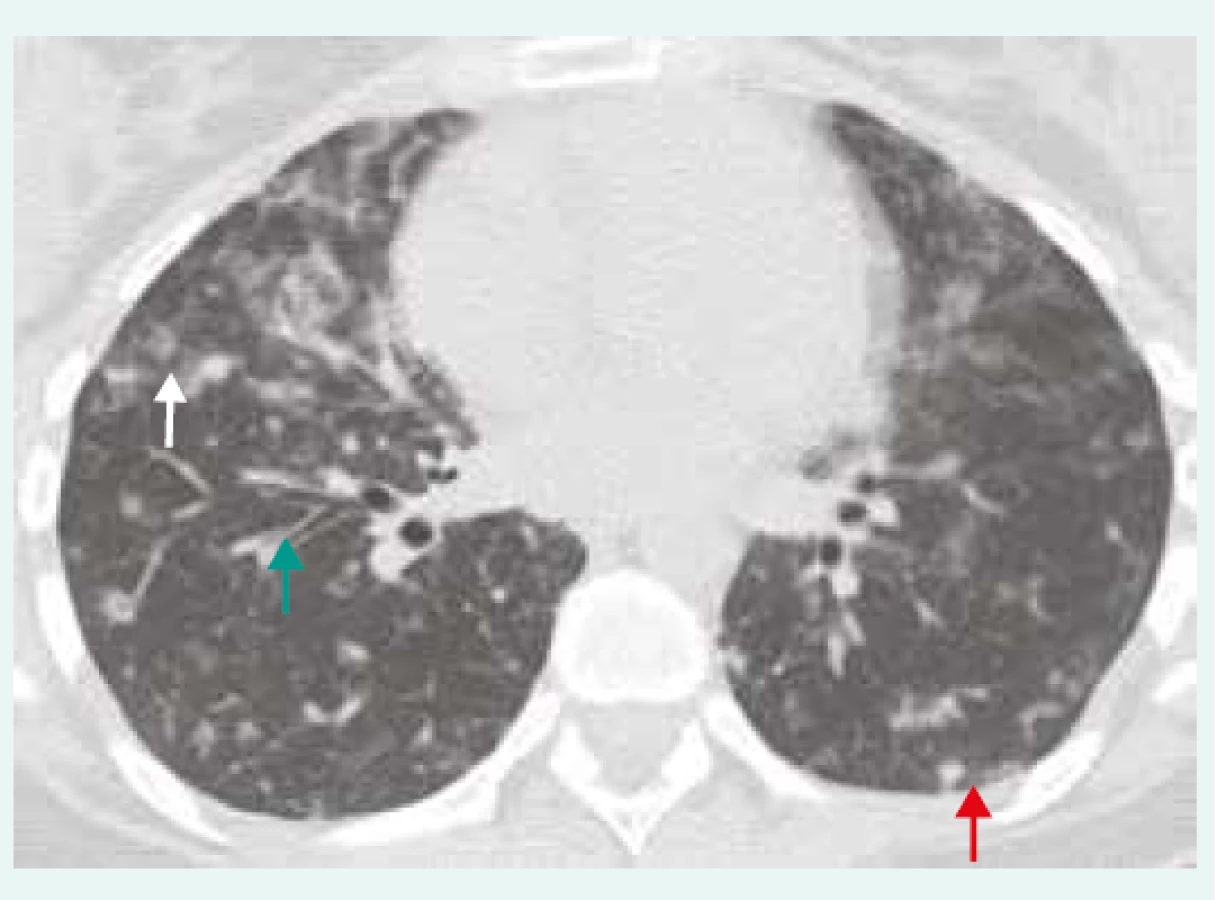
22letá pacientka (nekuřačka) byla přijata na plicní kliniku pro oboustrannou plicní infiltraci s podezřením na pneumonii. Její osobní anamnéza byla z pohledu sledovaných onemocnění bez pozoruhodností. Udávala však, že od 16 let trpěla častějšími infekcemi horních dýchacích cest. Nicméně otitidu, sinusitidu, meningitidu nebo pneumonii v minulosti nikdy neprodělala. Pracovala v obchodě s textilem. Kromě hormonální antikoncepce neužívala pravidelně žádné léky. Biochemické vyšetření a krevní obraz byly v normě. Po přeléčení pneumonie antibiotickou léčbou nedošlo k regresi radiologického nálezu. Na HRCT hrudníku byla stále přítomna oboustranná nehomogenní plicní infiltrace s hraniční hilovou a mediastinální lymfadenopatií (obr. 1). Bronchoskopie s bronchoalveolární laváží (BAL) ukázala lymfocytární alveolitidu (60 % lymfocytů v BAL; norma < 15 %), mikrobiologie byla negativní. Pro podezření na intersticiální plicní proces bylo u pacientky indikováno provedení plicní biopsie. Histopatologicky (chirurgická plicní biopsie) byl prokázán intersticiální plicní proces s bronchiolární akcentací lymfocytárních infiltrátů s mírnou plicní fibrózou a přítomností granulomů bez nekróz. V diferenciální diagnostice se zvažovala hypersenzitivní pneumonie, systémové onemocnění pojiva nebo polékové postižení. Vyvolávající příčina plicního postižení zjištěna nebyla. Pacientce byla nasazena imunosupresivní léčba systémovými kortikoidy (metylprednisolon v dávce 24 mg denně), při které došlo k téměř kompletní radiologické regresi. Snižování dávek kortikoidů však vedlo k relapsu onemocnění. Při doplnění základního imunologického laboratorního vyšetření byla prokázána hypogamaglobulinemie ve všech imunoglobulinových třídách [IgG 1,51 g/l (referenční rozmezí 7,51–15,6 g/l); IgM 0,10 g/l (0,4–2,3 g/l); IgA 0,08 g/l (0,7–4,0 g/l); IgE < 1 U/l (0–90 U/l)]. Proto bylo u pacientky vysloveno podezření na diagnózu běžné variabilní imunodeficience (CVID). U pacientky byly vyloučeny sekundární ztráty imunoglobulinů a zjištěna snížená koncentrace specifických protilátek proti proteinovým i polysacharidovým antigenům (protilátky ve třídě IgG proti tetanickému toxoidu 0,037 IU/ml – protektivní hodnoty > 0,12 IU/ml), proti pneumokokovým pouzderným polysacharidům < 3,3 mg/l (protektivní hodnoty > 15,4 mg/l) a proti Haemophilus influenzae typu b 0,011 mg/l (> 0,09 mg/l). U pacientky byla zahájena intravenózní substituční imunoglobulinová léčba v dávce 25 g měsíčně. Histopatologický plicní nález byl na podkladě výše uvedených skutečností přehodnocen a změněn na diagnózu granulomatózního lymfatického intersticiálního onemocnění plic (Granulomatous and Lymphocytic Interstitial Lung Disease – GLILD), které patří do obrazu možného intersticiálního postižení plicní tkáně u pacientů s CVID. V terapii IPP bude i nadále pokračováno s imunosupresivní léčbou kortikoidy v udržovací dávce mezi 5–15 mg/kg/den. Vzhledem k rozsahu IPP, funkčnímu postižení, nutnosti vyšších dávek kortikoidů (> 15 mg/kg/den) a netoleranci azatioprinu, je plánována kombinovaná léčba s antiCD20-monoklonální protilátkou.
Hraniční lymfadenopatie mediastinální a hilová,
nepravidelné centrilobulární opacity (bílá šipka),
nodularity zejména subpleurálně (červená
šipka), bronchiektázie/bronchioloektázie
(zelená šipka). IPP byla histologicky verifikována
jako GLILD/CVID. Z archivu autorky
Běžná variabilní imunodeficience (Common Variable Immunodeficiency – CVID) patří mezi nejčastější symptomatické protilátkové imunodeficience. Z této heterogenní skupiny onemocnění bylo do dnešní doby vyčleněno několik nových nozologických jednotek, protože byly identifikovány genetické mutace vedoucí ke klinickému a laboratornímu obrazu CVID [8]. Toto onemocnění je charakterizováno snížením koncentrace celkových imunoglobulinů ve třídě IgG a IgA anebo IgM; pacienti mají také porušenou protilátkovou odpověď po antigenní stimulaci a jsou vyloučeny ostatní příčiny hypogamaglobulinemie. V roce 2016 byla publikována nová diagnostická kritéria tohoto onemocnění (tab. 1) [9]. V popředí klinických příznaků pacientů s diagnózou CVID stojí opakované závažné infekce zejména ORL oblasti a dýchacích cest (otitidy, sinusitidy, pneumonie). Nejčastějším chronickým plicním nálezem jsou bronchiektazie jako výsledek opakujících se plicních zánětů. Do obrazu plicního postižení patří u těchto pacientů i kombinace granulomatózního plicního postižení v kombinaci s lymfoproliferací (GLILD). Výrazem dysregulace funkce imunitního systému těchto pacientů je vyšší frekvence výskytu autoimunitních onemocnění (zejména hematologických, ale i jiných), lymfoproliferativních poruch (generalizovaná lymfadenopatie, splenomegalie), ale také zvýšená frekvence maligních onemocnění zejména gastrointestinálního a lymfatického systému (tab. 2) [10]. Základem léčby těchto pacientů je substituční imunoglobulinová léčba, která je aplikována buď intravenózně nebo subkutánně, a dále symptomatická léčba infekcí a dalších přidružených komplikací tohoto onemocnění [11].
![Diagnostická ICON kritéria stanovení diagnózy běžné variabilní imunodeficience (CVID). Upraveno podle [9]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/0c888f5e0f3c4f92eb2d2868faf78fe4.png)

Kazuistika 2: Deficience cytotoxického T-lymfocytárního antigenu 4 (CTLA4)
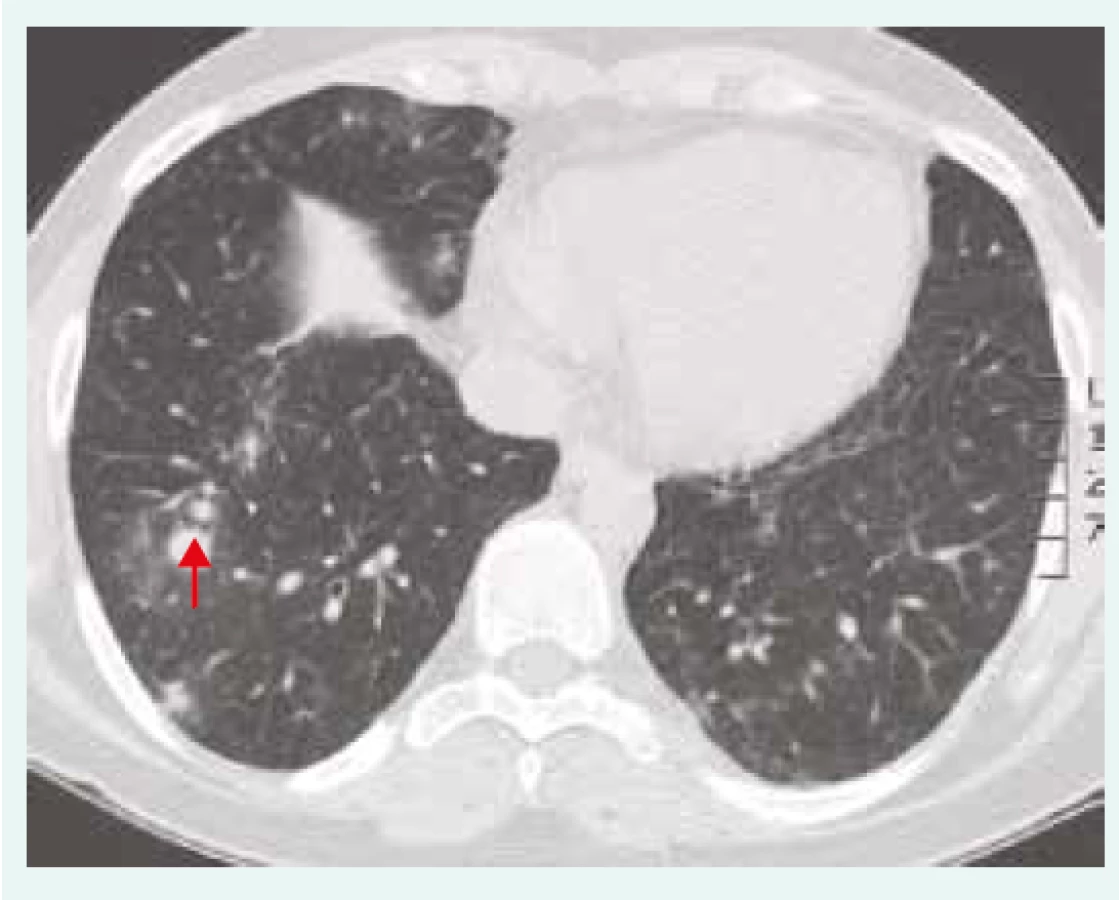
25letá pacientka (nekuřačka) dlouhodobě sledovaná pro juvenilní revmatoidní artritidu, opakované iridocyklitidy, kortikodependentní ložiskové postižení mozku manifestující se epileptickými záchvaty typu grand mal, neuropatii a zánětlivé střevní onemocnění, byla odeslána pro dušnost a podezření na intersticiální plicní proces dle zobrazovacích metod. Na HRCT hrudníku byla patrná nodulární plicní infiltrace s predilekcí vpravo (obr. 2). Bronchoskopicky z bronchoalveolární laváže byla potvrzena lymfocytární alveolitida a transbronchiální plicní biopsie prokázala lymfocytární celulizaci s mnohojadernými pěnitými buňkami. Nemocná užívala vzhledem ke své diagnóze juvenilní revmatoidní artritidy imunosupresivní léčbu (kortikoidy a cyklosporin). Plicní nález byl uzavřen jako intersticiální plicní proces v rámci zatím neklasifikovaného systémového onemocnění s multiorgánovým postižením (plíce, klouby, CNS, oči, gastrointestinální trakt). Při imunologickém laboratorním vyšetření byla zjištěna snížená koncentrace imunoglobulinů (IgG 2,71 g/l, IgM 0,22 g/l, IgA 0,08 g/l) a nemocné byla pro nespecifický imunodeficientní syndrom nejasné etiologie podávána intravenózní substituční imunoglobulinová terapie (IntraVenous ImmunoGlobulin – IVIG). V průběhu let došlo i přes zavedenou léčbu (IVIG, metotrexát, azatioprin, cyklosporin, leflunomid) k progresi intersticiálního plicního postižení, zhoršení neurologického nálezu a zánětlivého střevního onemocnění. Histologické vyšetření vzorků střevní tkáně nepotvrdilo diagnózu idiopatického zánětlivého střevního onemocnění. U pacientky bylo následně provedeno genetické vyšetření, které prokázalo deficit cytotoxického T-lymfocytárního antigenu 4 (CTLA4). Tato mutace by mohla být kauzální a vést k výše uvedeným autoimunitním projevům. Pacientka je indikována k terapii abataceptem (molekula CTLA4 fúzovaná s Fc částí imunoglobulinu IgG), ovlivňujícím činnost T-lymfocytů a působícím imunosupresivně.
Cytotoxický T-lymfocytární antigen 4 (CTLA4) je zásadním velmi významným regulátorem imunitní odpovědi. Je konstitutivně exprimován na povrchu regulačních T-lymfocytů (Treg), přičemž na ostatních T-lymfocytech se objevuje až po jejich stimulaci [12]. CTLA4 hraje zásadní roli v udržování imunitní homeostázy, protože představuje kontrolní bod (checkpoint) sloužící k regulaci imunitní odpovědi. CTLA4 jako inhibiční receptor na povrchu T-lymfocytů soutěží s aktivačním receptorem CD28 o vazbu 2 společných ligandů CD80 a CD86 na povrchu antigen prezentujících buněk, přičemž afinita a avidita vazby CTLA4 na CD80 a CD86 je v porovnání s CD28 vyšší [13]. Po vazbě ligandů CD80 a CD86 na receptor CTLA4 dochází k jejich internalizaci do nitra buňky, čímž dochází ke snížení aktivace T-lymfocytů zprostředkované antigen prezentujícími buňkami. Pacienti s CTLA4 deficiencí tedy vykazují klinické příznaky související s dysregulací imunitního systému. Mezi základní příznaky patří náchylnost k závažným respiračním infekcím, průjmy, rozvoj nejrůznějších autoimunitních onemocnění a přítomnost lymfocytárních infiltrátů a tvorby granulomů (tab. 2). Kromě náchylnosti k infekčním komplikacím pacienti trpí autoimunitním onemocněním (tyreoiditidy, cytopenie). Podobně jako u CVID i v tomto případě nacházíme IPP typu GLILD s plicními noduly a lymfocytárními infiltráty, splenomegalií a lymfadenopatií. Léčba těchto pacientů spočívá v řešení imunodeficience i dysregulace imunitního systému. Imunodeficience je zmírňována podáváním substituční imunoglobulinové léčby, někdy též s nutností antibiotické profylaxe. Pro léčbu dysregulace imunitního systému však neexistují žádné doporučené postupy. Nejčastěji se používají kortikoidy, jisté zkušenosti jsou i s používáním jiných imunosupresivních léčiv (rituximab, mykofenolát mofetil, cyklosporin A nebo anti-TNF léčba či vedoluzimab) [14,15]. Možností léčby těchto pacientů je také podávání abataceptu (CTLA4 fúzního proteinu, který je registrován pro léčbu revmatoidní artritidy), který by mohl sloužit jako substituce CTLA4 při jeho nedostatku u těchto pacientů [16]. U těžkých případů je ke zvážení též provedení transplantace hematopoetických krevních buněk [15].
Kazuistika 3: Deficit buněčné imunity (lymfocytopenie; podezření na idiopatickou CD4+ lymfocytopenii)
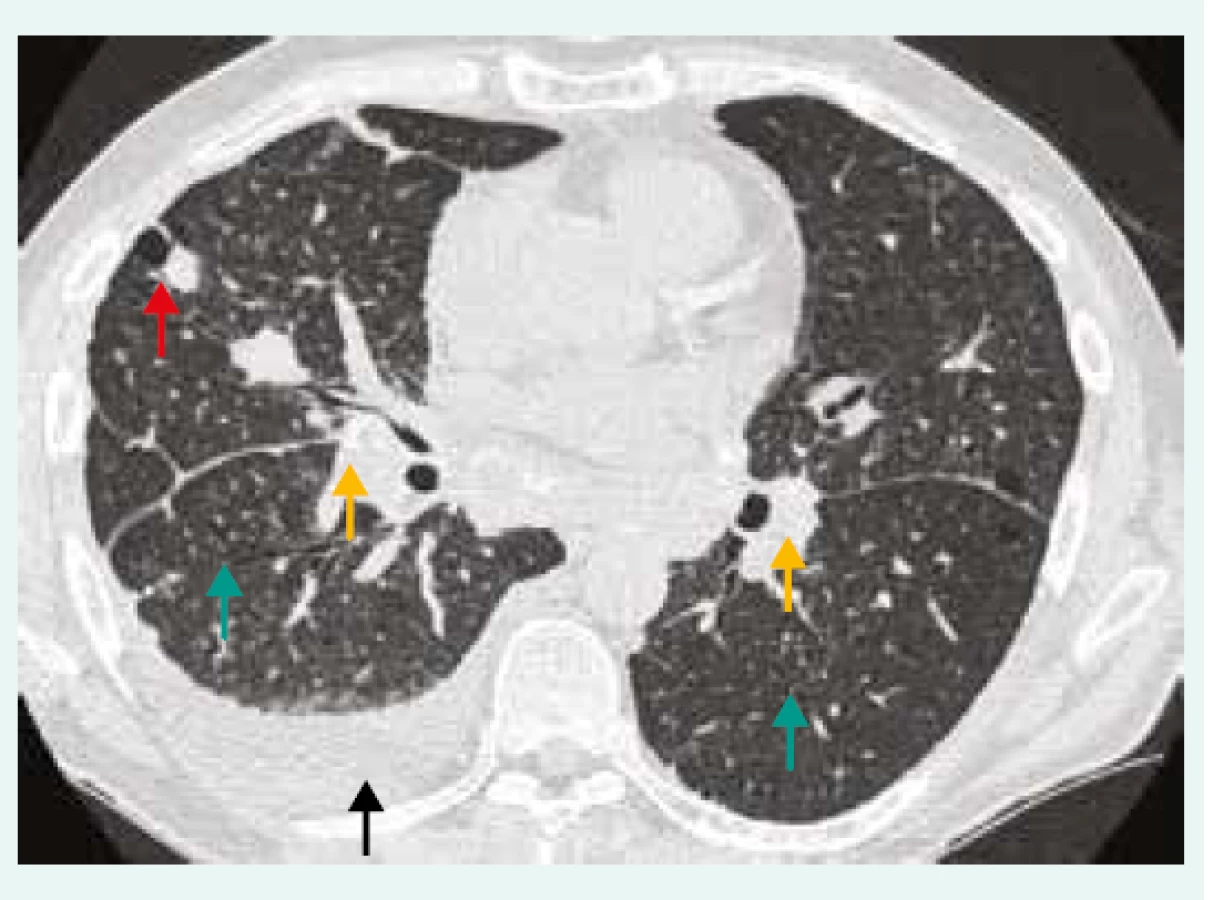
57letý pacient (bývalý kuřák) byl přijat na plicní kliniku pro rozsáhlou oboustrannou plicní infiltraci s pleurálním výpotkem charakteru exsudátu s mediastinální lymfadenopatií (obr. 3), teplotami a zánětlivou aktivitou (CRP 90 mg/l; referenční rozmezí 0–10 mg/l). Chronicky žádnou medikaci neužíval. Pracoval jako výtvarník a sochař (práce s pískovcem). V předchorobí netrpěl na opakované infekce. V diferenciálním rozpočtu leukocytů byl patrný pokles absolutního počtu lymfocytů (0,21 × 109/l; referenční rozmezí 0,8–4,0 × 109/l). Tato lymfocytopenie přetrvávala po celou dobu našeho sledování, a to i po přeléčení pneumonie s poklesem zánětlivých parametrů. Stav byl uzavřen jako oboustranná pneumonie, ale vzhledem k pouze částečné regresi plicního nálezu po antibiotické léčbě a podezření na IPP proces včetně pneumokoniózy byla s odstupem provedena plicní biopsie. Histologicky (chirurgická plicní biopsie) byla prokázána chronická forma silikózy s nevratnými fibrotickými změnami (vazivovatění jemné plicní tkáně). Při laboratorním vyšetření se zaměřením na imunologické parametry byly vyšetřeny základní buněčné lymfocytární subpopulace s nálezem sníženého absolutního i relativního počtu pomocných CD4+ Th lymfocytů: 0,01 × 109/l (referenční mez 0,30–1,40 × 109/l), 10 % (referenční mez 28–57 %). Koncentrace celkových imunoglobulinů byla v normě, vyšetřené autoprotilátky (ANA, anti-ENA, RF, anti-CCP, ANCA) byly negativní. Při pátrání po etiologii těžké CD4+ lymfocytopenie jsme vyloučili HIV infekci. V diferenciálním rozpočtu leukocytů odebraném praktickým lékařem před 2 a 7 lety byl též přítomen pokles absolutního počtu lymfocytů (0,10 × 109/l a 0,15 × 109/l). Další imunologické vyšetření již pacient nepodstoupil, protože zemřel měsíc po chirurgické plicní biopsii na virovou pneumonii (virus influenzae typu A). Při pitvě se nezjistila jiná patologie (autoimunita nebo malignita) než silikóza a oboustranná pneumonie. I když pacient trpěl několik let opakovaně prokázanou lymfopenií v absolutních počtech, diagnóza idiopatické CD4+ lymfocytopenie u něj nebyla jednoznačně prokázána, protože pro její stanovení je zapotřebí potvrdit pokles CD4+ více než jednou (tab. 3). Nicméně jako kazuistiku jej zde uvádíme, abychom upozornili i na méně běžné imunodeficity s plicními projevy.
Z archivu autorky
![Diagnostická kritéria idiopatické CD4
lymfocytopenie (ICL). Upraveno podle [17]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/149dcc1993a3421677098f41ef7feefd.png)
Idiopatická CD4+ lymfocytopenie (ICL) je vzácný heterogenní syndrom, popsaný poprvé v roce 1992 [17]. Příčina ICL není známa. ICL je charakterizována perzistujícím snížením počtu CD4+ T-lymfocytů. Diagnóza je stanovena per exclusionem po vyloučení přítomnosti HIV infekce a všech ostatních příčin sekundární lymfocytopenie (tab. 3) [18]. Celosvětově bylo popsáno méně než 300 případů tohoto onemocnění, ale skutečná prevalence, stejně tak jako patogeneze tohoto stavu zůstávají nezjištěny [19]. I když jsou někteří pacienti asymptomatičtí, většina pacientů trpí oportunními infekcemi připomínajícími infekce u HIV pozitivních pacientů a také mají vyšší riziko rozvoje zejména hematologických malignit [20]. Mezi další klinické příznaky patří rozvoj autoimunitních nebo neurologických onemocnění (tab. 2). Z autoimunitních onemocnění postihujících intersticium je to např. systémový lupus erythematodes nebo Sjögrenův syndrom. Kromě profylaktické antibiotické léčby neexistuje žádná standardní doporučená léčba a u symptomatických pacientů je doporučováno při prevenci oportunních infekcí postupovat podobně jako u HIV pacientů. Profylaktické podání antibiotika (nejčastěji sulfametoxazol/trimetoprim) je indikováno, pokud počet CD4+ T-lymfocytů poklesne pod 200 buněk/mm3 [19].
Patogeneze IPP
Přesný mechanizmus a charakteristika vyvolávajících příčin rozvoje řady IPP nejsou známy. Nicméně základním obecným imunopatologickým podkladem většiny IPP je zánět plicní tkáně, který je i základním profibrotickým procesem. Mechanizmus se u jednotlivých IPP liší. Všechny imunokompetentní buňky spolu se svými cytokiny, které se podílejí na vzniku a udržování zánětu (makrofágy, lymfocyty, polymorfonukleární leukocyty), mají potenciál vyvolat proces plicní fibrotické přestavby. Tradiční představa patogeneze většiny IPP je, že známé či neznámé agens vyvolá poškození pneumocytů I. typu nebo endotelií, a na toto poškození navazuje zánětlivá a imunitní odpověď organizmu (rozvoj alveolitidy s migrací alveolárních makrofágů, eozinofilních a neutrofilních leukocytů, lymfocytů, žírných buněk), která postupně při přetrvávající přítomnosti vyvolávajícího činitele vede k poškození alveolokapilární jednotky, k aktivaci fibroblastů spojené se zvýšenou produkcí extracelulární matrix a postupnému ukládání kolagenu do plicního intersticia [21].
U pacientů s primárními imunodeficiencemi vidíme často lymfoproliferativní onemocnění, která zasahují také plicní tkáň. Mezi nejčastější formy tohoto postižení patří folikulární bronchiolitida, nodulární lymfoidní hyperplazie, lymfocytární intersticiální pneumonie (LIP) a GLILD. Dále se u nich vyskytují nenekrotizující granulomatózní procesy, organizující se pneumonie a nespecifická intersticiální pneumonie [22–24]. LIP se částečně prolíná s pojmem folikulární bronchiolitida. Obě tyto jednotky zřejmě tvoří pouze různý obraz hyperplazie lymfoidní tkáně v plicním parenchymu. Vzhledem k riziku rozvoje klonality existuje těsná souvislost mezi hyperplazií lymfoidní tkáně a přechodem do plicního lymfomu. Proliferace reaktivních intrapulmonálních lymfatických folikulů může být podporována porušenou B-buněčnou homeostázou, zatímco tvorba granulomů se zdá být způsobena spíše T-lymfocyty. Nicméně někteří autoři uvádějí, že se v patogenezi LIP uplatňují také autoreaktivní T-lymfocyty [23,25]. Tomu odpovídá i to, že většina imunodeficiencí je asociována s porušením T-buněčné nebo B-buněčné homeostázy nebo porušením signalizace přes antigenní a cytokinové receptory [26].
Patofyziologické mechanizmy IPP jsou asi nejvíce prozkoumány u diagnózy CVID. Ektopická lymfoidní tkáň tvořící zárodečná centra je zodpovědná za terciární lymfoneogenezi v plicní tkáni pacientů s CVID. Lymfoidní hyperplazie se u těchto pacientů projevuje také perzistující lymfadenopatií a splenomegalií.
Četnější výskyt granulomů, autoimunitních projevů a splenomegalie byl nalezen u CVID pacientů s významným snížením počtu izotypově přesmyknutých paměťových B-lymfocytů [27,28]. Také u pacientů s CVID a expanzí CD21low B-lymfocytů byl zjištěn vyšší výskyt autoimunitní cytopenie a splenomegalie [29]. Bylo prokázáno, že se zvyšující se koncentrací celkových IgM imunoglobulinů v séru stoupá i riziko rozvoje polyklonální lymfocytární infiltrace nebo lymfoidní malignity (s každým 1 g/l IgM o 16 %, respektive 31 %). Navíc se snižujícím se relativním počtem CD8+ Tc-lymfocytů stoupá riziko rozvoje autoimunitních fenoménů [22].
Imunitní dysregulace nezávislá na přítomnosti infekce má pravděpodobně vliv na patogenezi lymfoproliferativních procesů a současně se podílí také na rozvoji IPP a autoimunitních onemocnění. Bylo prokázáno, že periferní paměťové a lymfatické folikulární CD4+ Th-lymfocyty pacientů s CVID tvoří ve srovnání s kontrolní populací více IFNγ [30]. Také u pacientů s CVID s vyšší mírou infekčních komplikací, v porovnání s ostatními pacienty s CVID, s X vázanou agamaglobulinemií (XLA) a kontrolní skupinou, byl zjištěn významně vyšší výskyt genů odpovídajících na stimulaci prostřednictvím interferonů [31]. U těchto pacientů byla též popsána porušená tvorba zárodečných center s nahromaděním T-box transkripčního faktoru (T-bet)+ B-lymfocytů v lymfatických uzlinách a T-bet+CD21low B-lymfocytů v periferní krvi [30], významné snížení absolutního počtu lymfocytů, cirkulujících B-lymfocytů a izotypově přesmyknutých paměťových B-lymfocytů [31]. Chronická upregulace interferonových signalizačních drah se mimo jiné vyskytuje také u autoimunitních onemocnění v důsledku aktivace Toll-like receptorů (TLR) a dalších zatím nepopsaných cytoplazmatických senzorů. Zdá se, že významná porucha adaptivní imunitní odpovědi u pacientů s CVID vede k chronické aktivaci interferonových mechanizmů vrozené imunity v odpovědi na antigeny prostředí [31]. Zvýšená tvorba IFNγ u pacientů s CVID a současně přítomnými zánětlivými a autoimunitními komplikacemi včetně IPP také koreluje s přítomností vysoce prozánětlivé populace přirozených lymfoidních buněk (ILC) v cirkulaci i slizničních tkáních [32]. U pacientů s CVID se současně přítomnými granulomy a autoimunitními onemocněními byl popsán zvýšený počet folikulárních pomocných T-lymfocytů (TFH) ve srovnání s CVID pacienty bez granulomatózních nebo autoimunitních projevů [33]. Folikulární pomocné T-lymfocyty (TFH) hrají ústřední roli v tvorbě zárodečných center, což je zásadní pro správný průběh humorální imunitní odpovědi. Funkce těchto buněk se zdá být u pacientů s CVID neporušená, nicméně vzhledem k jejich zvýšenému počtu u pacientů s granulomatózními a autoimunitními komplikacemi by se tyto buňky mohly nějakým způsobem podílet na patogenezi těchto komplikací [33]. Kromě výše popsaných faktorů se v rámci rozvoje IPP zvažuje také vliv infekčních příčin. Jedná se zejména o viry HHV8 a EBV, které vedou k zvýšené proliferaci lymfocytů, a tím k lymfocytární infiltraci orgánů [34,35].
Příčina rozvoje IPP a autoimunitních onemocnění není známá ani u pacientů s CTLA4 deficiencí nebo ICL. Na rozvoji IPP u pacientů s CTLA4 haploinsuficiencí se může podílet dysregulace regulačních T-lymfocytů a hyperaktivace efektorových T-lymfocytů [36]. U pacientů s ICL byl popsán relativně vyšší počet aktivovaných CD25+CD4+ a HLA-DR+CD4+ Th-lymfocytů. Tento hyperaktivovaný stav může být důsledkem CD4+ T-lymfocytenie (inverzní korelace mezi HLA-DR+CD4+ T-lymfocytů a CD4+ T-lymfocytů) a může být zodpovědný za autoimunitní manifestaci [37].
Výskyt IPP a jeho léčba u popsaných primárních imunodeficiencí
IPP jsou obvykle přítomny již v době diagnózy CVID [23]. CVID asociovaná s granulomatózním onemocněním (GD) se označuje jako CVID/GD [22,38,39]. Kombinace plicních granulomů a lymfoproliferace se pak nazývá granulomatózní lymfocytární intersticiální plicní onemocnění (Granulomatous Lymphocytic Interstitial Lung Disease – GLILD) [40–43]. Přítomnost GLILD u pacientů s CVID je spojena s významnou morbiditou [44,45]. Pojem lymfoproliferace zahrnuje lymfocytární intersticiální pneumonii (LIP), folikulární bronchiolitidu a lymfoidní hyperplazii. GLILD je často doprovázeno splenomegalií a difuzní lymfadenopatií se zvýšeným rizikem rozvoje lymfomu [42]. Granulomatózní zánět u sarkoidózy (systémové onemocnění spadající do IPP a postihující zejména respirační trakt) je podobný jako u GD/CVID. Nicméně určité klinické, radiologické, laboratorní a histologické odlišnosti pozorovat možné je [46,47]. Na rozdíl od sarkoidózy, u níž může být přítomna polyklonální hypergamaglobulinemie bez průkazu monoklonální gamapatie, jsou CVID/GD nebo GLILD provázeny hypogamaglobulinemií, často s progresivním vývojem plicního postižení bez tendence ke spontánní regresi a rezistencí na kortikoidní léčbu. Optimální terapie CVID/GD/GLILD není známa. Chybí totiž kontrolované studie věnující se léčbě CVID a IPP. Substituční imunoglobulinové léčba, která je pacientům pravidelně podávána ve snaze zmírnit dopady nedostatečné tvorby jejich vlastních protilátek, však nezabraňuje progresi IPP [48]. Většina pacientů je nejprve léčena perorálními kortikoidy, přičemž dávky se pohybují v rozmezí od 10 mg do 1–2 mg/kg denně, většina odborníků však užívá dávky 40 mg denně [45]. Pokud nejsou samotné kortikoidy dostatečně účinné, další terapie zahrnuje podávání azatioprinu, mykofenolátu mofetilu nebo rituximabu, samostatně nebo v kombinaci [23,45]. Pacienti s GLILD/CVID byly také úspěšně léčeni monoklonální protilátkou namířenou proti tumor nekrotizujícímu faktoru alfa (infliximabem), cyklosporinem nebo kombinací rituximabu a azatioprinu [49,50]. Bylo zjištěno, že u 6 pacientů s GLILD zlepšila kombinační léčba rituximabu s azatioprinem jak radiografické abnormality, tak pulmonální funkci [50]. Rituximab je chimérickou myší monoklonální protilátkou namířenou proti molekule CD20 na povrchu B-lymfocytů. Ovlivňuje však také funkci T-lymfocytů [51]. Léčba namířená proti B-lymfocytům může u těchto pacientů ovlivnit CVID asociovanou lymfocytární hyperplazii [24]. Hodnocení terapeutické odpovědi pacientů s IPP a zmíněnými primárními imunodeficiencemi sestává z opakovaných plicních funkčních vyšetření včetně plicní difuze a zobrazovacích metod (HRCT hrudníku nebo PET/CT).
Základní laboratorní vyšetření v rámci diferenciální diagnostiky u pacientů s IPP
Diagnóza IPP je sice definitivně stanovena až na základě korelace klinických, radiologických a histopatologických nálezů, nicméně laboratorní vyšetření tvoří nedílnou součást vyšetřovacího algoritmu. Kromě krevního obrazu s diferenciálním rozpočtem leukocytů, základního biochemického vyšetření a stanovení koncentrace C-reaktivního proteinu, který bývá zvýšen i u některých neinfekčních onemocnění včetně vaskulitid, má své nezastupitelné místo také vyšetření imunologické (tab. 4). Je důležité nejen pro diagnostiku autoimunitních onemocnění (ve vztahu k plicnímu postižení zejména vaskulitid nebo systémových onemocnění pojiva), ale i k diferenciální diagnostice IPP u primárních či sekundárních poruch imunitního systému. Na významnou poruchu funkce imunitního systému nás mohou anamnesticky upozornit opakující se infekce horních i dolních dýchacích cest (sinusitidy, otitidy, pneumonie), opakované meningitidy či další prodělané závažnější infekce.

V rámci imunologického vyšetření hodnotíme základní parametry humorální a buněčné imunity. Sérologické vyšetření se opírá o stanovení koncentrace jednotlivých imunoglobulinových tříd (IgG, IgA, IgM a IgE). Při rozvoji fibrotického postižení plic je nutno pomýšlet i na diagnózu IgG4 asociovaného onemocnění, k jehož diagnostice může dopomoci stanovení koncentrace podtřídy IgG4. Stanovení koncentrace celkových IgE imunoglobulinů a specifických IgE protilátek proti Aspergillus fumigatus se provádí při podezření na alergickou bronchopulmonální aspergilózu, která se projevuje přítomností oboustranných plicních infiltrátů. Určení specifických IgG protilátek proti příčinným antigenům pomůže při diagnostice hypersenzitivní pneumonie. K vyloučení monoklonální gamapatie je nutné doplnit imunoelektroforetické vyšetření séra. Systémová onemocnění pojiva doprovází polyklonální aktivace B-lymfocytů společně s hypergamaglobulinemií. Při podezření na systémové autoimunitní onemocnění pojiva se stanovují koncentrace autoprotilátek (tab. 5) [52]. Využívá se i detekce koncentrace složek komplementu (zejména C3 a C4) a cirkulujících imunokomplexů pro potvrzení konsumpce komplementu při jeho aktivaci v rámci autoimunitního onemocnění a imunokomplexových reakcí.

Buněčné imunologické vyšetření se opírá o stanovení absolutního a relativního zastoupení lymfocytárních subpopulací pomocí průtokové cytometrie buď z periferní krve nebo bronchoalveolární laváže (BAL). Jedná se o stanovení celkových CD3+ T-lymfocytů, CD3+CD4+ pomocných (Th) a CD3+CD8+ cytotoxických (Tc) T-lymfocytů, CD19+ B-lymfocytů a CD16+56+ přirozených zabíječů NK buněk (tab. 4). U sarkoidózy obvykle pozorujeme poměr CD4+/CD8+ v bronchoalveolární tekutině > 3,5, hodnoty poměru < 1,0 mohou být přítomny u hypersenzitivní pneumonie. Zvýšený počet Langerhansových buněk (CD1+ ≥ 5 %) nacházíme u kuřáků s plicní formou granulomatózy z Langerhansových buněk. Při poklesu CD4+ T-lymfocytů < 200 buněk/µl je pravděpodobnost infekce oportunními patogeny vyšší a je doporučována antibiotická profylaxe Pneumocystis jirovecii [21,53].
Závěr
Základní imunologické laboratorní vyšetření humorální a buněčné imunity je nezbytnou součástí diferenciálně diagnostického algoritmu intersticiálního plicního postižení. Na možný rozvoj intersticiálního plicního postižení musíme myslet nejen u pacientů s primárními imunodeficiencemi, ale na možnou přítomnost dosud neodhalené primární poruchy funkce imunitního systému musíme myslet i u pacientů s nově zjištěným intersticiálním plicním postižením. Některé poruchy se totiž, jak bylo ukázáno ve výše uvedených kazuistikách, manifestují až v dospělém věku.
Doručeno do redakce 22. 2. 2019
Přijato po recenzi 28. 4. 2019
MUDr. Martina Doubková, Ph.D.
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno, pracoviště Bohunice
Sources
- [American Thoracic Society; European Respiratory Society]. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002; 165(2): 277–304. Dostupné z DOI: <http://dx.doi.org/10.1164/ajrccm.165.2.ats01>.
- Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. I. Morphologic characteristics. Lab Invest 1973; 28(6): 686–692.
- Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. II. Functional characteristics. Lab Invest 1973; 28(6): 693–698.
- Pabst R, Tschernig T. Bronchus-associated lymphoid tissue: an entry site for antigens for successful mucosal vaccinations? Am J Respir Cell Mol Biol 2010; 43(2): 137–141. Dostupné z DOI: <http://dx.doi.org/10.1165/rcmb.2010–0152RT>.
- Neyt K, Perros F, Geurtsvan Kessel CH et al. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol 2012; 33(6): 297–305. Dostupné z DOI: <http://dx.doi.org/10.1016/j.it.2012.04.006>.
- Rangel-Moreno J, Hartson L, Navarro C et al. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006; 116(12): 3183–3194. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI28756>.
- Jordan M, Haczku A. Autoreactive bronchus-associated lymphoid tissue in interstitial lung disease: friend or foe? Am J Respir Cell Mol Biol 2013; 48(4): 397–398.
- Saikia B, Gupta S. Common Variable Immunodeficiency. Indian J Pediatr 2016; 83 : 338–344. Dostupné z DOI: <http://dx.doi.org/10.1007/s12098–016–2038-x>.
- Bonilla FA, Barlan I, Chapel H et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract 2016; 4(1): 38–59. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaip.2015.07.025>.
- Gathmann B, Mahlaoui N, Gérard L et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol 2014; 134(1): 116–126. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2013.12.1077>.
- Abbott JK, Gelfand EW. Common Variable Immunodeficiency: Diagnosis, Management, and Treatment. Immunol Allergy Clin North Am 2015; 35(4): 637–658. Dostupné z DOI: <http://dx.doi.org/10.1016/j.iac.2015.07.009>.
- Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol 2017; 49 : 14–19. Dostupné z DOI: <http://dx.doi.org/10.1016/j.coi.2017.07.014>.
- Rowshanravan B, Halliday N, Sansom D. CTLA-4: a moving target in immunotherapy. Blood 2018; 131(1): 58–67. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2017–06–741033>.
- Králíčková P, Kubcová Š, Kočová E et al. Successful rituximab treatment of granulomatous/lymphocytic interstitial lung disease in common variable immunodeficiency. Epidemiol Mikrobiol Imunol 2018; 67(3): 142–148.
- Verma N, Burns SO, Walker LSK et al. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol 2017; 190(1): 1–7. Dostupné z DOI: <http://dx.doi.org/10.1111/cei.12997>.
- Chitale S, Moots R. Abatacept: the first T lymphocyte co-stimulation modulator, for the treatment of rheumatoid arthritis. Expert Opin Biol Ther 2008; 8(1): 115–122. Dostupné z DOI: <http://dx.doi.org/10.1517/14712598.8.1.115>.
- [Centers for Disease Control (CDC)]. Unexplained CD4+ T-lymphocyte depletion in persons without evident HIV infection – United States. MMWR Morb Mortal Wkly Rep 1992; 41(30): 541–545.
- Yarmohammadi H, Cunningham-Rundles C. Idiopathic CD4 lymphocytopenia: Pathogenesis, etiologies, clinical presentations and treatment strategies. Ann Allergy Asthma Immunol 2017; 119(4): 374–378. Dostupné z DOI: <http://dx.doi.org/10.1016/j.anai.2017.07.021>.
- Asher I, Mahlab-Guri K, Elbirt D et al. Idiopathic CD4 Lymphopenia: Severe CD4 Lymphopenia in the Absence of Human Immunodeficiency Virus Infection. Isr Med Assoc J 2016; 18(10): 627–629.
- Walker UA, Warnatz K. Idiopathic CD4 lymphocytopenia. Curr Opin Rheumatol 2006; 18(4): 389–395. Dostupné z DOI: <http://dx.doi.org/10.1097/01.bor.0000231908.57913.2f>.
- Vašáková M, Polák J, Matěj R. Intersticiální plicní procesy: od etiopatogeneze přes radiologický obraz k histopatologické diagnóze. Maxdorf: Praha 2016. ISBN 978–80–7345–488–3.
- Chapel H, Lucas M, Lee M et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008; 112(2): 277–286. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2007–11–124545>.
- Prasse A, Kayser G, Warnatz K. Common variable immunodeficiency-associated granulomatous and interstitial lung disease. Curr Opin Pulm Med 2013; 19(5): 503–509. Dostupné z DOI: <http://dx.doi.org/10.1097/MCP.0b013e3283642c47>.
- Maglione PJ, Ko HM, Beasley MB et al. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol 2014; 133(2): 535–542. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2013.08.022>.
- Giovannetti A, Pierdominici M, Mazzetta F et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol 2007; 178(6): 3932–3943. Dostupné z DOI: <http://dx.doi.org/10.4049/jimmunol.178.6.3932>.
- Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol 2012; 3 : 210. Dostupné z DOI: <http://dx.doi.org/10.3389/fimmu.2012.00210>.
- Sánchez-Ramón S, Radigan L, Yu JE et al. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol 2008; 128(3): 314–321. Dostupné z DOI: <http://dx.doi.org/10.1016/j.clim.2008.02.013>.
- Wehr C, Kivioja T, Schmitt C et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 2008; 111(1): 77–85. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2007–06–091744>.
- Warnatz K, Wehr C, Dräger R et al. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology 2002; 206(5): 502–513. Dostupné z DOI: <http://dx.doi.org/10.1078/0171–2985–00198>.
- Unger S, Seidl M, van Schouwenburg P et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol 2018; 141(2): 730–740. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2017.04.041>.
- Park J, Munagala I, Xu H et al. Interferon signature in the blood in inflammatory common variable immune deficiency. PLoS One 2013; 8(9): e74893. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.0074893>.
- Cols M, Rahman A, Maglione PJ et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol 2016; 137(4): 1206–1215. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2015.09.013>.
- Coraglia A, Galassi N, Fernández Romero DS et al. Common Variable Immunodeficiency and Circulating TFH. J Immunol Res 2016; 2016 : 4951587. Dostupné z DOI: <http://dx.doi.org/10.1155/2016/4951587>.
- Wheat WH, Cool CD, Morimoto Y et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med 2005; 202(4): 479–484. Dostupné z DOI: <http://dx.doi.org/10.1084/jem.20050381>.
- Andiman WA, Eastman R, Martin K et al. Opportunistic lymphoproliferations associated with Epstein-Barr viral DNA in infants and children with AIDS. Lancet 1985; 2(8469–8470): 1390–1393. Dostupné z DOI: <http://dx.doi.org/10.1016/s0140–6736(85)92557–7>.
- Schussler E, Beasley MB, Maglione PJ. Lung Disease in Primary Antibody Deficiencies. J Allergy Clin Immunol Pract 2016; 4(6): 1039–1052. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaip.2016.08.005>.
- Régent A, Autran B, Carcelain G et al. Idiopathic CD4 lymphocytopenia: clinical and immunologic characteristics and follow-up of 40 patients. Medicine (Baltimore) 2014; 93(2): 61–72. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000000017>.
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999; 92(1): 34–48. Dostupné z DOI: <http://dx.doi.org/10.1006/clim.1999.4725>.
- Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol 2009; 133(2): 198–207. Dostupné z DOI: <http://dx.doi.org/10.1016/j.clim.2009.05.001>.
- Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 2010; 134(2): 97–103. Dostupné z DOI: <http://dx.doi.org/10.1016/j.clim.2009.10.002>.
- Bates CA, Ellison MC, Lynch DA et al. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114(2): 415–421. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2004.05.057>.
- Perez RL, Rivera-Marrero CA, Roman J. Pulmonary granulomatous inflammation: From sarcoidosis to tuberculosis. Semin Respir Infect 2003; 18(1): 23–32. Dostupné z DOI: <http://dx.doi.org/10.1053/srin.2003.50005>.
- Bouvry D, Mouthon L, Brillet PY et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J 2013; 41(1): 115–122. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.00189011>.
- Resnick ES, Moshier EL, Godbold JH et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119(7): 1650–1657. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–09–377945>.
- Boursiquot JN, Gérard L, Malphettes M et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol 2013; 33(1): 84–95. Dostupné z DOI: <http://dx.doi.org/10.1007/s10875–012–9778–9>.
- Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med 2014; 35(3): 330–335. <http://dx.doi.org/10.1055/s-0034–1376862>.
- Doubková M, Moulis M, Skřičková J. Intersticiální plicní procesy a granulomatózy asociované s běžným variabilním imunodeficitem. Vnitř Lék 2015; 61(2): 119–124.
- Quinti I, Soresina A, Spadaro G et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol 2007; 27(3): 308–316. Dostupné z DOI: <http://dx.doi.org/10.1007/s10875–007–9075–1>.
- Hatab AZ, Ballas ZK. Caseating granulomatous disease in common variable immunodeficiency treated with infliximab. J Allergy Clin Immunol 2005; 116(5): 1161–1162. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2005.08.041>.
- Chase NM, Verbsky JW, Hintermeyer MK et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013; 33(1): 30–39. Dostupné z DOI: <http://dx.doi.org/10.1007/s10875–012–9755–3>.
- Stasi R, Del Poeta G, Stipa E et al. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 2007; 110(8): 2924–2930. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2007–02–068999>.
- Doubková M, Pokorná J. Autoprotilátky u systémových onemocnění pojiva a ANCA asociovaných vaskulitid, jejich vztah k intersticiálním plicním procesům a prognóze. Vnitř Lék 2017; 63(2): 98–106.
- Boesecke C, Rockstroh JK, Thoden J. Opportunistic infections: What’s new?. Dtsch Med Wochenschr 2018; 143(24): 1755–1758. Dostupné z DOI: <http://dx.doi.org/10.1055/a-0641–9456>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2019 Issue 11
Most read in this issue
- Funkční onemocnění trávicího traktu a bolest
- Interferon-alfa v léčbě myeloproliferativních onemocnění
- Nedostatek vitaminu D a jeho zdravotní dopady
- Imunodeficience v rámci diferenciální diagnostiky intersticiálních plicních procesů