
Imitátory sepse
Sepsis mimics
Number of identical pathophysiological mechanisms is shared by sepsis and other clinical conditions and diseases. This could lead to their nearly similar clinical phenotype. However, the early discrimination of them is crucial – treatment of particular diseases differs significantly, and the mortality of the vast majority of them is considerable. The differential diagnostics possibilities together with brief description of selected clinical conditions are discussed within the review.
Keywords:
organ dysfunction – sepsis – sepsis mimics
Authors:
Thomas Karvunidis 1,2; Martin Matějovič 1,2
Authors‘ workplace:
I. interní klinika LF UK a FN Plzeň
1; Biomedicínské centrum LF UK, Plzeň
2
Published in:
Vnitř Lék 2019; 65(6): 440-448
Category:
Review
Overview
Sepse sdílí s celou řadou dalších klinických stavů a onemocnění množství identických patofyziologických mechanizmů. To je důvodem, který může vést k velmi podobnému klinickému obrazu. Včasné rozlišení je zcela zásadní – léčba jednotlivých onemocnění se významně liší a mortalita většiny z nich je vysoká. Tento přehledový článek diskutuje možnosti jejich diskriminace a stručně popisuje několik vybraných nozologických jednotek imitujících sepsi.
Klíčová slova:
mimikry sepse – orgánová dysfunkce – sepse
Úvod
Sepse je hlavní příčinou úmrtí na infekční nemoci. Každým rokem postihuje až 30 milionů jedinců na celém světě, z nichž zemře 25–30 % [1]. S ohledem na uvedenou incidenci a mortalitu lze sepsi považovat za jednu z nejvýznamnějších medicínských hrozeb současnosti.
Sepse je charakterizována komplexní patofyziologií a extrémně heterogenním fenotypem ve smyslu klinických symptomů, reakce na léčbu a výstupu. V současné době neexistuje zlatý standard její diagnostiky, kauzální léčba či spolehlivá predikce léčebného výstupu [2]. Recentní definice charakterizuje sepsi jako život ohrožující orgánovou dysfunkci způsobenou deregulovanou odezvou hostitelského organizmu na přítomnost infekce (SEPSIS-3) [3]. Uvedené lze zjednodušeně vyjádřit následovně:
sepse ≈ systémová infekce + orgánová dysfunkce
V tomto novém pojetí, lépe respektujícím patofyziologické mechanizmy, je definice sepse relativně senzitivní. Specifická je však pouze v případech, v nichž jsou klinické a laboratorní známky orgánového postižení skutečně způsobeny infekcí (syndrom sepse). Zatímco identifikace orgánových dysfunkcí většinou nečiní potíže, jednoznačný průkaz infekce však nebývá vždy snadný. Zejména v prvním kontaktu s nemocným pracujeme s určitou mírou podezření na infekční onemocnění (pracovní diagnóza), jež je založeno na klinických projevech, dostupných paraklinických vyšetřeních, průběhu a našich znalostech a zkušenostech.
Moderní mikrobiologická diagnostika s užitím vysoce citlivých metod (např. hmotnostní spektrometrie či celogenomová sekvenace) umožňuje časnou a přesnou identifikaci patogenů ve stále větším počtu případů. Je-li kauzální souvislost detekovaného agens, místa vzniku infekce a klinického stavu alespoň minimálně pravděpodobná, je výše uvedená rovnice kompletní a diagnóza sepse obvykle nečiní potíže. Pochyby o správnosti diagnózy mohou nastat v případě atypického průběhu onemocnění, při absenci průkazu infekčního agens nebo neadekvátní terapeutické odpovědi na zavedenou léčbu. Dle recentních dat nebývá kauzální patogen identifikován ve 30–45 % případů sepse [4,5]. Pravděpodobnost „kultivačně negativní“ sepse je vetší u již hospitalizovaných pacientů, jedinců předléčených antimikrobiologiky a nemocných s hematologickými malignitami. V těchto případech je potřeba pomýšlet i na stavy, syndromy a nozologické jednotky, které se manifestují jako sepse, ale jejich etiologie, a tedy i léčba jsou odlišné. Shodují se v klinických projevech, tedy v tom, co bylo dříve označováno jako syndrom systémové zánětlivé odpovědi (Systemic Inflammatory Response Syndrome – SIRS). Společné jsou také rysy orgánových dysfunkcí a případného šoku. Přehled vybraných stavů, které mohou imitovat sepsi případně septický šok, je uveden v tab. 1.
Hledání svatého grálu
Podstata záměny sepse a jiného onemocnění spočívá v obdobných patofyziologických mechanizmech. Iniciální imunitní odpověď organizmu na mikrobiální infekci, ale i na sterilní stimuly (trauma, popáleniny, hemoragický šok) je fenotypicky vysoce stereotypní a uniformní a v rámci zmíněných stavů navzájem v podstatě klinicky neodlišitelná. Studie genových expresí a transkripčních profilů cirkulujících leukocytů nemocných s tupými traumaty, popáleninami a infekcí dokládají globální změny v intracelulárních funkcích a signalizacích zaujímajících > 80 % genů [6–8].
Laboratorní parametry nejsou pro sepsi rovněž specifické. Jakákoliv forma fyziologického stresu vede ke zvýšení počtu leukocytů s dominancí mladých forem granulocytů (posun doleva) [9,10]. Trombocytopenie, koagulopatie a elevace fibrin-degradačních produktů jsou rovněž častými nálezy; obdobně nespolehlivě však neodlišují sepsi a neinfekční systémovou inflamaci.
C-reaktivní protein (CRP) je syntetizován v játrech po indukci interleukiny; zvýšení jeho hladin nastává během 6–8 hod a vrcholu dosahuje obvykle po 35–50 hod [11]. Je senzitivním, ale velmi málo specifickým proteinem akutní fáze. Jeho zvýšené hladiny můžeme zaznamenat u infekcí, nádorových onemocnění, autoimunitních chorob, traumat, popálenin a obecně u jakýchkoliv inflamatorních stavů [12].
Prokalcitonin (PCT) je při systémové inflamaci produkován neuroendokrinními buňkami v plicích a střevech. Interval mezi stimulem a zvýšením plazmatických hladin je 3–5 hod a biologický poločas 25–30 hod. I přes relativně vysokou senzitivitu (77 %) i specificitu (79 %) není však ani PCT spolehlivým biomarkerem k diskriminaci sepse a systémové inflamace neinfekční etiologie [13,14]. I přesto je jeho senzitivita větší než CRP, interleukinu 6 (IL6), proteinu vázajícímu lipopolysacharid (lipopolysacharid-binding protein), solubilního receptoru exprimovaného na myeloidních buňkách 1 (soluble triggering receptor expressed on myeloid cells 1 – sTREM1) či solubilního receptoru pro urokinázový aktivátor plazminogenu (soluble urokinase plasminogen activator receptor – suPAR) [12,15–19].
Presepsin (sCD14-ST – PSEP) je solubilní fragment membránové molekuly CD14, která je exprimována na makrofázích a monocytech a v přítomnosti bakteriálních lipopolysacharidů se účastní aktivace Toll-like receptoru 4 s následnou produkcí tumor nekrotizujícího faktoru α (TNFα). Solubilní CD14 molekuly jsou uvolňovány v průběhu sepse; jejich fyziologická role není zcela objasněna, předpokládá se jejich zapojení v procesu fagocytózy a lyzosomální degradace mikroorganizmů [20]. Elevace plazmatických hladin sCD14-ST je mnohem časnější než PCT, IL6 i TNFα [21]. Dle recentně publikovaných metaanalýz je senzitivita PSEP 83–86 %, specificita 78 %. Přesto jej i přes řadu slibných signálů prozatím nelze izolovaně doporučit jako jediný biomarker k diskriminaci sepse a neinfekční inflamace [22,23]. Zvýšené hladiny presepsinu byly také pozorovány u systémového lupus erytematosus (SLE) a korelovaly s aktivitou nemoci [24].
Feritin je globulární protein produkovaný retikuloendoteliálním systémem (játra, slezina, kostní dřeň, ledvin) zodpovědný za intracelulární uchování zásoby železa v organizmu. Extracelulární, cirkulující feritin je tvořen glykosylovaným feritinem (60–80 %) fyziologicky secernovaným buňkami a tkáňovým feritinem (20–40 %) uvolněným z buněk při jejich postižení [25]. Normální sérové hladiny feritinu jsou 30–300 μg/l [25]. Hyperferitinemie je relativně častým laboratorním nálezem u celé řady onemocnění včetně sepse (tab. 2). Nejčastějšími příčinami elevace feritinu bez současného zvýšení saturace transferinu jsou: systémová infekce, alkoholizmus, poškození jater a kosterních svalů (cytolýza), metabolický syndrom a autoimunitní onemocnění. Pro diagnózu sepse je tedy feritin opět velmi málo specifický. Jeho extrémní hladiny (> 5 000–10 000 μg/l) jsou však pro „prostou“ systémovou infekci málo pravděpodobné. V těchto případech s klinickými projevy sepse je tato extrémní hyperferitinemie relativně diskriminující – je nezbytné uvažovat o SLE, Stillově chorobě dospělých (Still‘s Disease of Adults – SDA), hemofagocytární lymfohistiocytóze/syndromu aktivovaných makrofágů či hemochromatóze (při současně zvýšené saturaci transferinu) [25].
![Příčiny hyperferitinemie. Upraveno podle [25]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/98b834c02f46c4e97ca7b66c5f1849ac.png)
Souhrnně lze konstatovat, že žádný z uvedených ani z žádných dalších dosud popsaných biomarkerů sepse (jednotlivě ani v jejich kombinaci) není schopen spolehlivě rozlišit systémovou infekci od neinfekčních příčin SIRS. Již jen důvodné klinické podezření na rozvoj sepse však opravňuje k aktivním terapeutickým krokům (antimikrobiální léčba, kontrola/odstranění zdroje infekce, tekutiny) bez ohledu na naplnění či nenaplnění kritérií definice. Atypický průběh by měl vždy vést k opakovanému přehodnocení diagnózy, zamyšlení nad možnou jinou etiologií a aktivním krokům k jejich odhalení.
Vybrané stavy imitující sepsi
Hemofagocytární lymfohistiocytóza
Hemofagocytární lymfohistiocytóza (HLH) je vzácným agresivním a život ohrožujícím hyperinflamatorním syndromem charakterizovaným excesivní aktivací imunitního systému s následným tkáňovým postižením. Dospělé postihuje méně často než děti, jsou známy familiární formy (primární; familiární hemofagocytární lymfohistiocytóza – FHL) i formy sporadické (sekundární, získané) [26,27]. Samostatnou jednotkou je výskyt HLH u autoimunitních, resp. revmatických onemocnění – v těchto případech se označuje jako syndrom aktivovaných makrofágů (Macrophage Activation Syndrome – MAS). Nejčastěji je popisován u SLE, SDA a systémové juvenilní idiopatické artritidy [28]. Incidence v dospělé populaci není vzhledem k vzácnému výskytu známa; u dětí se odhaduje na 1 případ na 50 000–100 000 jedinců [29,30]. Bude však nepochybně významně podhodnocena vzhledem ke klinickému průběhu velmi věrně imitujícímu sepsi či septický šok.
Klíčovými hráči začarovaného kruhu patofyziologie HLH jsou:
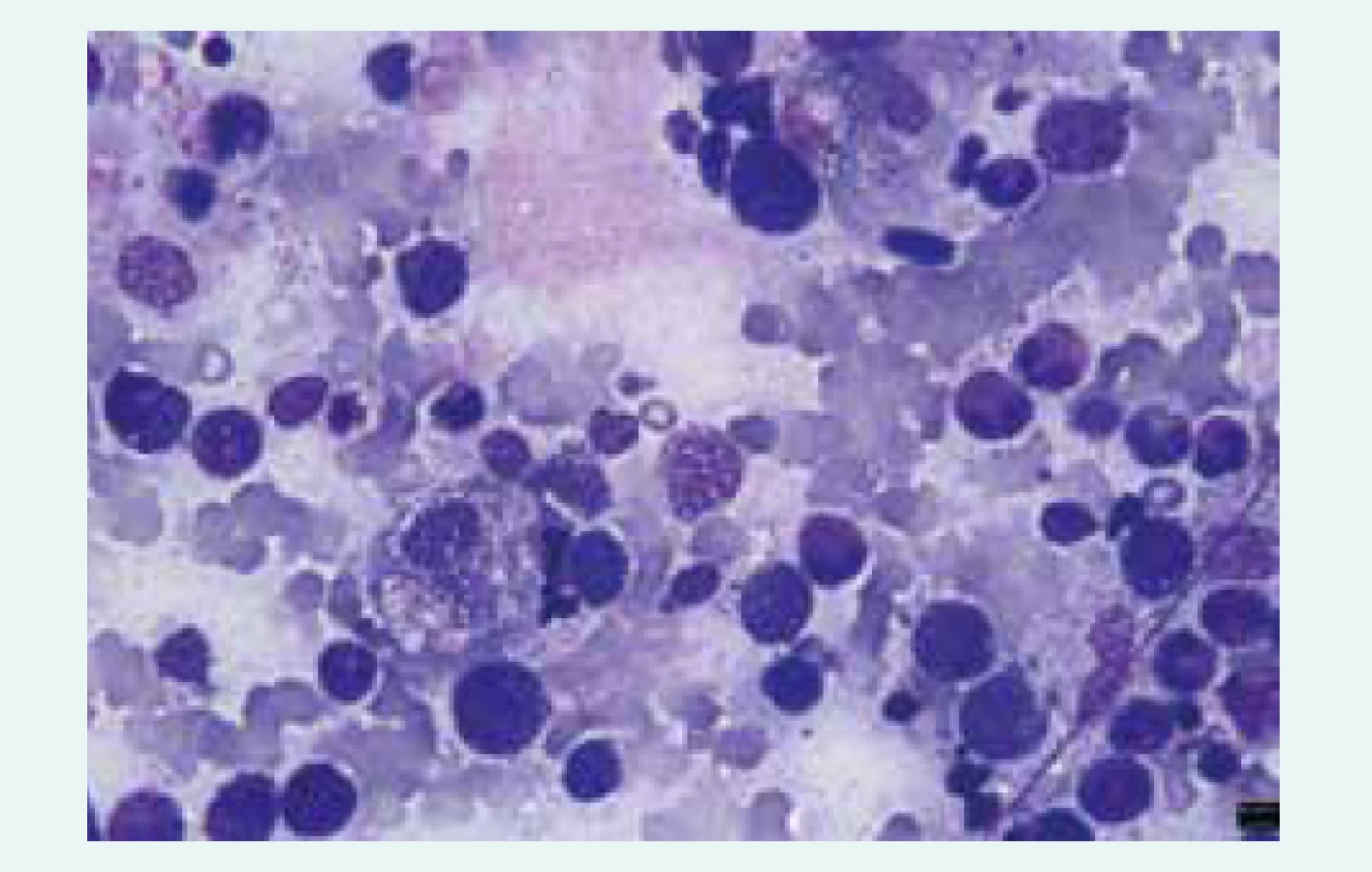
- makrofágy extenzivně aktivované cytokinovou bouří a s vystupňovanou fagocytární aktivitou (fagocytóza všech krevních elementů či jejich fragmentů – hemofagocytóza – ve tkáních), obr
- NK-buňky (lymfocyty) a cytotoxické T-lymfocyty (CD8+ T-lymfocyty) s porušenou cytotoxickou funkcí (perforin-dependentní cytotoxicita) a selháním eliminace aberantně aktivovaných makrofágů
- excesivní produkce cytokinů (cytokinová bouře; IFNγ, TNFα, IL6, IL10, IL12 a sCD25/sIL2R) výše uvedenými buněčnými elementy [31–34]
Spouštěčem epizody primární i sekundární HLH je obvykle infekce či defekt imunitní homeostázy – imunodeficience (malignity, autoimunitní onemocnění, infekce virem lidského imunodeficitu – HIV aj). Z infekcí jsou nejčastější virová onemocnění, typicky infekce virem Epsteina-Barrové (EBV). EBV navozuje komplexní imunoalteraci, která může i v nepřítomnosti genetické predispozice vést k poruchám signalizačních drah imunitní odpovědi a defektu cytotoxické aktivity T-lymfocytů a NK-buněk. Dalšími častými patogeny mohou být: cytomegalovirus (CMV), parvovirus B16, lidský herpesvirus 6 (HHV6) a HIV [30,31]. Mnohem méně často se uplatňují bakteriální, parazitární a mykotické infekce (mykobakteria, leischmanie, malárie aj) [35].
Jak již bylo zmíněno, HLH patří ve svých klinických projevech mezi špičkové virtuózy v napodobení sepse. Typicky se projevuje jako febrilní onemocnění s rychlou deteriorací a progresí do multiorgánového postižení (Multiple Organ Dysfunction Syndrome – MODS). Iniciální příznaky tedy mohou připomínat běžné infekce, horečku nejasné etiologie (Fever of Unknown Origin – FUO), hepatitidu, encefalitidu aj. Ve 2 studiích zahrnujících dohromady 371 nemocných byly prominentními klinickými příznaky: splenomegalie (97 %), hepatomegalie (95 %), horečka (95 %), lymfadenopatie (33 %), neurologické symptomy (33 %) a exantém (31 %) [36,37]. K typickým laboratorním nálezům patří cytopenie, zejména anémie a trombocytopenie (až 80 % nemocných) [29,37] a hyperferitinemie (> 3 000 μg/l) [37]. Zdrojem feritinu jsou aberantně aktivované makrofágy [38]. Extrémní hyperferitinemie (> 10 000 μg/l) vykazuje 90% senzitivitu a 96% specificitu pro HLH [39], nicméně ani nízká hladina feritinu (< 500 μg/l) nevylučuje HLH. Vzhledem k velmi frekventnímu postižení jater při HLH je abnormální elevace jaterních enzymů (ALT, AST, GGT), laktátdehydrogenázy, bilirubinu, triglyceridů a přítomnost koagulopatie typická [40]. Relativně časté neurologické postižení se může manifestovat jako encefalopatie, kvantitativní porucha vědomí, křeče či ataxie. Nemocní s HLH jsou také ve vysokém riziku rozvoje syndromu reverzibilní posteriorní leukoencefalopatie (reversible posterior leukoencephalopathy syndrome – RPLS/PRES) charakterizovaném cefaleou, poruchou vědomí, poruchami vizu a křečemi [41,42].
Diagnóza HLH není jednoduchá, klíčem k úspěchu je na diagnózu HLH myslet. K podezření by nás měl většinou přivést abnormální průběh stonání a absence klinické odpovědi na zavedenou léčbu (nejčastěji antimikrobiální léčba při primárním podezření na systémovou infekci/sepsi/septický šok). Diagnostická kritéria spočívají v molekulárně genetickém průkazu mutace asociované s HLH či naplnění minimálně 5 z 8 následujících nálezů: horečky, splenomegalie, cytopenie postihující 2 a více krvetvorných řad, hypertriglyceridemie, hypofibrinogenemie, hyperferitinemie, průkaz tkáňové hemofagocytózy (kostní dřeň, slezina, lymfatické uzliny), poruchy funkce NK-buněk a elevace hladin solubilního receptoru pro IL2 (sIL2R, sCD25) [43]. Je potřeba upozornit, že uvedená kritéria byla primárně vytvořena pro potřeby studie pediatrických nemocných a extrapolována na dospělou populaci [44]. S ohledem na často progresivní průběh onemocnění, vedoucí k deterioraci klinického stavu s rozvojem MODS, není nutné při významném podezření ne HLH splnit všechna požadovaná kritéria. V těchto případech je po zvážení poměru rizika a benefitu indikováno časné zahájení specifické léčby.
HLH bez zahájení specifické terapie vede k rapidnímu zhoršení klinického stavu s orgánovým selháním a smrti v horizontu týdnů. Včasné zahájení léčby je tedy klíčové. Již při podezření na HLH by nemocní měli být neodkladně referováni a transportováni do zdravotnického centra se zázemím specializované hematoonkologické péče a současně i péče intenzivní. Základním mechanizmem terapie HLH je přerušení deregulované imunitní odpovědi supresí T-lymfocytů a NK-buněk. Pacienti ve stabilním klinickém stavu mohou nejprve podstoupit léčbu základního či spouštějícího onemocnění (malignita, infekce, autoimunita). U akutně stonajících a nestabilních jedinců je indikováno zahájení HLH specifické léčby dle léčebného protokolu HLH-94 [37,45,46]. Základem tohoto terapeutického schématu je etopozid a dexametazon aplikované ve snižujících se dávkách během 8 týdnů. Při známkách postižení centrálního nervového systému je doporučeno současně intratekálně aplikovat metotrexát a hydrokortizon [47]. Současně bývá většinou nezbytná specializovaná intenzivní a podpůrná péče (náhrady a podpora orgánových funkcí, transfuze, léčba infekčních komplikací aj). Terapeutický efekt lze hodnotit dle vývoje klinického stavu (orgánových dysfunkcí) a laboratorních parametrů (ideálně feritin a sCD25). Odpověď na popsanou indukční léčbu je determinantou pro další postup. Při známkách selhání či nedostatečného efektu tím může být eskalace dávek etopozidu a dexametazonu či záchranná léčba alemtuzumabem (anti-CD52 protilátka, „panleukocytární“ antigen) [46,48,49]. Alemtuzumab může být i lékem první volby (v kombinaci s dexametazonem) u jedinců s významným postižením jater a ledvin, u nichž by byla léčba etopozidem zatížena významnou toxicitou. V současné době probíhají studie zkoumající efekt tocilizumabu (inhibitor IL6) a ruxolitinibu (inhibitor Janus-kinázy 1 a 2). Záchrannou léčebnou modalitou je pak transplantace kmenových buněk krvetvorby (vyhrazená pro jedince v relativně dobrém klinickém stavu a s dobrými fyziologickými rezervami).
Odhadované celkové přežití akutně nemocných jedinců s léčenou HLH je 25–50 % (55–80 % ve 30 dnech). Pětileté přežití pacientů po transplantaci kmenových buněk krvetvorby je přibližně 65 % [50,51].
Syndromy trombotických mikroangiopatií
Trombotické mikroangiopatie (TMA) označují široké spektrum onemocnění s obdobným klinickým průběhem a patofyziologickými mechanizmy. Mezi typické charakteristiky patří mikroangiopatická hemolytická anémie, trombocytopenie a orgánové poškození. Klinické rysy mohou nezřídka imitovat sepsi a septický šok [52]. Přehled a základní charakteristiky získaných TMA jsou shrnuty v tab. 3. Současně je zde také uveden přehled nejčastějších onemocnění prezentujících se určitými rysy TMA, jejichž patofyziologie a projevy se mohou vzájemně prolínat. Nejčastějšími klinickými příznaky jsou slabost, gastrointestinální symptomy (bolesti břicha, průjmy), purpura a neurologické abnormality. Většina nemocných má také známky poškození ledvin, mikroangiopatickou hemolytickou anémii (známky intravaskulární hemolýzy: hyperbilirubinemie, elevace laktátdehydrogenázy, snížení haptoglobinu, přítomnost signifikantního počtu schistocytů) a trombocytopenii. Relativně diagnostická pro trombotickou trombocytopenickou purpuru (TTP) je detekce snížení aktivity metaloproteinázy ADAMTS13 < 5–10 %. Průkaz shiga-toxin produkující Escherichia coli (STEC; kmen O157:H7) v souvislosti s popsanými klinickými příznaky směřuje diagnózu k shiga-toxin mediovanému hemolyticko-uremickému syndromu (ST-HUS). Abnormality ve složkách komplementu (C3, C4, H, B, I aj) mohou dohromady s relativně normální aktivitou ADAMTS13 a nepřítomností STEC charakterizovat komplementem zprostředkovanou TMA (C-TMA; dříve tzv. atypický HUS). Separátní skupinou jsou imunitně i neimunitně (idiosynkrazie) zprostředkované polékové TMA.
![Přehled, základní charakteristiky a asociovaná onemocnění získaných TMA. Upraveno podle [52]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/14a1b60659a83e157f978d9a4d021f51.jpeg)
Mortalita TMA je obecně velmi vysoká. U neléčených případů TTP může dosahovat až > 90 %. Léčba se u jednotlivých typů TMA liší a je stručně v principech uvedena v tab. 3.
Katastrofický antifosfolipidový syndrom
Katastrofický antifosfolipidový syndrom (Catastrophic Antiphospholipid Syndrome – CAPS) je raritní, život ohrožující autoimunitní onemocnění charakterizované diseminovanou intravaskulární trombotizací (thrombotic storm) rezultující v multiorgánové selhání. Jde o trombotickou mikroangiopatii postihující dominantně cévy malého a středního kalibru [53,54]. CAPS je akcelerovanou formou antifosfolipidového syndromu (APS). Vyskytuje se přibližně u 1 % nemocných s APS, izolovaně nebo současně s jiným autoimunitním onemocněním, zejména SLE. Až 72 % postižených jedinců tvoří ženy [54,55]. Patofyziologickým podkladem APS/CAPS je inhibice přirozené antikoagulační a fibrinolytické aktivity přítomností antifosfolipidových protilátek (antikardiolipinové protilátky, anti-β2-glykoprotein-I protilátky, lupus antikoagulans) a aktivace endotelu a komplementu [54]. Vše uvedené pak v důsledku vede ke zmíněné intravaskulární trombóze s následným tkáňovým poškozením. Precipitujícími faktory jsou infekční onemocnění, operační výkon, těhotenství, ukončení antikoagulační léčby či přítomnost nádorového či jiného autoimunitního onemocnění [54].
Klinickým projevem CAPS je již zmíněné multiorgánové postižení, ne nepodobné postižení při sepsi či septickém šoku. K nejčastěji postiženým orgánům patří v sestupném pořadí: ledviny, plíce, centrální nervový systém, srdce, kůže, játra aj.
Diagnostika spočívá v detekci antifosfolipidových protilátek, současném postižení 3 a více orgánů v průběhu jednoho týdne a případném histologickém průkazu intravaskulární trombózy s okluzí cév v postižených orgánech [54]. Z laboratorních výsledků je typické prodloužení aPTT nekorigovatelné přidáním normální lidské plazmy (test korekce patologicky prodlouženého aPTT), trombocytopenie (konsumpční), přítomnost schistocytů a případně i dalších parametrů intravaskulární hemolýzy. Diferenciálně diagnosticky je nezbytné odlišit jiné trombotické mikroangiopatie: hemolyticko-uremický syndrom, trombotickou trombocytopenickou purpuru, diseminovanou intravaskulární koagulaci či heparinem indukovanou trombocytopenii.
Vzhledem k vysoké mortalitě CAPS (35–55 %) je časné zahájení adekvátní terapie nezbytné. Ta spočívá v antikoagulační léčbě a eliminaci autoprotilátek pomocí výměnných plazmaferéz (plasma Exchange – PEX) či intravenózních imunoglobulinů (IVIG). Klasická imunosupresivní léčba (kortikosteroidy, cyklofosfamid aj) nebývá v případě akutní fáze samotného CAPS účinná (protrahovaný nástup účinku), lze ji zvážit v případě chronického APS anebo konkomitantního autoimunitního onemocnění (SLE aj) [54,56,57]. Slibným směrem terapie CAPS včetně refrakterních forem je užití ekulizumabu (protilátky proti složce komplementu C5) či rituximabu (protilátce proti membránové molekule CD20 B-lymfocytů) [58–60].
Syndrom uvolnění cytokinů
Syndrom uvolnění cytokinů (Cytokine Release Syndrome – CRS) je nejvýznamnější a potenciálně život ohrožující komplikací moderní cílené léčby (imunoterapie/targeted therapy) malignit, zejména relabujících či refrakterních akutních lymfoblastických leukemií [61,62]. Incidence CRS variabilní tíže se pohybuje od 50 % (blinatumomab) do 100 % (chimeric antigen receptor T-cells – CAR-TC) [63]. Patofyziologickým mechanizmem je aktivace T-lymfocytů navozená léčbou s extenzivním uvolněním proinflamatorních cytokinů (INFγ, IL6, IL10 a sIL2R aj). Současně dochází k elevaci obecných markerů systémové inflamace (CRP, PCT, feritin aj).
Klinické příznaky jsou od sepse/septického šoku či HLH/MAS obtížně odlišitelné [63,64].
Léčba CRS po blinatumomabu či CAR-TC se významně odlišuje. Aplikaci blinatumomabu lze již při prvních známkách CRS (zpravidla horečky) kdykoliv přerušit a zabránit tak plnému rozvoji CRS s šokem a orgánovým postižením. Po odeznění příznaků je možné v podání pokračovat. CAR-TC jsou extrémně sofistikovanou a unikátní léčbou; snaha předcházet, mitigovat a léčit známky CRS významně limituje jejich protinádorovou aktivitu [63]. Vždy je nezbytné vyloučit jiné alternativy CRS – nejčastěji tedy infekční komplikaci, sepsi, septický šok. To však bývá ve většině případů vzhledem k identickým patofyziologickým mechanizmům nemožné. Na druhou stranu, CRS může být život ohrožujícím stavem, není tedy na místě se zahájením adekvátní léčby dlouho otálet. Ideální je ji zahájit ještě před rozvojem orgánového postižení. V případě CAR-TC asociovaného CRS je lékem volby tocilizumab, protilátka proti IL6 [63]. Tento postup umožňuje relativně zachovat efektivitu CAR-TC při současné kontrole symptomů CRS v porovnání s léčbou kortikosteroidy [63]. Kortikosteroidy (dexametazon) jsou pak lékem volby v prevenci a terapii CRS indukovaného blinatumomabem. U těžké, život ohrožující a kortikorezistentní formy lze také zvážit aplikaci tocilizumabu [63].
Idiopatický syndrom systémové zvýšené propustnosti kapilár
Idiopatický syndrom zvýšené propustnosti kapilár (Idiopathic Systemic Capillary Leak Syndrome – ISCLS) je vzácným onemocněním charakterizovaným epizodami těžké hypotenze, hypoalbuminemie a hemokoncentrace. Literatura dokumentuje celosvětově přibližně 150–200 popsaných případů [65]. Patofyziologické mechanizmy vzniku a rozvoje ISCLS nejsou zcela objasněny. Několik studií odhalilo monoklonální gamapatii u většiny nemocných s projevy ISCLS. Lze pouze spekulovat o přímém postižení endotelu paraproteinem. Pravděpodobně je však paraproteinemie spíše epifenoménem nepoznaného patofyziologického procesu/onemocnění než přímým spouštěcím faktorem [66,67]. Apoptóza endoteliálních buněk je také zvažovaným mechanizmem ISCLS. Tato hypotéza je podporována histologickými nálezy těchto změn v biopsiích u nemocných s akutní atakou ISCLS [68,69]. Procesu ztráty kapilární integrity se také účastní množství mediátorů jako leukotrieny, TNFα, IL2 či VEGF aj [70,71].
Ataky ISCLS se klinicky manifestují obvykle ve 3 fázích:
- prodromální symptomy: slabost, malátnost, bolest břicha, myalgie, artralgie, nevolnost aj; až 30 % nemocných udává předchozí příznaky infekce horních dýchacích cest či flu-like symptomy se subfebriliemi či febriliemi, ataky ISCLS může rovněž spouštět výrazná fyzická zátěž [72]
- fáze extravazace: zvýšená kapilární propustnost se objevuje přibližně 1–4 dny po prodromální periodě, typická je triáda příznaků – hypotenze, hemokoncentrace a hypoalbuminemie [66]; dalšími příznaky jsou generalizované otoky, ascites, fluidotorax, perikardiální výpotek, otok mozku a encefalopatie, současně také známky, projevy a důsledky hypovolemie, hypotenze, hypoperfuze a orgánového poškození; průměrná doba trvání této fáze jsou 3–4 dny
- fáze rezoluce: dochází k úpravě propustnosti endotelu a redistribuci tekutin zpět do cirkulace
Frekvence a tíže atak ISCLS je signifikantně individuální; dle dostupných dat dochází u postižených jedinců průměrně ke 3 exacerbacím ročně [73].
ISCLS má mnoho společných klinických i laboratorních rysů se sepsí a septickým šokem. Diagnóza je nesnadná a převážně možná vyloučením jiných častějších příčin, zejména infekčních, anafylaxe či HLH/MAS. Nápomocnou může být detekovaná monoklonální paraproteinemie.
Léčba ISCLS je extrapolována z obecných principů léčby šokových stavů. Základem je rychlá stabilizace vitálních funkcí, zejména hemodynamiky s udržením tkáňové perfuze a zabráněním orgánového postižení. Balancované krystaloidní roztoky jsou jednoznačně preferovány ve smyslu volumexpanze a udržení intravaskulárního cirkulujícího objemu. Ve vybraných případech (těžká hypoalbuminemie) lze zvážit substituci albuminem. V případě potřeby je vazopresorem volby noradrenalin. Ve stadiu daty neověřené léčby a kontroverzních výsledků je případné podání intravenózního imunoglobulinu [74–76] bevacizumabu (anti-VEGF protilátky) [77] či terbutalinu s aminofylinem (zvýšení intracelulárního cyklického adenozinmonofosfátu vedoucí ke snížení kapilární propustnosti) [65–67]. Nejzávažnějšími komplikacemi ISCLS je syndrom kompartmentu, plicní otok, srdeční tamponáda, pankreatitida a hluboká žilní trombóza [72]. První 4 uvedené mohou být významně exacerbovány nekontrolovanou tekutinovou resuscitací. Lze tak doporučit frekventní kontrolu intravaskulární volémie pomocí ultrazvuku, nejlépe pak se sledováním trendu a s užitím dynamických manévrů.
Prognóza ISCLS je relativně dobrá; při adekvátní léčbě je 5leté přežití více než 70 % [66].
Závěr
Sepse je bezesporu hlavní a nejčastější příčinou stavů spojených s rychlým rozvojem systémové zánětově odpovědi, multiorgánového poškození a vazoplegického šoku. Stavy, které sepsi více či méně napodobují, jsou však také v řadě případů velmi závažné, často postihují pacienty v produktivním věku a bez včasné a adekvátní léčby jsou rovněž zatížené vysokou smrtností. Jejich znalost je tudíž klíčová navzdory jejich nízké incidenci. Vzhledem k nárokům na diagnostické a léčebné postupy je vždy výhodou dobrá mezioborová spolupráce a centralizace péče o tyto pacienty na pracovištích se zkušenostmi a adekvátním zázemím včetně intenzivní péče.
Podpořeno: Programem rozvoje vědních oborů Univerzity Karlovy (PROGRES – projekt Q39), Projektem CZ.02.1.01/0.0/0.0/16_019/0000787, Centrum výzkumu infekčních onemocnění, udělený MŠMT, financovaný EFRR, Projektem institucionálního výzkumu MZČR – FNPl 00669806.
Doručeno do redakce 14. 9. 2018
Přijato po recenzi 28. 1. 2019
MUDr. Thomas Karvunidis, Ph.D.
I. interní klinika LF UK a FN Plzeň
Sources
- Fleischmann C, Scherag A, Adhikari NK et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193(3): 259–272. Dostupné z DOI: <http://dx.doi.org/10.1164/rccm.201504–0781OC>.
- Cohen J, Vincent JL, Adhikari NK et al. Sepsis: a roadmap for future research. Lancet Infect Dis 2015; 15(5): 581–614. Dostupné z DOI: <http://dx.doi.org/10.1016/S1473–3099(15)70112-X>. Erratum in Corrections. [Lancet Infect Dis 2015].
- Singer M, Deutschman CS, Seymour CW et al. The third international consensus definitions for sepsis and septic shock. JAMA 2016; 315(8): 801–810. Dostupné z DOI: <http://dx.doi.org/10.1001/jama.2016.0287>.
- Gupta S, Sakhuja A, Kumar G et al. Culture-negative severe sepsis: nationwide trends and outcomes. Chest 2016; 150(6): 1251–1259. Dostupné z DOI: <http://dx.doi.org/10.1016/j.chest.2016.08.1460>.
- Tillmann B, Wunsch H. Epidemiology and outcomes. Crit Care Clin 2018; 34(1): 15–27. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ccc.2017.08.001>.
- Calvano SE, Xiao W, Richards DR et al. A network-based analysis of systemic inflammation in humans. Nature 2005; 437(7061): 1032–1037. Dostupné z DOI: <http://dx.doi.org/10.1038/nature03985>. Erratum in Nature 2005; 438(7068): 696.
- Xiao W, Mindrinos MN, Seok J et al. A genomic storm in critically ill injured humans. J Exp Med 2011; 208(13): 2581–2590. Dostupné z DOI: <http://dx.doi.org/10.1084/jem.20111354>.
- Larosa SP, Opal SM. Immune aspects of sepsis and hope for new therapeutics. Curr Infect Dis Rep 2012; 14(5): 474–483. Dostupné z DOI: <http://dx.doi.org/10.1007/s11908–012–0276–2>.
- Kaukonen KM, Bailey M, Pilcher D et al. Systemic inflammatory response syndrome criteria in defining severe sepsis. N Engl J Med 2015; 372(17): 1629–1638. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1415236>.
- Long B, Koyfman A. Clinical mimics: an emergency medicine-focused review of sepsis mimics. J Emerg Med 2017; 52(1): 34–42. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jemermed.2016.07.102>.
- Larsen FF, Petersen JA. Novel biomarkers for sepsis: a narrative review. Eur J Intern Med 2017; 45 : 46–50. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejim.2017.09.030>.
- Simon L, Gauvin F, Amre DK et al. Serum prokalcitonin and C-reactive protein levels as markers of bacterial infection: a systematic review and meta-analysis. Clin Infect Dis 2004; 39(2): 206–217. Dostupné z DOI: <http://dx.doi.org/10.1086/421997>. Erratum in Clin Infect Dis 2005; 40(9): 1386–1388.
- Wacker C, PrknoA, Brunkhorst FM et al. Procalcitonin as a diagnostic marker for sepsis: a systematic review and meta-analysis. Lancet Infect Dis 2013; 13(5): 426–435. Dostupné z DOI: <http://dx.doi.org/10.1016/S1473–3099(12)70323–7>.
- van Engelen TSR, Wiersinga WJ, Scicluna BP et al. Biomarkers of sepsis. Crit Care Clin 2018; 34(1): 139–152. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ccc.2017.08.010>.
- Reinhart K, Meisner M. Biomarkers in the critically ill patient: procalcitonin. Crit Care Clin 2011; 27(2): 253–263. <http://dx.doi.org/10.1016/j.ccc.2011.01.002>.
- Sakr Y, Burgett U, Nacul FE et al. Lipopolysacharide binding protein in a surgical intensive care unit: a marker of sepsis? Crit Care Med 2008; 36(7): 2014–2022. Dostupné z DOI: <http://dx.doi.org/10.1097/CCM.0b013e31817b86e3>.
- Bouchon A, Facchetti F, Weingand MA et al. sTREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001; 410(6832): 1103–1107. Dostupné z DOI: <http://dx.doi.org/10.1038/35074114>.
- Wu Y, Wang F, Fan X et al. Accuracy of plasma sTREM-1 for sepsis diagnosis in systemic inflammatory patients: a systematic review and meta-analysis. Crit Care 2012; 16(6): R229. Dostupné z DOI: <http://dx.doi.org/10.1186/cc11884>.
- Backes Y, van der Sluijs KF, Mackie DP et al. Usefulness of suPAR as a biological marker in patients with systemic inflammation of infection: a systematic review. Intensive Care Med 2012; 38(9): 1418–1428. Dostupné z DOI: <http://dx.doi.org/10.1007/s00134–012–2613–1>.
- Yaegashi Y, Shirakawa K, Sato N et al. Evaluation of a newly identified soluble CD14 subtype as a marker for sepsis. J Infect Chemother 2005; 11(5): 234–238. Dostupné z DOI: <http://dx.doi.org/10.1007/s10156–005–0400–4>.
- Shozushima T, Takahashi G, Matsumoto N at al. Usefulness of presepsin (sCD14-ST) measurements as a marker for the diagnosis and severity of sepsis that satisfied diagnostic criteria of systemic inflammatory response system. J Infect Chemother 2011; 17(6): 764–749. Dostupné z DOI: <http://dx.doi.org/10.1007/s10156–011–0254-x>.
- Zhang J, Hu ZD, Song J et al. Diagnostic value of presepsin for sepsis: a systematic review and meta-analysis. Medicine (Baltimore) 2015; 94(47): e2158. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000002158>.
- Brodska H, Valenta J, Pelinkova K et al. Diagnostic and prognostic value of presepsin vs. established biomarkers in critically ill patients with sepsis of systemic inflammatory response syndrome. Clin Chem Lab Med 2018; 56(4): 658–668. Dostupné z DOI: <http://dx.doi.org/10.1515/cclm-2017–0839>.
- Tanimura S, Fujieda Y, Kono M et al. Clinical significance of plasma presepsin levels in patients with systemic lupus erythematosus. Mod Rheumatol 2018; 28(5): 865–871. Dostupné z DOI: <http://dx.doi.org/10.1080/14397595.2017.1408755>.
- Lorcerie B, Audia S, Samson M et al. Diagnosis of hyperferritinemia in routine clinical practice. Presse Med 2017; 46(12 Pt 2): e329-e338. Dostupné z DOI: <http://dx.doi.org/10.1016/j.lpm.2017.09.028>.
- Lachmann G, Spies C, Schenk T et al. Hemophagocytic lymphohistiocytosis: potentially underdiagnosed in intensive care units. Shock 2018; 50(2): 149–155. Dostupné z DOI: <http://dx.doi.org/10.1097/SHK.0000000000001048>.
- Šrámek J, Karvunidis T, Lysák D et al. Hemofagocytární lymfohistiocytóza u dospělých: review a kazuistika. Vnitř Lék 2018; 64(3): 300–307.
- Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001; 85(5): 421–426. Dostupné z DOI: <http://dx.doi.org/10.1136/adc.85.5.421>.
- Niece JA, Rogers ZR, Ahmad N et al. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer 2010; 54(3): 424–428. Dostupné z DOI: <http://dx.doi.org/10.1002/pbc.22359>.
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A et al. Adult haemophagocytic syndrome. Lancet 2014; 383(9927): 1503–15116. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(13)61048-X>. Erratum in Lancet 2014; 383(9927): 1464.
- Filipovich A, McClain K, Grom A et al. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant 2010; 16(1 Suppl): S82-S89. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bbmt.2009.11.014>.
- Dalal BI, Vakil AP, Khare NS et al. Abnormalities of the lymphocyte subsets and their immunophenotype, and their prognostic significance in adult patients with hemophagocytic lymphohistiocytosis. Ann Hematol 2015; 94(7): 1111–1117. Dostupné z DOI: <http://dx.doi.org/10.1007/s00277–015–2350-y>.
- Henter JI, Elinder G, Soder O et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991; 78(11): 2918–2922.
- Osugi Y, Hara J, Tagawa S et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997; 89(11): 4100–4103.
- Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Imunol 2006; 6(12): 940–952. Dostupné z DOI: <http://dx.doi.org/10.1038/nri1983>.
- Arico M, Janka G, Fischer A et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International registry. FHL study group of the Histiocyte society. Leukemia 1996; 10(2): 197–203.
- Trottestam H, Horne A, Arico M et al. Chemoimmunotherapy for the hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 2011; 118(17): 4577–4584. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–06–356261>.
- Cohen LA, Gutierrez L, Weiss A et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010; 116(9): 1574–1584. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2009–11–253815>.
- Allen CE, Yu X, Kozinetz CA et al. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008; 50(6): 1227–1235. Dostupné z DOI: <http://dx.doi.org/10.1002/pbc.21423>.
- Fukaya S, Yasuda S, Hashimoto T et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008; 47(11): 1686–1691. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/ken342>.
- Deiva K, Mahlaoui N, Beaudonnet F et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology 2012; 78(15): 1150–1156. Dostupné z DOI: <http://dx.doi.org/10.1212/WNL.0b013e31824f800a>.
- Jovanovic A, Kuzmanovic M, Kravljanac R et al. Central nervous system involvement in hemophagocytic lymphohistiocytosis: a single-center experience. Pediatr Neurol 2014; 50(3): 233–237. Dostupné z DOI: <http://dx.doi.org/10.1016/j.pediatrneurol.2013.10.014>.
- Henter JI, Horne A, Arico M et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48(2): 124–131. Dostupné z DOI: <http://dx.doi.org/10.1002/pbc.21039>.
- Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015; 125(19): 2908–2914. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2015–01–551622>.
- Henter JI, Samuelsson-Horne A, Arico M et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunotherapy and bone marrow transplantation. Blood 2002; 100(7): 2367–2373. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2002–01–0172>.
- Jordan MB, Allen CE, Weitzman S et al. How I treat hemophagocytic lymphohistiocytosis. Blood 2011; 118(15): 4041–4052. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–03–278127>.
- Henter JI, Arico M, Egeler RM et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study group of the Histiocyte society. Med Pediatr Oncol 1997; 28(5): 342–347.
- Jiang L, Yuan CM, Hubacheck J et al. Variable CD52 expression in mature T cell and NK cell malignancies: implications for alemtuzumab therapy. Br J Haemathol 2009; 145(2): 173–179. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2009.07606.x>.
- Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol 2010; 7(7): 415–420. Dostupné z DOI: <http://dx.doi.org/10.1038/nrclinonc.2010.40>.
- Parikh SA, Kapoor P, Letendre L et al. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014; 89(4): 484–492. Dostupné z DOI: <http://dx.doi.org/10.1016/j.mayocp.2013.12.012>.
- Li J, Wang Q, Zheng W et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 patients. Medicine (Baltimore) 2014; 93(2): 100–105. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000000022>.
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014; 371(7): 654–666. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMra1312353>.
- Erkan D, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: updated diagnostic algorithms. Autoimmun Rev 2010; 10(2): 74–79. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2010.08.005>.
- Nayer A, Ortega LM. Catastrophic antiphospholipid syndrome: a clinical review. J Nephropathol 2014; 3(1): 9–17. Dostupné z DOI: <http://dx.doi.org/10.12860/jnp.2014.03>.
- Cervera R, Piette JC, Font J et al. Antiphospholipid syndrome: clinical and immunlogic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46(4): 1019–1027.
- Bucciarelli S, Espinosa G, Cervera R et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum 2006; 54(8): 2568–2576. Dostupné z DOI: <http://dx.doi.org/10.1002/art.22018>.
- Cervera R, Bucciarelli S, Plasín MA et al. Catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the „CAPS Registry“. J Autoimmun 2009; 32(3–4): 240–245. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaut.2009.02.008>.
- Zikos TA, Sokolove J, Ahuja N et al. Eculizumab Induces Sustained Remission in a Patient With Refractory Primary Catastrophic Antiphospholipid Syndrome. J Clin Rheumatol 2015; 21(6): 311–313. Dostupné z DOI: <http://dx.doi.org/10.1097/RHU.0000000000000290>.
- Espinosa G, Rodríquez-Pintó I, Cervera R. Catastrophic antiphospholipid syndrome: an update. Panminerva Med 2017; 59(3): 254–268. Dostupné z DOI: <http://dx.doi.org/10.23736/S0031–0808.17.03324–9>.
- Cervera R, Rodríquez-Pintó I, Espinosa G. The diagnosis and clinical management of the catastrophic antiphospholipid syndrome: A comprehensive review. J Autoimmun 2018; 92 : 1–11. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaut.2018.05.007>.
- Maude SL, Frey N, Shaw PA et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371(16): 1507–1517. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1407222>.
- Topp MS, Gokbuget N, Stein AS et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 2015; 16(1): 57–66. Dostupné z DOI: <http://dx.doi.org/10.1016/S1470–2045(14)71170–2>.
- Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2016; 2016(1): 567–572. Dostupné z DOI: <http://dx.doi.org/10.1182/asheducation-2016.1.567>.
- Karvunidis T, Matějovič M. Vybrané akutní stavy v moderní onkologii. Vnitř Lék 2019; 65(6): 405–415.
- Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med 2010; 153(2): 90–98. Dostupné z DOI: <http://dx.doi.org/10.7326/0003–4819–153–2-201007200–00005>.
- Dhir V, Aria V, Malav IC et al. Idiopathic systemic capillary leak syndrome (SCLS): case report and systematic review of cases reported in the last 16 years. Intern Med 2007; 46(12): 899–904. Dostupné z DOI: <http://dx.doi.org/10.2169/internalmedicine.46.6129>.
- Dowden AM, Rullo OJ, Aziz N et al. Idiopathic systemic capillary leak syndrome: novel therapy for acute attacks. J Allergy Clin Immunol 2009; 124(5): 1111–1113. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jaci.2009.06.043>.
- Johansson BR, Lofdahl CG. Ultrastructure of the microvessels in skeletal muscle in a case of systemic capillary leak syndrome. Acta Med Scand 1979; 206(5): 413–416.
- Assaly R, Olson D, Hammersley J et al. Initial evidence of endothelial cell apoptosis as a mechanism of systemic capillary leak syndrome. Chest 2001; 120(4): 1301–1308. Dostupné z DOI: <http://dx.doi.org/10.1378/chest.120.4.1301>.
- Xie Z, Ghosg CC, Patel R et al. Vascular endothelial hyperpermeability induces the clinical symptoms of Clarkson disease (the systemic capillary leak syndrome). Blood 2012; 119(18): 4321–4332. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–08–375816>.
- Nagao Y, Harada H, Yamanaka H et al. Possible mediators for systemic capillary leak syndrome. Am J Med 2011; 124(1): e7-e9. Dostupné z DOI: <http://dx.doi.org/10.1016/j.amjmed.2010.04.024>.
- Gousseff M, Arnaud L, Lambert M et al. The systemic capillary leak syndrome: a case series of 28 patients from a European registry. Ann Intern Med 2011; 154(7): 464–471. Dostupné z DOI: <http://dx.doi.org/10.7326/0003–4819–154–7-201104050–00004>.
- Kapoor P, Greipp PT, Schaefer EW et al. Idiopathic systemic capillary leak syndrome (Clarkson‘s disease): the Mayo clinic experience. Mayo Clin Proc 2010; 85(10): 905–912. Dostupné z DOI: <http://dx.doi.org/10.4065/mcp.2010.0159>.
- Lambert M, Launay D, Hachulla E et al. High-dose intravenous immunoglobulins dramatically reverse systemic capillary leak syndrome. Crit Care Med 2008; 36(7): 2184–2187. Dostupné z DOI: <http://dx.doi.org/10.1097/CCM.0b013e31817d7c71>.
- Pineton de Chambrun M, Luyt CE, Beloncle F et al. The Clinical Picture of Severe Systemic Capillary-Leak Syndrome Episodes Requiring ICU Admission. Crit Care Med 2017; 45(7): 1216–1223. Dostupné z DOI: <http://dx.doi.org/10.1097/CCM.0000000000002496>.
- Pineton de Chambrun M, Gousseff M, Mauhin W et al. Intravenous Immunoglobulins Improve Survival in Monoclonal Gammopathy-Associated Systemic Capillary-Leak Syndrome. Am J Med 2017; 130(10): 1219.e19–1219.e27. Dostupné z DOI: <http://dx.doi.org/10.1016/j.amjmed.2017.05.023>.
- Yabe H, Yabe M, Koike T et al. Rapid improvement of life-threatening capillary leak syndrome after stem cell transplantation by bevacizumab. Blood 2010; 115(13): 2723–2724. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2009–11–247056>.
Labels
Diabetology Endocrinology Internal medicine Intensive Care Medicine Trauma surgeryArticle was published in
Internal Medicine

2019 Issue 6
Most read in this issue
- Diagnostika akutních poruch acidobazické rovnováhy
- Delirium v intenzivní péči
- Mimotělní eliminační metody v toxikologii: 1. část
- Vybrané akutní stavy v moderní onkologii