
Náhodne zistený feochromocytóm u 33-ročného pacienta s Lynchovým syndrómom
An incidental finding of pheochromocytoma in a 33-year-old patient with Lynch syndrome
Pheochromocytoma is a catecholamine-producing neuroendocrine tumor arising from chromaffin cells of the adrenal medulla. The detection of these tumors is extremely important because they are associated with high cardiovascular morbidity and mortality. Progress in molecular genetics has revealed that up to 35% of pheochromocytomas are inhereted. Lynch syndrome (hereditary nonpolypous colorectal cancer – HNPCC) is an autosomal dominant genetic condition that is associated with a high risk of colorectal cancer or other extracolonic tumors (adenocarcinoma of endometrium, stomach, ovarian carcinoma, carcinoma of urinary tract, small intestine, brain tumors and skin cancer). Foreign medical journals are reporting an increasing number of cases on coexistence of HNPCC and neuroendocrine tumors, including pheochromocytoma. It increases the likelihood that this type of tumor could represent an additional extracolonic manifestation of Lynch syndrome.
Keywords:
pheochromocytoma – Lynch syndrome – extracolonic manifestation
Authors:
Emília Mojtová 1; Jana Hanajíková 2; Olívia Hamidová 3; Gabriel Bognár 4; Daniel Dyttert 5; Marianna Grigerová 1; Štefan Kečkéš 6; Ján Podoba 1
Authors‘ workplace:
Klinika endokrinológie LF SZU a OUSA, Bratislava
1; II. rádiologická klinika LF UK a OUSA, Bratislava
2; Klinika lekárskej genetiky, OUSA Bratislava
3; Ústav patológie LF SZU a OUSA, Bratislava
4; Klinika onkologickej chirurgie LF UK a OUSA, Bratislava
5; Oddelenie imunodiagnostiky OUSA a Vysoká škola zdravotníctva a sociálnej práce sv. Alžbety, Bratislava
6
Published in:
Vnitř Lék 2020; 66(5): 80-84
Category:
Case Report
Overview
Feochromocytóm je katecholamíny produkujúci neuroendokrinný nádor vychádzajúci z chromafinných buniek drene nadobličky. Záchyt týchto nádorov je mimoriadne dôležitý, pretože sú spojené s vysokou kardiovaskulárnou morbiditou a mortalitou. Vďaka pokrokom v molekulárnej genetike sa vie, že až 35 % feochromocytómov je podmienených geneticky. Lynchov syndróm (hereditárny nepolypózny karcinóm kolorekta – HNPCC) patrí medzi autozomálne dominantné dedičné ochorenia, pričom nositelia patogénnych variantov majú predispozíciu na vznik kolorektálneho karcinómu alebo ďalších extrakolonických nádorov (adenokarcinómy endometria, žalúdka, karcinómy vaječníkov, močového traktu, tenkého čreva, nádory mozgu a kože). V zahraničných periodikách pribúdajú kazuistiky súbežného výskytu pacientov s HNPCC a neuroendokrinnými nádormi včítane feochromocytómu, predostierajúce otázku, či by tento typ nádoru mohol predstavovať ďalší extrakolonický prejav Lynchovho syndrómu.
Klíčová slova:
feochromocytóm – Lynchov syndróm – extrakolonický prejav
Úvod
Feochromocytóm (FEO) je nádor vychádzajúci z chromafinných buniek drene nadobličky. Pokiaľ má tumor pôvod v chromafinnom tkanive sympatikových ganglií, nazýva sa paraganglióm. Sú to zriedkavé nádory s incidenciou 2–6/1 000 000/rok/. Z epidemiologického hľadiska ich možno charakterizovať ako tumory, na ktoré sa „často myslí, avšak zriedka sa diagnostikujú“. Potvrdzuje to aj fakt, že v pitevných nálezoch sa zisťujú v 0,05% prípadov, pričom samotný patológ je často ich prvým diagnostikom v prípade úmrtia na niektorú z internistických emergencií. Maximálny záchyt ochorenia je medzi 40.–50. rokom života. V literatúre sú opísané aj prípady výskytu týchto nádorov u mladších pacientov, u ktorých sa väčšinou dokázala geneticky podmienená forma (1, 2).
Väčšina FEO je sporadických. Vďaka pokrokom v molekulárnej genetike sa vie, že až 35% FEO je podmiených geneticky. Približne 10 % vzniká v rámci známych genetických syndrómov. Syndróm mnohopočetnej endokrinnej neoplázie typu 2 (MEN 2) je spôsobený mutáciou (patogénnym variantom) RET protoonkogénu. Obojstranný feochromocytóm postihuje 50% nositeľov mutovaného génu, vzniká približne 10 rokov po diagnostikovaní medulárneho karcinómu štítnej žľazy, ktorý je u 98% nositeľov v poradí prvou neopláziou. Von Hippelov – Lindauov syndróm je asociovaný s patogénnym variantom VHL génu. VHL proteín reguluje hypoxiou indikovateľný faktor 2α (HIF 2α) a rôzne bunkové procesy, včítane angiogenézy. Pri VHL syndróme 1 typu vznikajú mnohopočetné benígne a malígne nádory retiny, CNS, obličiek, endokrinného pankreasu, avšak bez FEO. Pacienti s VHL syndrómom typu 2 majú vždy FEO, najčastejšie v kombinácii s ďalšími nádormi. Neurofibromatóza typu 1 (NF 1) má bohatú a variabilnú manifestáciu ktorá zahrňuje FEO v spojení s nádormi pošiev periférnych nervov, medulárnym karcinómom štítnej žľazy, karcinoidom, tumormi prištítnych teliesok a chronickou myeloidnou leukémiou. Carneyova triáda (komplex) je tumorózny syndróm postihujúci aspoň 5 orgánov. Vyskytujú sa GIST žalúdka, pľúcne chordómy, paragangliómy, adrenálne adenómy, feochromocytómy a leiomyómy pažeráka. Etiológia Carneyovho komplexu nie je známa, zatiaľ sa neodhalil kandidátsky gén. Carneyov – Stratakisov syndróm je autozomálne dominantné dedičné ochorenie zahrňujúce FEO, gastrointestinálne stromálne nádory (GIST) žalúdka a pľúcne chordómy. Je spôsobený mutáciou SDH génov kódujúcich podjednotky sukcinátdehydrogenázového komplexu (4, 5).
SDH komplex je proteín, ktorý sa podieľa na elektrónovom transporte v Krebsovom cykle a v mitochondriálnom dýchacom reťazci. Enzým sa skladá zo 4 podjednotiek (SDHA, SDHB, SDHC a SDHD), ktoré katalyzujú oxidáciu sukcinátu na fumarát. SDHx gény majú aj úlohu tumoróznych supresorov (5, 6).
Najvýznamnejšie sú patogénne varianty SDHB génu asociované s familiárnymi paragangliómami brušnej dutiny, malej panvy a hrudníka. Menej často sú spojené s feochromocytómom a paragangliómom parasympatiku. Nádory majú agresívny klinický priebeh. Riziko malignity u nositeľov patogénneho variantu je veľké. U 20% nositeľov patogénnych variantov sa počas sledovania vyvinul malígny paraganglióm alebo feochromocytóm a u viac ako 50% pacientov s malígnym paragangliómom sa potvrdil patogénny variant SDHB génu (6, 7). Okrem nádorov chromafinného tkaniva sú patogénne varianty SDHB génov asociované aj s karcinómami obličiek, papilárnym karcinómom štítnej žľazy, neuroblastómom a GISTy, Carneyovým komplexom a Carneyov – Stratakisov syndrómom (7, 8).
Symptómy a objektívne príznaky FEO sú spôsobené účinkom vysokých koncentrácií cirkulujúcich katecholamínov. Najdôležitejším príznakom je sekundárna artériová hypertenzia, v polovici prípadov trvalá, v tretine paroxyzmálna. Zvyšok pacientov má normotenziu. Typická labilita krvného tlaku je spôsobená charakterom sekrécie (kontinuálna, paroxyzmálna), konkrétnym typom secernovaného hormónu, prípadne sekréciou ďalších vazoaktívnych látok. Noradrenalín secernujúce tumory spôsobujú trvalú hypertenziu, kým tumory produkujúce adrenalín s kosekréciou noradrenalínu spôsobujú paroxyzmálnu hypertenziu. FEO produkujúce iba adrenalín sa môžu prejavovať naopak hypotenziou, alebo ortostatickými kolapsami na pozadí hypertenzie (1, 2, 9).
Výskyt nádorov hrubého čreva a konečníka (kolorekta) celosvetovo narastá a v krajinách strednej Európy predstavuje jednu z najčastejších malignít. Kolorektálne karcinómy sa vyskytujú v dvoch formách: sporadické a dedične podmienené, pričom väčšinu tvoria sporadické formy. Asi 5 – 10 % zo všetkých prípadov nádorov hrubého čreva a konečníka patrí k dedičným typom nádorov (10).
Lynchov syndróm (LS), známy aj ako hereditárny nepolypózny karcinóm kolorekta (HNPCC), predstavuje približne 3 % zo všetkých kolorektálnych karcinómov a je najčastejším zo syndrómov s dedičnou predispozíciou ku kolorektálnemu karcinómu (11).
Klinická diagnóza syndrómu sa stanovuje na základe interpretácie výsledkov molekulárno genetických vyšetrení. Diagnózu potvrdzuje prítomnosť mikrosatelitovej instability v tumore, strata expresie MMR génov („mismatch“ reparačný systém) stanovená imunohistochemickým vyšetrovaním (IHC) a identifikácia zárodočnej mutácie (patogénneho variantu) v niektorom z MMR génov sekvenovaním. LS je spôsobený patogénnymi germinatívnymi variantami v génoch „mismatch“ reparačného systému (MLH1, MSH2, MSH6, PMS1, PMS2, MLH3, MSH3), ktorý sa zúčastňuje na oprave chybne zaradených báz, tzv. „mismatchov“. Najčastejšie postihnutými génmi sú MLH1 (41 %), MSH2 (40 %), MSH6 (12 %) a minoritne PMS2 (7 %). Nositelia týchto patogénnych variantov majú zvýšené riziko vzniku kolorektálneho karcinómu a tiež ďalších nádorov (endometria, ovárií, žalúdka, hepato - biliárneho traktu, tenkého čreva, pankreasu, karcinómu obličkovej panvičky, mozgu a kože) (10, 11). V zahraničných periodikách pribúdajú kazuistiky so súbežným výskytom HNPCC a neuroendokrinných nádorov vrátane feochromocytómu, čo zvyšuje pravdepodobnosť, že tento typ nádoru by mohol predstavovať jeho ďalší extrakolonický prejav (12, 13). Zároveň pribúdajú aj kazuistiky súčasného výskytu adrenokortikálneho karcinómu (ACC) u pacientov s Lynchovým syndrómom. Spájajú sa so zárodočnou mutáciou (patogénnym variantom) MSH6 i MSH2 génu (14, 15). Podľa Kauura bol ACC hlásený až u 3.2 % pacientov s LS (14).
Vlastný prípad
33ročný pacient bol pôvodne vyšetrený pre enterorágiu chirurgom v rajónnej chirurgickej ambulancii. Palpačne na dosah prsta bol zachytený laločnatý tumorózny útvar rekta. Z endoskopickej excízie bol histologicky potvrdený stredne diferencovaný invazívny tubulo‑papilárny adenokarcinóm Gr II. V rámci stagingu novozisteného nádorového ochorenia sa realizovala počítačová tomografia (CT) brušnej dutiny, ktorá okrem tumoru rekta s akcentovanou lokálnou lymfadenopatiou zobrazila aj hypervaskularizovanú tumoróznu expanziu pravej nadobličky veľkosti 32 mm. Rádiológ nález hodnotil ako pravdepodobnú metastázu klinicky známeho onkologického ochorenia (pracovný klinický staging ochorenia bol T3N1M1). V pravej nadobličke bolo prítomné oválne ložisko rozmerov 32x28mm, natívnej denzity do 25 HU, s nehomogénnym postkontrastným vysycovaním –suspektná metastáza (Obr. 1, 2).
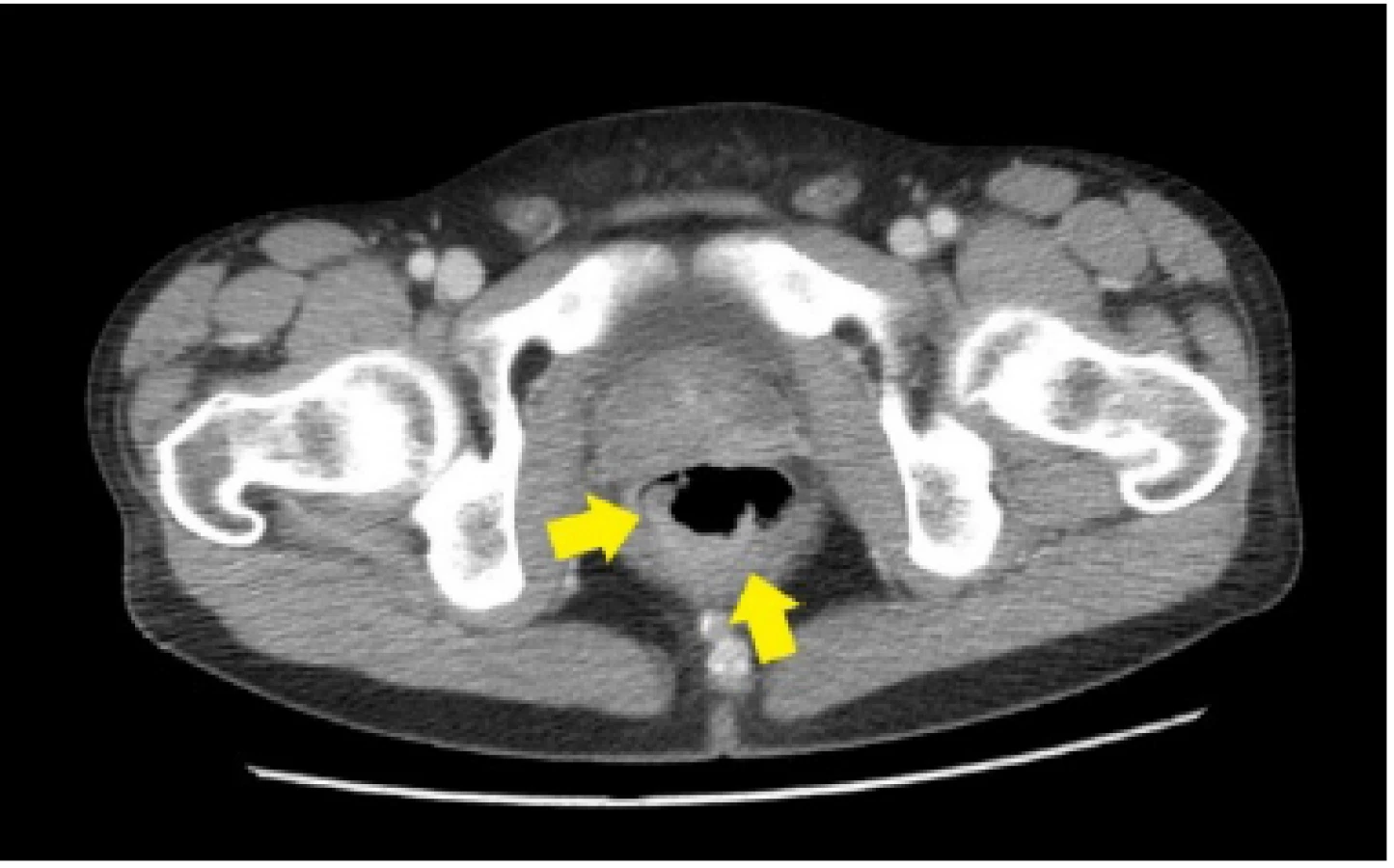
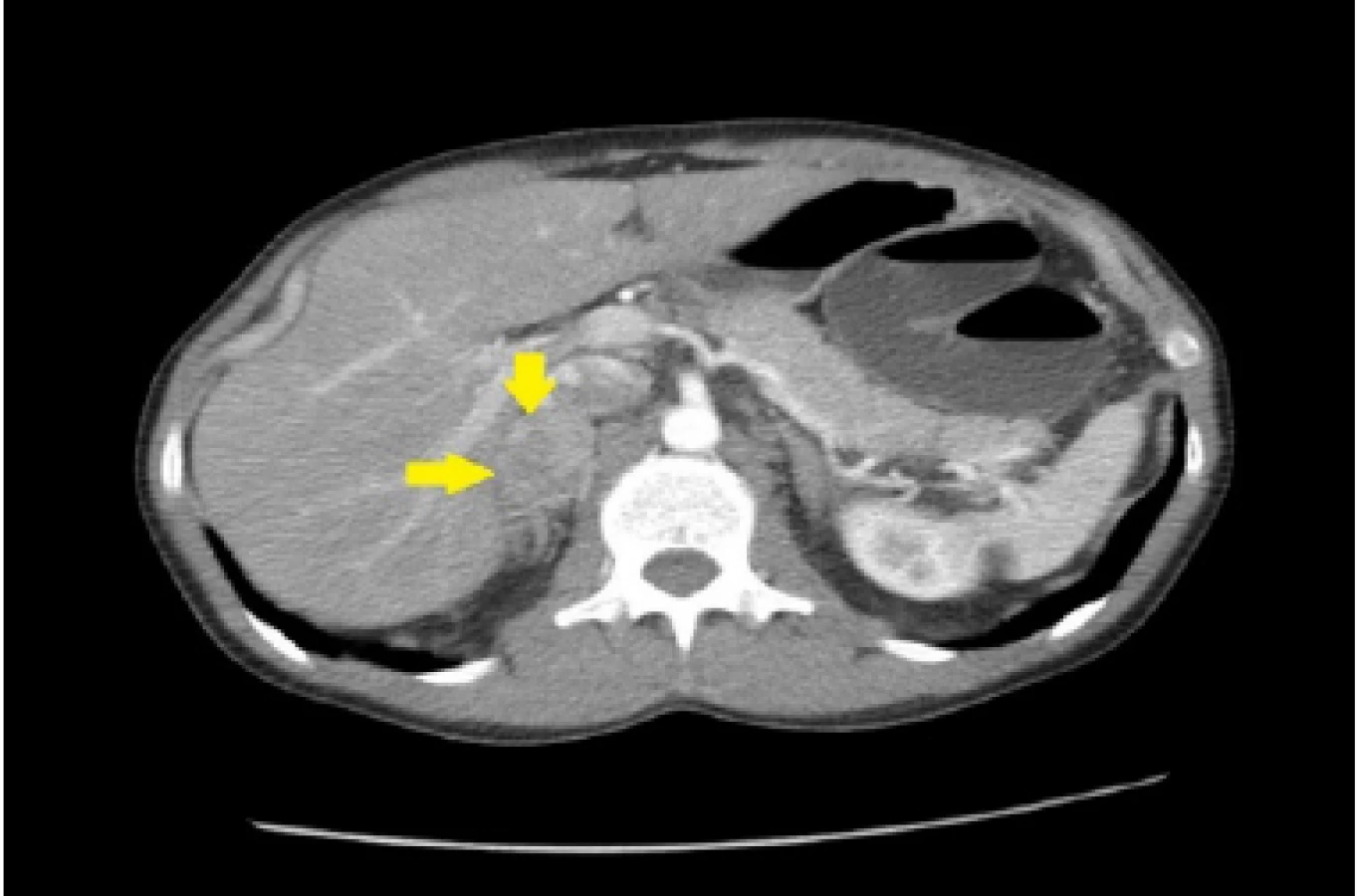
Pacienta chirurg odoslal na vyššie pracovisko za účelom zváženia neoadjuvatnej liečby. Realizované bolo onkologické konziliárne vyšetrenie v Onkologickom ústave sv. Alžbety (OUSA), odporučená predoperačná rádioterapia + konkomitantne podávaná chemoterapia (kapecitabín). Počas hospitalizácie na Klinike radiačnej onkológie bol pacient odoslaný na endokrinologické vyšetrenie v zmysle medzinárodných odporúčaní diagnostiky adrenálnych incidentalómov.
Základné anamnestické údaje neodhalili nič podozrivé, pacient doteraz nebol na nič liečený, krvný tlak mával v norme, ale bol meraný veľmi sporadicky, rodinná anamnéza bez pozoruhodností, užíval xelodu, algifen, hylak, espumisan a loperamid. Udával problém s početnými hnačkovitými stolicami (4× až 6× denne) a občasné záchvaty búchania srdca, ktoré pripisoval veľkému strachu o svoju budúcnosť. V klinickom obraze dominoval sklon k tachykardii, tremor, potenie a strach. Doplnilo sa rádiologické konziliárne vyšetrenie prinesenej CT dokumentácie, so záverom hypervaskularizovanej tumoróznej expanzie pravej nadobličky s prítomnou centrálnou nekrózou, v diferenciálnej diagnostike prichádzal do úvahy primárny tumor (nie lipid‑rich adenóm, a ani myelolipóm) alebo metastáza známeho ochorenia.
Pacientova telesná hmotnosť pri vyšetrení bola 50 kg, telesná výška 162 cm, tlak krvi 140/85mmHg, srdcová frekvencia 89/min.
V rámci endokrinologického testovania hormonálnej aktivity incidentalómu pravej nadobličky sa realizovali nasledovné vyšetrenia:
- SODÍK(*) [mmol/l] 139,0 136,0–145,0
- DRASLÍK(*) [mmol/l] 4,82 3,30–5,20
- ALDOSTERON [ng/dL] 6,5 3,7–43,2
- RENIN PRIAMY [mlU/L] 89,9 5,3–99,1, ARR 0,06
- KORTIZOL(*) [nmol/l] 405 240–620
KORTIZOL 1 mg DXM 35nmol/l - DHEAS [μmol/l] 4,6 0,96–11,5
- P‑F METANEF 426,10 ng/l (norma do 90)
- P‑F N‑METANEF 622,80 ng/l (norma do 180)
- P‑adrenalin 284,0 ng/l (norma do 84)
- p‑noradrenalin 1372,0 ng/l (norma do 420)
- P‑dopamin menej ako 30
Vzhľadom na CT nález tumoru nadobličky s jednoznačným záchytom zvýšenej hladiny adrenalínu, noradrenalínu a metanefrínov v sére sme u pacienta potvrdili prítomnosť FEO. Indikovali sme predoperačnú liečbu alfa‑blokátorom, neskôr sme pre sklon k tachykardii pridali aj malú dávku beta‑blokátora. Odporučili sme prvotne vykonať pravostrannú adrenalektómiu počas plánovanej laparotómie pred radikálnym odstránením tumoru rekta. Manipulácia s tumorom rekta s okolitými štruktúrami v brušnej dutine pri nepoznanom FEO by mohla vyvolať nečakané internistické akútne stavy a ohroziť život pacienta.
Počas veľkej brušnej operácie sa realizovala v prvom kroku adrenalektómia vpravo a následne resekcia rekta sec. Miles. Histologicky z nádoru nadobličky sa potvrdil feochromocytóm PASS score podľa „Pheochromocytoma of the Adrenal gland Scaled Score“ podľa AJSP 2002 1 bod, hodnotený bol ako benígny feochromocytóm, z tumoru rekta sa potvrdil adenokarcinóm kolonického typu.
Vzhľadom na mladý vek a výskyt duplexnej malignity pacient absolvoval molekulárno‑genetické vyšetrenie s cieľom objasnenia súvislostí onkologických ochorení s dedičnou predispozíciou. Metódou priameho sekvenovania DNA sa vyšetrili celé kódujúce sekvencie génov RET, VHL, SDHAF2, SDHB, SDHC a SDHD. Na základe údajov z medzinárodných databáz a predikčných softvérov v uvedených génoch nebola dokázaná prítomnosť patogénneho ani potenciálne patogénneho variantu (mutácie) asociovaného so zvýšeným rizikom vývoja feochromocytómu alebo paragangliómu. Z nádorového tkaniva rekta sa realizovalo vyšetrenie mikrosatelitovej instability (MSI), ktoré je vysoko senzitívnym testom v prípade podozrenia na prítomnosť hereditárneho nepolypózneho kolorektálneho karcinómu (HNPCC), ktorý je asociovaný s defektom v MMR génoch. Výsledok MSI testu bol pozitívny, DNA nádorového tkaniva vykazovala instabilitu v 4 z 5 mikrosatelitových markerov analyzovaných fluorescenčnou analýzou v porovnaní s DNA zdravého tkaniva. Imunohistochemickým vyšetrením realizovaným z tkaniva nádoru rekta bola preukázaná strata nukleárnej expresie MLH1 a PMS2 proteínu, pri súčasnej intaktnej nukleárnej pozitivite MSH2 a MSH6 proteínu. Pozitivita MSI testu ako aj nález straty nukleárnej expresie MLH1 a PMS2 v nádorových bunkách poukazovala na Lynchov syndróm. Následne bola vzorka periférnej krvi analyzovaná metódou priameho sekvenovania DNA, počas ktorého bola vyšetrená celá kódujúca sekvencia MLH1 génu. Uvedeným vyšetrením (aj opakovanou analýzou z nezávislej vzorky krvi) bola u pacienta potvrdená prítomnosť pravdepodobne patogénneho variantu c.35_46del (p. Asp12_Val16del), ktorý spôsobuje zámenu aminokyseliny v kodóne 12 s následným skrátením proteínu o 4 aminokyseliny. Uvedený variant nie je opísaný v dostupných databázach (InSight a ClinVar). Vzhľadom na charakter variantu (vedie ku skráteniu proteínu) a výsledkov IHC (strata expresie MLH1/PMS2) a MSI analýzy (MSI‑H tumor) ide vysoko pravdepodobne o patogénny variant asociovaný so zvýšeným rizikom vzniku onkologických ochorení asociovaných s Lynchovým syndrómom. Pacientovi bola na základe výsledku vyšetrenia odporúčaná pravidelná dispenzarizácia zameraná na detekciu kolorektálnych aj extrakolonických tumorov asociovaných s Lynchovým syndrómom (pravidelné kolonoskopické kontroly, gastrofibroskopické kontroly, vyšetrenia USG brušnej dutiny so zameraním na žlčové cesty, obličky a močový mechúr, vyšetrenia moču, urologický skríning zameraný na detekciu karcinómu prostaty, dermatologické kontroly a neurologické vyšetrenie). Vzhľadom na prítomnosť nálezu pravdepodobne patogénneho variantu asociovaného s rizikom vzniku hereditárnych onkologických ochorení bolo odporučené vyšetrenie konkrétneho variantu MLH1 génu aj u všetkých prvostupňových príbuzných (rodičia a sestra, následne ďalší príbuzní podľa výsledku).
Diskusia
Kazuistika poukazuje na dôležitosť hormonálneho prešetrenia každého novozisteného tumoru nadobličky. V niektorých prípadoch ani erudovaný rádiológ nemôže na základe zobrazovacieho vyšetrenia jednoznačne určiť dignitu tumoru. V prípade rádiologického nálezu onkologických pacientov je popisujúci lekár – rádiológ už ovplyvnený preexistujúcou klinickou informáciou o prítomnosti malignity. Feochromocytóm, podobne ako aj metastáza, majú v CT obraze podobné zobrazovacie charakteristiky, medzi ktoré patria: heterogénna textúra, oválny tvar, nehomogénna hustota, vysoké natívne hodnoty denzity CT (> 20 HU), oneskorený wash out (< 50 % za 10 minút), zakrvácania a nekrózy (16). Rozdielnosť býva vo veľkosti, FEO býva zvyčajne väčší a metastázy častejšie obojstranné (Tab. 1). Práve pri menších rozmeroch FEO bývajú časté rádiologické pochybnosti.
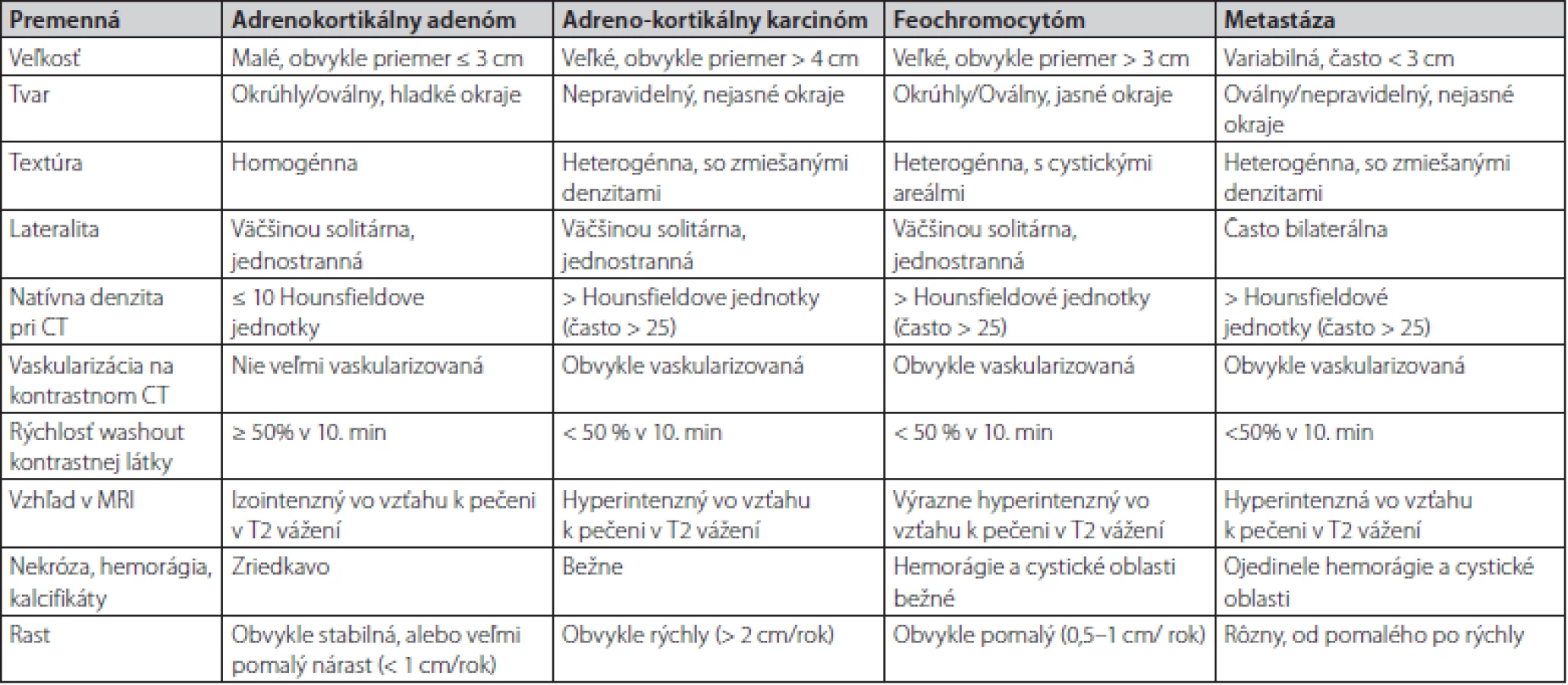
Natívne denzity adenómov sú nízke z dôvodu vyššieho obsahu lipidov v cytoplazme, zatiaľ čo natívne denzity metastáz, adrenokortikálnych karcinómov a feochromocytómov sú vyššie, pretože výskyt lipidov v týchto léziách je zriedkavý. Natívna denzita ≤ 10 HU sa všeobecne považuje za hraničnú hodnotu medzi adrenálnymi adenómami a nádormi neadénomového typu. Približne 30% adenómov má však natívnu denzitu vyššiu ako 10 HU. V takýchto prípadoch nie je možné spoľahlivo odlíšiť adenóm chudobný na lipidy (lipid poor adenoma) od feochromocytómu, adrenokortikálneho karcinómu alebo metastázy. Správna diagnóza je obzvlášť dôležitá u feochromocytómov a adrenokortikálnych karcinómov, pretože hormonálna aktivita alebo rýchle metastatické šírenie môžu viesť k významným komplikáciám a úmrtnosti. Multicentrická retrospektívna štúdia realizovaná na viacerých endokrinologických pracoviskách vrátane našeho pracoviska pod vedením MUDr. Čtvrtlíka, PhD. z roku 2019 sa zamerala na výskyt rádiologických špecifických znakov pre feochromocytómy.Na základe výsledkov štúdie sa vytvoril model výpočtu pravdepodobnosti feochromocytómu pomocou analýzy regresného modelu pomocou týchto prediktorov: maximálny priemer, tvar, prítomnosť centrálnej nekrózy a prítomnosť „kruhového znaku“. Model dosiahol vysokú citlivosť a špecifickosť identifikácie feochromocytómu 80% a 95% a spĺňal ich aj náš pacient (17).
Základom diagnostiky FEO je biochemický dôkaz zvýšenej koncentrácie metanefrínov (MNF), metylovaných degradačných produktov katecholamínov v plazme a ich zvýšeného odpadu v moči. Z ďalších markerov sa vyšetruje koncentrácia chromogranínu, ktorá je zvýšená u 91% pacientov. CT alebo MRI sú dostatočným vyšetrením pre detekciu tumoru. Ak je FEO menší ako 3 cm, súčasne má pacient menej ako 40 rokov a nemá pozitívnu rodinnú anamnézu FEO, nie sú potrebné ďalšie doplňujúce vyšetrenia v rámci diagnostiky. V nejasných prípadoch sa indikuje funkčné zobrazenie. 18 F‑DOPA PET/CT je momentálne vyšetrením prvej voľby pri podozrení na FEO. Scintigrafia s MIBG si ponecháva svoju indikáciu pri rozhodovaní o stratégii liečby u pacientov s metastatickým FEO, kde liečebné podávanie MIBG môže byť jednou z možností terapie (18).
Jedinou kauzálnou liečbou feochromocytómu je chirurgická liečba. U pacientov s dokázaným FEO treba hneď začať pre toto ochorenie špecifickú antihypertenzívnu liečbu, ktorej ťažiskom sú alfablokátory. Dávka sa postupne zvyšuje do dosiahnutia normotenzie alebo miernej hypertenzie. Pri nedostatočnom efekte, alebo pretrvávajúcej tachykardii sa pridávajú betablokátory. Ďalej možno pridať blokátory kalciových kanálov (amlodipín). V liečbe sa kladie dôraz na to, že sa vždy musí začať alfablokádou a nie betablokádou.
Chirurgická liečba je jedinou kuratívnou terapeutickou modalitou FEO. Dnes sa odporúča laparoskopická minimálne invazívna adrenalektómia. Laparoskopicky je možné operovať i pacientov s nádorom väčším ako 9 cm. Otvorená resekcia sa odporúča pre veľké alebo invazívne feochromocytómy, aby sa zabezpečila kompletnosť resekcie a prevencia ruptúry nádoru. Manažment FEO je multidisciplinárny, a mal by sa uskutočniť v centrách, kde má každý člen tímu dostatočné skúsenosti. Mimoriadne dôležitá je prítomnosť skúseného anestéziológa.
V zahraničnej literatúre sa uvádza potenciálne nová súvislosť Lynchovho syndrómu s FEO spôsobená mutáciou (patogénnym variantom) MSH6 génu (12,13). V našej kazuistike sa FEO vyskytol u pacienta s patogénnym variantom MLH1 génu. Imunohistochemické vyšetrenie MMR proteínov sme realizovali aj z tumorového tkaniva feochromocytómu, výsledkom však na rozdiel od nádorového tkaniva z tumoru rekta bola intaktná nukleárna expresia všetkých štyroch MMR proteínov (MLH1, MSH2, MSH6 aj PMS2). Uvedeným vyšetrením sa nám nepodarilo potvrdiť súvislosť feochromocytómu a Lynchovho syndrómu u nášho pacienta, môžeme zatiaľ iba konštatovať koincidenciu ochorení. V prípade nových poznatkov ohľadom kauzálnej súvislosti Lynchovho syndrómu a FEO by bolo prospešné dispenzarizáciu Lynchovho syndrómu doplniť o skríning na zachytenie včasných štádií FEO s možnosťou ich liečby a v prípade simultánneho výskytu, upraviť poradie operačných zákrokov. Zvýšený záchyt prípadov FEO u pacientov s Lynchovým syndrómom oproti bežnej populácii však môže byť spôsobený i zvýšeným využívaním zobrazovacích vyšetrení za účelom skríningu nosičov HNPCC (10, 11).
Záver
Feochromocytóm sa môže prejavovať rôznymi klinickými príznakmi v dôsledku nepretržitého alebo paroxyzmálneho uvoľňovania katecholamínov. V niektorých prípadoch sa môžu vyskytnúť závažné kardiovaskulárne komplikácie, ako sú hypertenzná kríza, srdcový infarkt, srdcové zlyhanie ale aj šokový stav. Pretože mnoho z týchto komplikácií môže byť život ohrozujúcich, jedinou prevenciou je včasná diagnostika FEO a jeho správna liečba. Kazuistika poukazuje na dôležitosť hormonálneho prešetrenia každého incidentalómu nadobličky i u onkologických pacientov. Lynchov syndróm je najčastejším zo syndrómov s dedičnou predispozíciou ku kolorektálnemu karcinómu. Každý pacient s LS vyžaduje genetickú konzultáciu, genetické testovanie MMR génov, špeciálnu zdravotnú starostlivosť zameranú na skoré rozpoznanie s LS asociovaných malignít a celoživotnú dispenzarizáciu.
V zahraničnej literatúre pribúdajú kazuistiky vzájomného výskytu LS a feochromocytómu (12, 13), podobne aj LS a adrenokortikálneho karcinómu (14, 15). Nakoľko molekulárna medicína i genetika idú dopredu míľovými krokmi, v budúcnosti sa možno odhalia spojitosti medzi nimi.
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Emília Mojtová, emojtova@ousa.sk
Endokrinologická ambulancia, Onkologický ústav sv. Alžbety, Heydukova 10, Bratislava 812 50
Článek přijat redakcí: 9. 9. 2019
Článek přijat po recenzích k publikaci: 11. 12. 2019
Sources
1. Zelinka T, Widimský J. Pheochromocytoma – Why is its early diagnosis so important for patient? | Feochromocytom – Proč je jeho časná diagnóza pro pacienta dôležitá? Vnitř Lék., 2015; 61 (5) : 487–491.
2. Pacák K, Lazúrová I. Endokrinní nádory nadledvin v současné klinické praxi. Galen; 2011 : 240 s.
3. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. Journal of Clinical Oncology, 34, 2005; 34 : 8812–8818.
4. Martucci VL, Pacak K. Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, Management and Treatment. Curr Probl Cancer, 2014; 38 : 7–41.
5. Jochmanova I, Wolf KI, King KS, et al. SDHB ‑ related pheochromocytoma and paraganglioma penetrance and genotype‑phenotype correletions. J Cancer Res Clin Oncol, 2017; 143 : 1421–1435.
6. Costa MHS, Ortiga‑Carvalho TM, Violante AD, et al. Pheochromocytomas and paragangliomas: clinical and genetic approach. Frontiers in Endocrinology. 2015, 6 : 1–9.
7. Lenders JWM, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet 2005; 366 : 665–675.
8. Jochmanová I, Lazúrová I. Diagnostika a manažment metastatického feochromocytómu a paragangliómu. Vnitř Lék, 2017; 63(9): 580–588.
9. Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2014; 99 : 1915–1942.
10. Závodná K, Vávrová Ľ, Hamidová O. Diagnostika pacientov s Lynchovým syndrómom. Onkológia, 2017; 12(6): 414–420.
11. Ilenčíková D, Bartošová Z, Babál P. Lynchov syndróm – novinky v diagnostike a liečbe. Onkológia, 2010; 5 (2): 70–77.
12. Perrier RL, Van Galen P, Pasieka JL, et al. An unusual tumor spectrum in Lynch syndrome caused by MSH6 mutation. Hereditary Cancer in Clinical Practice. 2010; 8 : 17.
13. Riff BP, Katona BW, Wilkerson M, et al. HNPCC‑associated pheochromocytoma: expanding the tumor spectrum. Pancreas 2014; 44 : 676–678.
14. Kaur RJ, Pichurin PN, Hines JM. Adrenal Cortical Carcinoma Associated With Lynch Syndrome: A Case Report and Review of Literature. Journal of the Endocrine Society, 2019; 3(4): 784–790.
15. Benjamin G. Challis, Narayanan Kandasamy, Andrew S. Powlson, et al. Familial Adrenocortical Carcinoma in Association With Lynch Syndrome, J Clin Endocrinol Metab. 2016; 101(6): 2269–2272.
16. Young WF. The incidentally discovered adrenal mass. Clinical practice. The New England journal of medicine, 2007; 356 : 601–610.
17. Čtvrtlík F, Tudos Z, Szász P, et al. Characteristic CT features of pheochromocytomas – probability model calculation tool based on a multicentric study. Biomedical Papers, 2019; 163 (3): 212–219.
18. Balogova S, Talbot JN, Nataf V, et al. 18 F‑Fluorodihydroxyphenylalanine vs other radiopharmaceuticals for imaging neuroendocrine tumours according to their type. European Journal of Nuclear Medicine and Molecular Imaging, 2013; 40 : 943–966.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2020 Issue 5
Most read in this issue
- Osteolytická ložiska, hyperkalcemie a paraprotein, ale myelom to není
- Onemocnění jater související s alkoholem (ALD)
- Chronické cholestatické jaterní choroby, primární biliární cholangitida a primární sklerozující cholangitida
- Doporučená očkování pro diabetiky