
Tangierská nemoc v rodině s fenotypem familiární hypercholesterolemie
Tangier disease in family with the phenotype of familial hypercholesterolemia
Within the project MedPed (Make Early Diagnosis to Prevent Deaths) we have examined patient with familial hypercholesterolemia in our lipid ambulance. During the following investigation of the patient‘s family we found out that her sister has on the contrary very low levels of total and LDL-cholesterol. Concentration of HDL-cholesterol was extreamly low (almost immeasurable). Differential diagnosis uttered a suspicion of rare form of familial hypoalfalipoproteinemia so-called Tangier disease. This suspicion was then confirmed by molecular genetic examination. Tangier disease is a rare lipoprotein metabolism disorder characterized biochemically by almost complete absence of plasmatic HDL- cholesterol, extremely low level of apolipoprotein A-I and accumulation of cholesterol esters in macrophages. The first case was recorded on the Tangier island in 1961. In our research we describe the first case of a patient with homozygous form of Tangier disease in the history of the Czech Republic.
Keywords:
familial hypercholesterolemia – low ApoA -I – low HDL -cholesterol – primary hypoalfalipoproteinemia – project MEDPED – Tangier disease
Authors:
Robin Urbánek 1; Lukáš Tichý 2; Tomáš Freiberger 3
Authors‘ workplace:
Lipidová a obezitologická ambulance, Zlín
1; Interní hematoonkologická klinika, Centrum molekulární biologie a genové terapie, FN Brno
2; Centrum kardiovaskulární a transplantační chirurgie. Genetická laboratoř, FN u sv. Anny Brno
3
Published in:
Vnitř Lék 2020; 66(7): 443-446
Category:
Case Report
Overview
V rámci projektu MedPed (Make Early Diagnosis to Prevent Early Deaths) byla v naší lipidové ambulanci vyšetřena pacientka s familiární hypercholesterolemií. Při dalším vyšetření rodiny byly u její sestry zjištěny naopak velmi nízké hodnoty celkového i LDL- cholesterolu a velmi nízká (prakticky neměřitelná) koncentrace HDL- cholesterolu. Diferenciálně diagnosticky bylo vysloveno podezření na vzácnou formu familiární hypoalfalipoproteinemie, tzv. Tangierskou nemoc. Toto podezření pak bylo potvrzeno molekulárně genetickým vyšetřením. Tangierská nemoc je vrozené onemocnění lipidového metabolismu charakterizované extrémně nízkou koncentrací HDL -cholesterolu, apolipoproteinu A -I a akumulací esterů cholesterolu v makrofázích. První případ Tangierské nemoci byl popsán v roce 1961 na ostrově Tangier. V naší práci popisujeme prvního pacienta s homozygotní formou Tangierské nemoci v České republice.
Klíčová slova:
familiární hypercholesterolemie – nízký ApoA -I – nízký HDL -cholesterol – primární hypoalfalipoproteinemie – projekt MedPed – Tangierská nemoc
Úvod
Tangierská nemoc (TN) (OMIM katalogové číslo #205400) je raritní vrozené onemocnění lipidového metabolismu charakterizované extrémně nízkou hladinou HDL -cholesterolu (HDL -C), extrémně nízkou hladinou apolipoproteinu A- I (ApoA- I) a akumulací esterů cholesterolu v makrofázích. Poprvé bylo toto onemocnění popsáno Fredricsonem a spol. v roce 1961 u dvou sourozenců žijících na ostrově Tangier (1). Příčinou onemocnění je mutace v genu pro ABCA1 (fosfolipidy transportující ATPasa- ATP binding cassette transporter1). U TN se klinický fenotyp přenáší autozomálně recesivně, zatímco lipidový fenotyp se přenáší kodominantně (2). Typickým biochemickým nálezem u homozygotů s TN je plazmatická hladina HDL-C pod 0,13 mmol/l, apoA -I pod 0,16 g/l, ApoA -II dosahuje 5–10 % normy. Plazmatická hladina celkového cholesterolu (T- Chol) je pod 3,9 mmol/l. Hodnota triacylglycerolů (Tg) je v plazmě buď normální, nebo jen mírně zvýšena v rozmezí 2,0–4,5 mmol/l (2, 3, 4). Extrémně nízké hodnoty HDL-C a ApoA -I nejsou běžným nálezem v naší klinické praxi. Tento nález vždy vyžaduje podrobné vyšetření, zda se jedná o primární či sekundární příčinu. U naší pacientky byla potvrzena diagnóza TN, která představuje jednu z nejtěžších forem familiární hypoalfalipoproteinemie. U dvou členů její rodiny byl velmi zajímavý laboratorní nález typický pro familiární hypercholesterolemii.
Tangierská nemoc
TN byla poprvé popsána Fredricksonem a jeho kolegy v roce 1961 u 5letého chlapce žijícího na ostrově Tangier v zálivu Chesapeake, 13 mil vzdáleného od státu Virginie (USA). Podstoupil tehdy tonsilektomii, při které mu byly odstraněny hypertrofické, oranžovo -žlutavě zbarvené mandle. Stejně zbarvené tonsily pak byly zjištěny i u jeho starší sestry. Navíc měli oba mírnou hepatomegalii, opacity v oční rohovce a makrofágy bohaté na cholesterol byly nalezeny i mimo tonsily (kostní dřeň, periferní nervová tkáň a buňky hladkého svalstva). Co však bylo zásadní, u obou sourozenců byla téměř neměřitelná hodnota HDL- C v plazmě. Rodiče těchto dětí měli hladiny HDL- C asi na poloviční hodnotě normálu (1, 5, 6, 7). Příčinou TN je mutace v genu pro ABCA1 (ATP- binding cassette transporter1); GenBank accession no. NP_005493, který se nachází na chromozomu 9q22-q31. ABCA1 je transmembránový protein, který hraje klíčovou roli v effluxu volného cholesterolu z makrofágů tkání a cévní stěny do nativně vznikajících HDL částic. Na biochemické úrovni jsou pro TN typické extrémně nízké hladiny HDL -C a ApoA -I. Na buněčné úrovni se estery cholesterolu hromadí v tkáních bohatých na makrofágy. U homozygotní TN s recesivním přenosem se jedná o mutaci obou alel genu pro ABCA1 (2). Homozygoti s TN mají v plazmě přítomny pouze pre beta-1 HDL částice, zatímco heterozygoti mají nedostatek velkých alfa-1 a alfa-2 HDL částic a normální hladiny prebeta-1 HDL (5, 6, 7). Heterozygotní pacienti s mutací jedné alely ABCA1 vykazují intermediární fenotyp nízkého HDL -C, označovaný jako familiální HDL -C deficience a jejich ABCA1 zprostředkovaný efflux cholesterolu z buněk je pouze 50 %. Klinický obraz nedostatečného effluxu cholesterolu z buněk není u nich tak závažný (8, 9). Předpokládá se, že nedostatečná absorpce cholesterolu z buněk pomocí HDL částic vede u homozygotů s TN k relativnímu obohacení jádra LDL beta -karotenem a po jejich zvýšeném vychytávání retikuloendoteliálními buňkami tak dochází k typickému oranžovému zbarvení těchto tkání. (7). S výjimkou malých populací „zakladatelů“ (ostrov Tangier, Virginie) je TN velmi vzácná. Na základě evidence frekvencí alel s tzv. variantou loss of function dle databáze ExAC (1/400 jedinců s heterozygotní formou TN) se odhaduje prevalence homozygotní formy Tangierské nemoci na 1/640 000 (2, 10). Skutečný počet nositelů homozygotní formy TN v světě není přesně znám. Podrobně se tomu věnují jen dva autoři. V první práci Schaefer a spol. uvádí 185 nositelů homozygotní formy TN (7). O dva roky později publikuje podobný přehled Muratsu a ten udává jen 133 nositelů TN na světě (11). Obojí jen potvrzuje extrémně raritní výskyt této nemoci. Až do současnosti bylo popsáno celkem 236 ABCA1 mutací u pacientů s TN (missense, nonsense, inserce a delece) (2, 4, 12, 13) (http://www. hgmd.cf.ac.uk/ac/index.php).
Typickým biochemickým nálezem u homozygotů s TN je plazmatická hladina HDL- C pod 0,13 mmol/l, apoA- I pod 0,16 g/l, ApoA- II dosahuje 5–10% normy. Plazmatická hladina T- Chol je pod 3,9 mmol/l.
Hodnota Tg je v plazmě buď normální, nebo jen mírně zvýšena v rozmezí 2,0–4,5 mmol/l (2–4). Toto je zcela v souladu s laboratorními hodnotami naměřenými u naší pacientky. V klinickém obraze jsou pro TN typickým nálezem hyperplastické, oranžově- žluté tonsily, hepatosplenomegalie, lymfadenopatie, zákal rohovky, trombocytopenie, anémie, stomatocytoza a zvýšené riziko ischemické choroby srdeční (14). Periferní neuropatie, demyelinizační či axonální se nachází u více jak 50 % pacientů s TN. Velmi vzácným bývá nález syringomyelia- like neuropatie vedoucí k úbytku svaloviny v oblasti horní části trupu včetně obličeje, která přechází postupně na dolní končetiny a je spojena s únavou a kachektizací (15, 16). U každého pacienta s TN se však mohou tyto klinické znaky různě manifestovat, kombinovat, nebo i chybět. (17). V literatuře existuje významný spor o tom, zda jsou pacienti s homozygotní TN ohroženi výskytem předčasné ischemické choroby srdeční (ICHS). V dřívějších pracích se popisuje u homozygotních či složených heterozygotních nositelů nemoci starších 30 let šestkrát vyšší riziko ICHS (18), zatímco u heterozygotů TN je frekvence výskytu ICHS trojnásobná (9). Zajímavý vztah popsali u svých 185 pacientů Schaefer a spol. Uvádí dva hlavní typy pacientů s homozygotní či složenou heterozygotní formou TN. První skupinu tvořili pacienti se značnou hepatosplenomegalií, anémií a nízkými hladinami non- HDL- C (< 1,8 mmol/l). U této skupiny pacientů předčasný výskyt ICHS pozorován nebyl. Druhou skupinu pak představovali pacienti bez výrazné hepatosplenomegalie nebo anémie, s normálními nebo téměř normálními hladinami non- HDL-C (> 1,8 mmol/l) a u nich byl pozorován předčasný výskyt ICHS. Autor se domnívá, že přítomnost nebo nepřítomnost výrazné splenomegalie a různé hladiny non- HDL- C se zdají být odpovědny za variabilitu rizika ICHS u homozygotních pacientů s Tangierskou chorobou (7). Nicméně vloni publikovaný konsensus panel o raritních nemocech považuje riziko předčasného výskytu aterosklerotické ischemické choroby srdeční u TN stále za kontroverzní (19).
Kazuistika
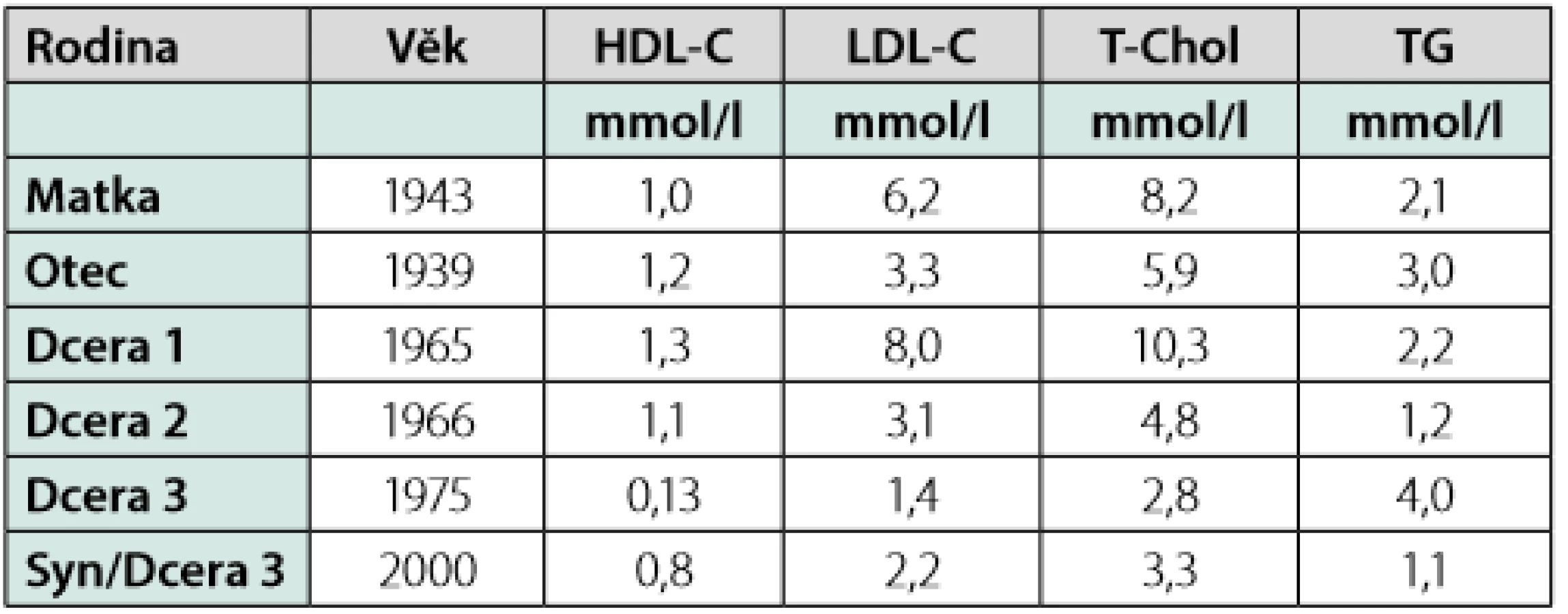
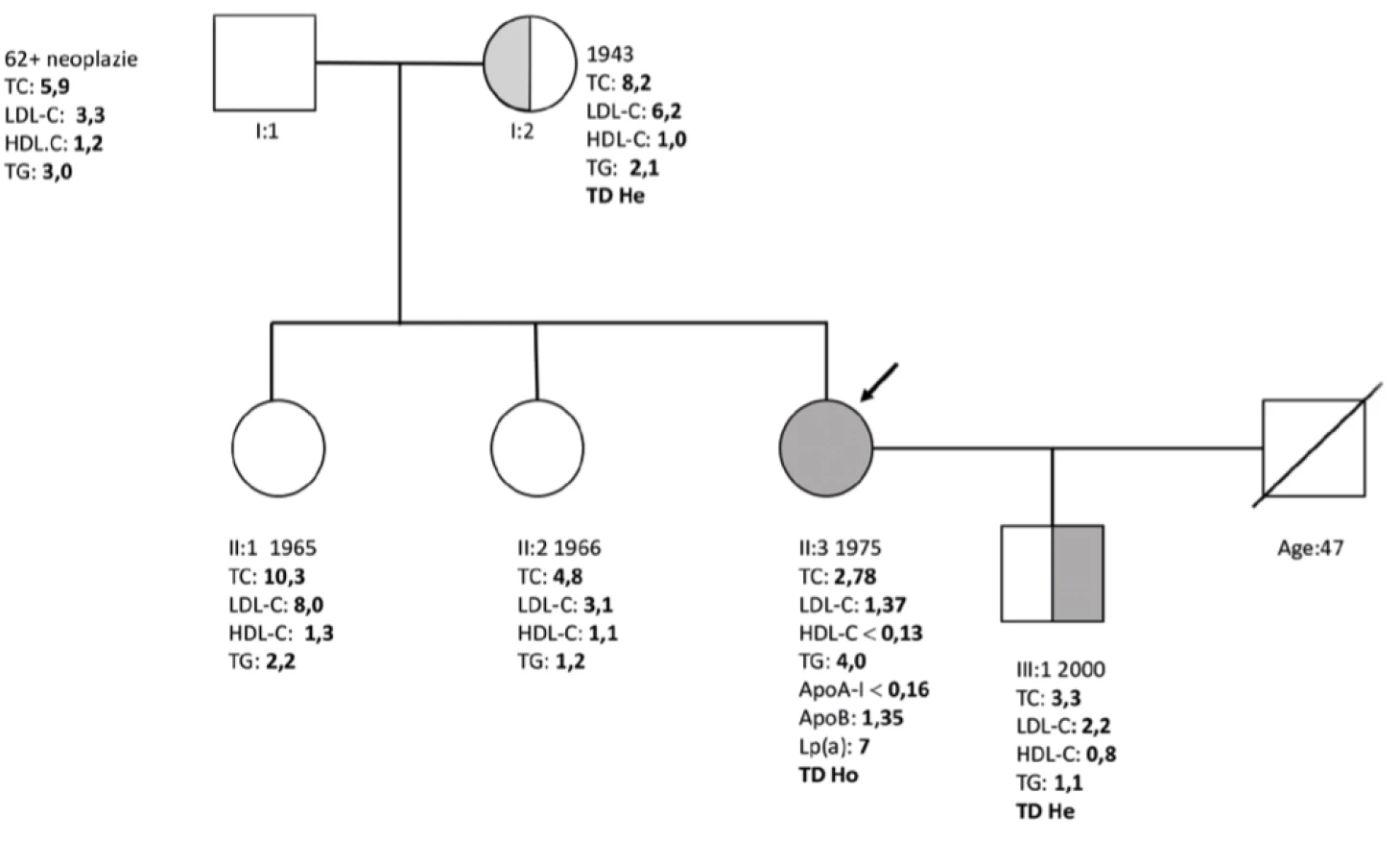
Do lipidové ambulance byla poslána žena (1965) k dispenzarizaci pro těžkou hypercholesterolemii. Pozvali jsme poté k vyšetření i její matku (1943) a dvě sestry (1966, 1975). U matky byl podobný laboratorní nález. Lipidový profil u jedné ze sester (1966) byl zcela v normě. Žádná z výše uvedených pacientek neměla klinické známky aterosklerózy. Co nás však překvapilo, byl lipidový profil u nejmladší z tří sester (1975), u které byla zjištěna téměř neměřitelná koncentrace HDL -C. Všechna měření byla provedena na analyzátoru Modular SWA (Roche, Švýcarsko) s použitím komerčně dostupných kitů. Hodnota LDL- C byla vypočítána dle Friedewaldovy rovnice. Lipidový profil celé rodiny je podrobně uveden v (Tab. 1, Graf 1).
Naše další pozornost se zaměřila hlavně na pacientku (1975). V osobní anamnéze byla uvedena tonsilektomie v 8 letech. Při fyzikálním vyšetření jsme nenalezli u pacientky žádnou patologii. Pouze při ultrazvuku karotid bylo oboustranně v oblasti bifurkace popsáno ztluštění IMT (1,2 mm) a semicirkulární homogenní plát 3 mm v levé bifurkaci. Extrémně nízké hodnoty HDL- C spolu s ApoA- I nás vedly k podezření na geneticky podmíněné onemocnění metabolismu HDL. V rámci diferenciální diagnostiky jsme zvažovali nejčastější primární či sekundární příčiny spojené s nízkou hladinou HDL -C. Mezi nejčastější sekundární příčiny uváděné v literatuře patří malignity, sekundární dyslipidemie, paraproteinemie (mnohočetný myelom), těžká zánětlivá onemocnění, malabsorpce, malnutrice, konečné stadium renálního selhání, infekce, cystická fibróza, hypothyreoza, obezita, pohybová inaktivita, hypertriglyceridemie, diabetes mellitus 2. typu, léčba glukokortikoidy, anabolické steroidy, estrogeny, probucol, vysoké dávky thiazidových diuretik, vysoké dávky beta- blokátorů, kouření a abúzus alkoholu (3, 16, 20). Všechny výše vyjmenované patologické jednotky byly postupným vyšetřením krok za krokem vyloučeny.
V metabolismu HDL- C hrají zásadní roli tři klíčové geny, a to gen pro ApoA -I, ABCA1 a gen pro LCAT (lecithin cholesterol acyl transferáza). Extrémně nízké hodnoty HDL- C a ApoA- I vedly k vysokému podezření na primární, monogenetické onemocnění, jako jsou deficit ApoA -I, řada tzv. missense mutací ApoA- I (např. ApoA- I Milano, ApoA- I Paris a další), Tangierská nemoc, familiární HDL deficit či familiární/částečný deficit LCAT (7, 20). Zejména typický laboratorní nález nás však od začátku vedl k podezření na Tangierskou nemoc. Ke stanovení této diagnózy jen laboratorní hodnoty nestačí, diagnózu může potvrdit jedině molekulárně genetické vyšetření. Byl odebrán krevní vzorek, nejen u pacientky, ale také u její nejstarší sestry (1965) a matky. Informovaný souhlas byl získán, jak je zvykem u pacientů vyšetřovaných v rámci MedPed projektu (21). U otce nebylo možné provést stejné vyšetření, neboť zemřel v roce 2006. K dispozici byl jen jeho lipidogram z roku 2005, viz Graf 1.
Provedené molekulárně genetické vyšetření neprokázalo žádnou mutaci v genu LCAT a ApoA- I. Naopak, suspektní diagnóza TN byla potvrzena nálezem dvou patogenních sekvenčních variant v genu kódujícím ABCA1. U pacientky byly nalezeny dvě doposud nepopsané mutace c.1789C>T; p. (Gln597*) a c.4449delG; p. (Leu1484Cysfs*17). Kauzalita mutací byla posouzena na základě hodnocení dle ACMG kritérií a obě varianty byly vyhodnoceny jako patogenní. Jde tedy o složeného heterozygota TN (22). Stejný typ mutace c.4449delG; p. (Leu1484Cysfs*17) byl nalezen také u její matky, nikoli však u starší sestry (1965). U matky byla navíc nalezenea jedna varianta neznámého významu v genu pro APOB (heterozygot), c.4449delG; p. (Leu1484Cysfs*17).
Diskuze
U ženy (1975) jsme molekulárně genetickým vyšetřením prokázali složeného heterozygota pro mutaci genu ABCA1. Odpovídal tomu i typický laboratorní nález téměř neměřitelných hodnot HDL- C a ApoA- I, který je prakticky identický s laboratorní diagnostikou TN (2–4).
Nejtypičtějším nálezem u homozygotů TN jsou zbytnělé, oranžovo-žlutě zbarvené tonsily. Tento nález jsme však u naší pacientky nemohli potvrdit, protože již v dětství prodělala tonsilektomii a operační protokol se nepodařilo dohledat. Co však bylo zajímavé, pacientka nevykazovala žádné klinické znaky typické pro TN. Nebyla přítomna hepato- či splenomegalie. Pacientka měla normální krevní obraz bez stomatocytózy, anémie či trombocytopenie. Neurologické vyšetření zaměřené na periferní či jiné formy neuropatií uváděné v souvislosti s TN bylo negativní. Oftalmologické vyšetření bylo zcela v normě, bez známek opacit typických pro TN. Jedinou patologii představoval semicirkulární homogenní AS plát v levé bifurkaci, který můžeme považovat za známky preklinické aterosklerózy (9, 18, 19). I přes určitou kontroverzi ohledně rizika předčasné ICHS u pacientů s TN jsme se na základě tohoto nálezu rozhodli pro hypolipidemickou léčbu (rosuvastatin 20 mg), přestože měla velmi nízkou hodnotu LDL- C (1,37 mmol/l). Obecně lze tedy řící, že klinický nález byl velmi netypický pro TN, ale i toto je v souladu s literaturou. Fenotyp homozygota s TN může být u jeho nositelů velmi variabilní a řada typických klinických znaků může chybět (17). Naše pacientka je složeným heterozygotem pro dvě sekvenční varianty v genu kodujícím ABCA1(c.1789C>T; p. (Gln597*) a c.4449delG; p. (Leu1484Cysfs*17). U pacientky se jedná o dvě doposud nepopsané mutace. Kauzalita mutací byla posouzena na základě vyhodnocení ACMG kriterií a obě varianty byly vyhodnoceny jako patogenní. Transpozice těchto variant byla potvrzena při genetickém vyšetření ostatních členů rodiny. Tzv. nonsense či frameshift mutace představují obvykle těžší formu mutace (22). Stejný typ mutace (c.4449delG; p. (Leu1484Cysfs*17) jedné z alel genu pro ABCA1 byl nalezen také u její matky(1943), nikoli však u sester (1965, 1966). Doplnili jsme i laboratorní vyšetření syna pacientky s TN (2000), kde byl očekávaně laboratorní nález typický pro heterozygotní formu TN (Tab. 1, Graf 1).
Pro co však nemáme zatím vysvětlení, je klinický nález těžké hypercholesterolemie u matky (1943) a jedné ze sester (1965). U matky
byl použit běžný panel molekulárně genetických vyšetření s 97 geny, u kterých je známa spojitost s dyslipidemií. Nenalezli jsme žádnou známou sekvenční variantu, která by byla jasně zodpovědná za její laboratorní fenotyp. Byla nalezena pouze jedna varianta neznámého významu v genu pro ApoB (heterozygot), c.4449delG; p. (Leu1484Cysfs*17). Domníváme se, že tato varianta není kauzální, neboť není lokalizována v tzv. ApoB „hot spot“ oblasti a vedle toho je nosičem této varianty i naše pacientka s TN. Její LDL- C není při tom zvýšen. V literatuře existují případy heterozygotních jedinců s mutací ABCA1, kteří mají i mírně zvýšené hladiny cholesterolu (https://www.ncbi.nlm.nih.gov/pubmed/11086027 a https://www.ncbi.nlm.nih.gov/pubmed/30503498). Žádný z těchto pacientů však neměl tak těžký fenotyp hypercholesterolemie jako matka naší pacientky s TN. Zdá se, že současná přítomnost hypercholesterolemie není pro pacienty s Tangierskou nemoci typická, ale vzácné výjimky u heterozygotních nosičů s mutací ABCA1 existují.
Závěr
V naší práci popisujeme kazuistiku pacientky s velmi raritním onemocněním metabolismu lipidů, tzv. Tangierskou nemocí. Jedná se o první pacientku s TN diagnostikovánou v České republice. Diagnóza byla stanovana na základě velmi suspektního laboratorního nálezu a následně pak potvrzena molekulárně genetickým vyšetřením. Při něm byly navíc zjištěny dvě doposud nepopsané mutace genu ABCA1. Extrémně nízké hladiny HDL- C nepatří k běžnému laboratornímu nálezu v našich ambulancích. Pokud se však objeví, je třeba pomýšlet i na vzácná, monogeneticky vázaná onemocnění. Díky screeningovému vyšetřování pacientů v rodinách s familiární hypercholesterolemií (projekt MedPed) se tak podařilo jedno takové raritní onemocnění objevit. Vzhledem k přítomné preklinické ateroskleroze jsme zahájili hypolipidemickou léčbu.
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Robin Urbánek,
Lipidová a obezitologická ambulance Zlín,
www.obezita-vyziva.cz
Cit. zkr: Vnitř Lék 2020; 66(7): 443–446
Článek přijat redakcí: 19. 3. 2020
Článek přijat po recenzích k publikaci: 27. 8. 2020
Sources
1. Fredrickson DS, Altrocchi PH, Avioli L, et al. Tangier Disease- combined clinical staff conference at the National Institute of Health. Ann Intern Med 1961; 55: 1016.
2. Puntoni M, Sbrana F, Bigazzi F, Sampietro T. Tangier disease: epidemiology, pathophysiology and management. Am J Cardiovasc Drugs 2012 (5): 303–313.
3. Luchci T, Calandra S, Rabacchi C, Conti G, Ardolino G, Assolari L, Arosio B, Vergani C. A man with low cholesterol and weakness of the lower limbs: Intern Emerg. Med (2014) 9: 449–453.
4. Tall AR, Breslow JL, Rubin EM. Genetic disorders affecting plasma high- density lipoproteins. In: Scriver CR, Beaudot AL, Valle D, Sly WS (eds) The metabolic and molecular bases of inherited disease, 8th edition New York, Mc Graw -Hill, 2001, pp 2915–2936.
5. Santos RD, Asztalos BF, Martinez LR, et al. Clinical presentation, laboratory values, and coronary heart disease risk in marked high- density lipoprotein- deficiency states. J Clin Lipidol. 2008; 2: 237–247.
6. Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010; 21: 289–297.
7. Schaefer EJ, Anthanont P, Diffenderfer MR, Polisecki E, Asztalos BF. Diagnosis and tretment of high density lipoprotein deficiency. Prog Cardiovasc, DiS. 2016 Sep- Oct; 59(2): 97–106.
8. Fasano T, Zanoni P, Rabacchi C, Pisciotta L, Favari E, Adorni MP, Deegan PB, Park A, Hlaing T, Feher MD, Jones B, Uzak AS, Kardas F, Dardis A, Sechi A, Bembi B, Minuz P, Bertolini S, Bernini F, Calandra S. Novel mutations of ABCA1 transporter in patients with Tangier disease and familial deficiency. Mol Genet Metab. 2012 Nov; 107(3): 534–541.
9. Clee SM, Kastelein J, van Dam M, Marcil M, Roomp K, Zwarts KY, Collins JA, Ceska R, Stulc T,Roelants R, Tamasawa N, Frohlich J, Hayden MR, et al. Age and residual cholesterol efflux affect HDL cholesterol levels and coronary artery disease in ABCA1 heterozygotes, J.Clin Invest 106 (2000) 1263–1270.
10. Hooper AJ, McCormick SPA, Hegele RA, Burnett JR. Clinical utility gene card for: Tangier disease.Europeran Journal of Human Genetics (2017)25.
11. Muratsu J, Koseki M, Masuda D,Yasuga Y, Tomoyama S, Keiji S,Ataka K, Yagi Y, Nakagawa A, Hamada H, Fujita S, Hattori H, Ohama T, Nishida M, Hiraoka H, Matsuzawa Y, Yamashita S Accelerated atherogenicity in Tangier disease. A case accompanied by extensive atherosclerotic lesions, Leriche syndrom and bleeding tendency, and Review of the literature. J Atheroscler Thromb 2018 Oct 1; 25(10): 1076–1085.
12. Stocchi L, Giardina E, Varriale L, Sechi A, Vagnini A, Parri G, Valentini M, Capalbo M. Can Tangier disease cause male infertility? A case report and an overview on genetic causes of male infertility and hormonal axis involved. Molec.Gene. and Metab 123 (2018) 43–49.
13. Brooks- Wilson A, Marcil MClee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne- Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frochlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR. Mutations in ABCA in Tangier disease and familial high -density lipoprotein deficiency, Nat Genet 22 (1999) 336–345).
14. Hobbs HH, Rader DJ. ABCA1: connecting yellow tonsils, neuropathy, and very low HDL. J Clin Invest.104 (1999) 1015–1017.
15. Petrini V, Rizzuto N, Vergani C, et al (1985) Neuropthy in Tangier disease: a clinicopathologic study and a review of the literature. Acta Neurol Scand 72(5): 495–505.
16. Rader DJ, deGoma EM. Aproach to the patient with extremely low HDL -cholesterol, J Clin Endocrinol Metab, (2012),97 (10): 3399–3407.
17. Negi SI, Brautbar A, Virani SS, Anand A, Polisecki E, Asztalos BF et al. A novel mutation in the ABCA1 gene causing an atypical phenotype of Tangier disease. J Clin Lipidol 7 (2012) 82–87.
18. Oram JF, Vaughan AM. ATP- Binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006; 99(10): 1031–1043.
19. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CHJ, Calabresi L, Chapman MJ, Cuche Ml, Eckardstein A, Frikke -Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer HG, Raal FJ, Ray KK, Remaley AT, Stock JK, S Stroes ES, Tokgözoğlu L, Catapano AL. Rare dyslipidemias, from phenotype to genotype to management: a European Atherosclerosis Society task force concensus statement. Lancet Diabetes Endocrinol 2019 S2213–8587(19)
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2020 Issue 7
Most read in this issue
- Hypoglykemie u nediabetiků
- Metformin -asociovaná laktátová acidóza
- Koagulopatie asociovaná s onemocněním COVID-19
- CAR T-lymfocyty: horká novinka v léčbě nádorů