
Branchio–oto–renální syndrom
Branchio–Oto–Renal Syndrome
Branchio-oto-renal syndrome is a disease characterized by branchial malformations, anomalies of external, middle or internal ear and kidney damage. The disease inheritance is autosomal dominant with very variable expressivity in clinical picture even in a single family.
The authors present a case report of a mother and child with different expressivity of clinical picture. The mother fulfils the criteria of branchio-oto-renal syndrome, the child that of the branchio-oto syndrome. The diagnosis of the disease was established on the basis of a clinical examination. The genetic analysis of this syndrome is not available in the Czech Republic. The diagnosis of the mother was established at her adult age, when the ear defect was searched for and bilateral partial hearing loss was determined. Based on the family anamnesis the authors searched specifically for ear and kidney damage, which made it possible to detect the hearing loss in the child and compensate the insufficiency with a hearing aid.
Key words:
branchio-oto-renal syndrome branchio-oto syndrome, genetic examination.
Authors:
L. Školoudík 1; D. Kalfeřt 1; J. Růžička 1; J. Kopřiva 2
Authors‘ workplace:
Klinika ušní, nosní a krční LF UK a FN, Hradec Králové
; přednosta prof. MUDr. V. Chrobok, CSc., Ph. D.
Radiologická klinika LF UK a FN, Hradec Králové
1; přednosta prof. MUDr. P. Eliáš, CSc.
2
Published in:
Otorinolaryngol Foniatr, 60, 2011, No. 2, pp. 99-102.
Category:
Case Reports
Overview
Branchio-oto-renální syndrom je onemocnění charakterizované branchiálními malformacemi, anomáliemi zevního, středního nebo vnitřního ucha a ledvinným postižením. Onemocnění je autosomálně dominantně dědičné s velmi variabilní expresivitou klinického obrazu i v rámci jedné rodiny.
Autoři prezentují kazuistiku matky a dítěte s rozdílnou expresivitou klinického obrazu. Matka splňuje kritéria branchio-oto-renálního syndromu, dítě branchio-otického. Diagnóza onemocnění je stanovena na podkladě klinického vyšetření. Genetická analýza tohoto syndromu není v ČR dostupná. U matky byla diagnóza stanovena až v dospělosti, kdy byla pátráná po ušním postižení a zjištěna oboustranná středně těžká nedoslýchavost. U dítěte se na základě rodinné anamnézy cíleně pátralo po ušním a ledvinném postižení, což umožnilo včasný záchyt nedoslýchavosti dítěte a kompenzaci sluchové vady sluchadlem.
Klíčová slova:
branchio-oto-renální syndrom, branchio-otický syndrom, genetické vyšetření.
ÚVOD
Branchio-oto-renální syndrom je autosomálně dominantní onemocnění, charakterizované malformacemi zevního, středního nebo vnitřního ucha, branchiálními malformacemi a ledvinovými anomáliemi. Syndrom má výrazně variabilní expresivitu klinického obrazu i v rámci jedné rodiny. Postižení mívají percepční, ale i smíšenou nebo čistě převodní nedoslýchavost, malformace ušního boltce a cysty či píštěle v oblasti 2. žaberní štěrbiny.
Postižení ledvin může být od mírné hypoplazie bez funkčního postižení až po oboustrannou renální agenezi. Do skupiny branchio-oto-renálního spektra patří i syndrom branchio-otický s absencí renálního postižení.
KAZUISTIKY
V srpnu 2010 byl na Kliniku ušní, nosní a krční FN Hradec Králové přijat 18měsíční chlapec s postupně narůstajícím zduřením vpravo na krku v typické lokalizaci pro laterální krční cystu (obr. 1).
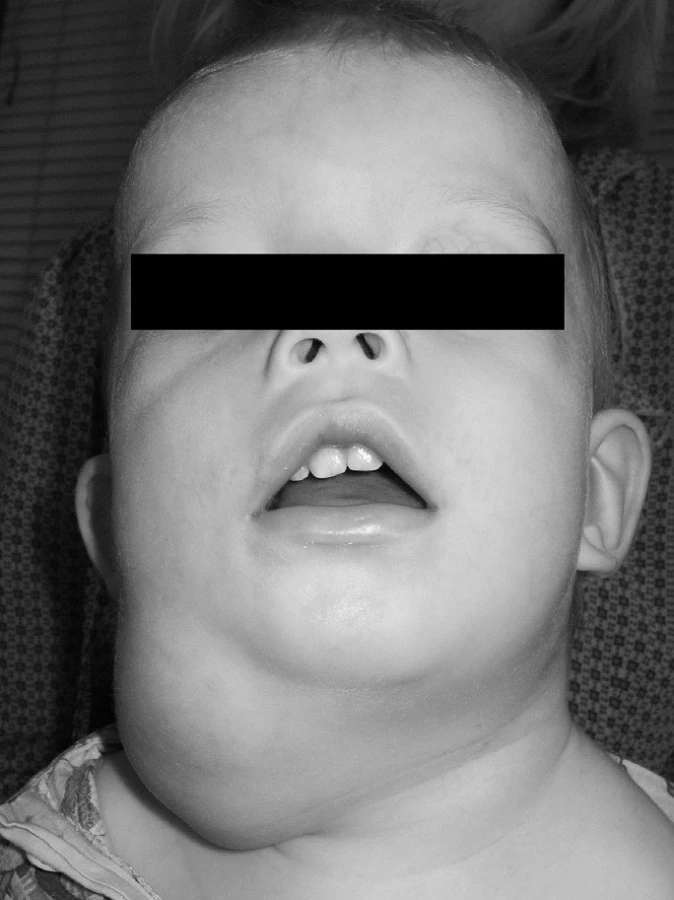
Matkou dítěte je 36letá žena, která je sledována nefrologem pro vrozenou vývojovou vadu ledvin. V dětském věku podstoupila opakované operace na Chirurgické klinice FN v Hradci Králové pro refluxní nefropatii s hypoplastickou, téměř afunkční, levou ledvinou a operace oboustranných krčních a preaurikulárních píštělí. Branchiální krční píštěle byly operovány exstirpací zevní části píštěle bez preparace jejího dalšího průběhu, bez tonzilektomie. Po operaci nedošlo k recidivě píštěle. Od narození hůře slyší, ale nikdy nebyla vyšetřena otolaryngologem.
Po přijetí na kliniku bylo doplněno audiometrické vyšetření matky prokazující oboustranně symetrickou percepční nedoslýchavost se ztrátami 20-60 dB s maximem v nízkých frekvencích (obr. 2).
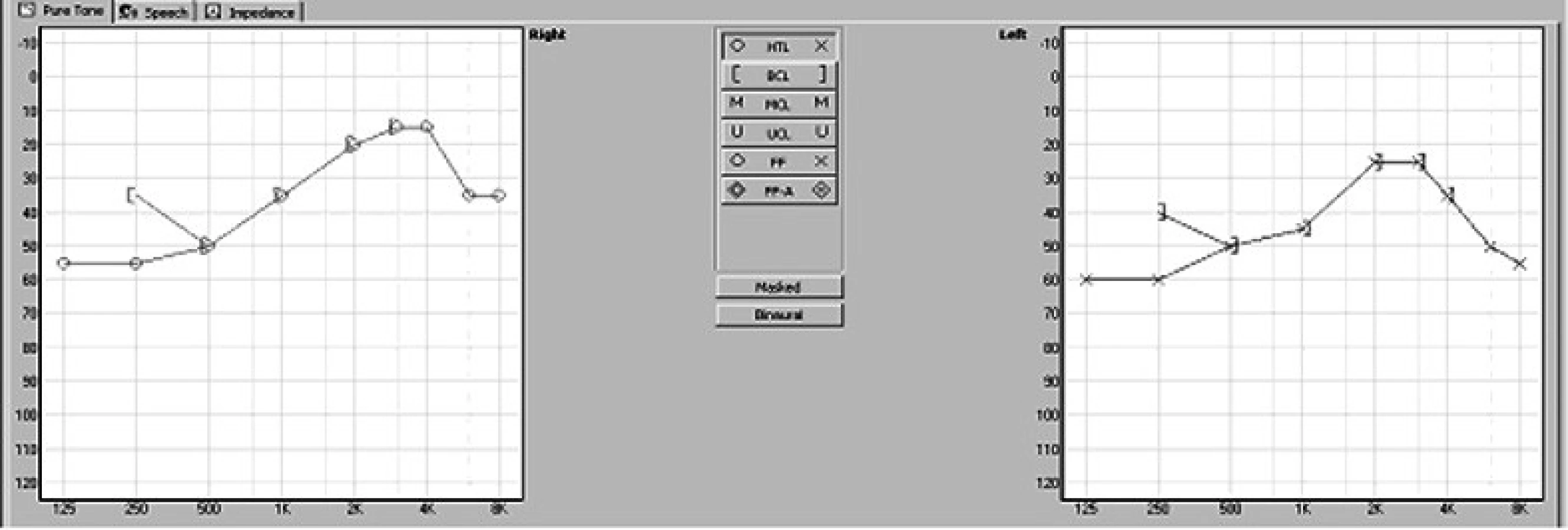
Matka splňuje klinická kritéria branchio – oto–renálního syndromu.
Dítě mělo od narození zjevné zevní ústí laterální krční píštěle oboustranně ve střední části kývače. Vlevo byla ze zevního ústí patrná intermitentní hlenovitá sekrece. Vpravo nebyla sekrece nikdy pozorována, avšak v úrovni zevního ústí píštěle postupně narůstala nebolestivá rezistence. Poslední 4 dny před přijetím k hospitalizaci došlo k akceleraci růstu krční rezistence se zarudnutím, palpační bolestivostí a febriliemi. Ultrazvukové vyšetření potvrdilo ohraničenou dvoukompartmentovou expanzi vpravo na krku velikosti 35x26 mm, centrálně bez detekovatelných dopplerovských toků se zahuštěným obsahem. Vedlejším nálezem byly oboustranné nesecernující preaurikulární píštěle. Dítě nemělo žádnou pravidelnou medikaci, nebylo sledováno pro žádné chronické onemocnění.
Dítě bylo přijato k hospitalizaci k antibiotické terapii zánětlivě změněné laterální krční cysty vpravo. K objasnění rozsahu a vnitřního ústí píštěle byla provedena RTG fistulografie levostranné píštěle. Vyšetření potvrdilo píštěl ústící do hltanu v místě tonzilárního lůžka (obr. 3).
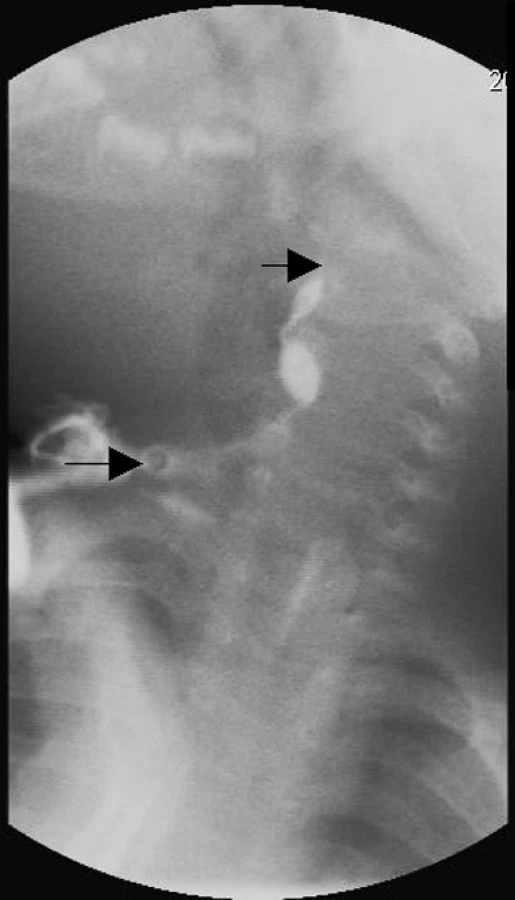
Po zklidnění akutního zánětu byla indikována operace branchiogenní malformace vpravo. Vzhledem k výsledku fistulografie vlevo jsme předpokládali obdobné vnitřní ústí i pravostranné branchiogenní malformace, kde uzavření zevního ústí píštěle vedlo ke vzniku cysty. Při operaci bylo postupováno od zevního ústí k cystickému útvaru a byl vypreparován provazec vedoucí k tonzilárnímu lůžku. Transorálně byla provedena tonzilektomie s identifikací vnitřního ústí píštěle a odstranění vnitřní porce branchiogenní malformace. Operační výkon v celkové anestezii i pooperační průběh byl bez komplikací. Dle histologického vyšetření byla píštěl, včetně cystické části,vystlána nerohovějícím dlaždicobuněčným epitelem.
Vzhledem k rodinné anamnéze bylo pomýšleno na branchio-oto-renální syndrom, proto bylo doplněno vyšetření ledvin a sluchu. Dle ultrazvukového vyšetření nebyly nalezeny žádné renální malformace. Tranzientní evokované otoakustické emise byly oboustranně nevýbavné. Tympanometrické vyšetření prokazuje vpravo B křivku, vlevo A křivku s nevýbavným stapediálním reflexem. Vyšetření BERA svědčí pro sluchový práh vpravo na hladině intenzity 60 dB, vlevo 40 dB. Po dobu následujících 3 měsíců byly opakovaně potvrzeny ploché B tympanometrické křivky oboustranně s otoskopickým nálezem odpovídajícím oboustranně sekretorické otitidě. Vzhledem k rinoepifaryngoskopickému průkazu adenoidních vegetací s klinickými projevy nosní obstrukce a přetrvávající chronické sekretorické otitidě dítě podstoupilo v březnu 2011 endoskopickou adenotomii, oboustrannou paracentézu a při nálezu mukotympana byly oboustranně zavedeny ventilační trubičky vnitřního průměru 0,76 mm. Následně bylo provedeno vyšetření frekvenčně specifických evokovaných kmenových potenciálů Notched-Noise-BERA, které prokázalo sluchový práh vpravo 50-60-80-70 dB a vlevo 50-50-30-50 dB na frekvencích 0,5, 1, 2 a 4 kHz. Vzhledem k tíži sluchové vady bylo indikováno sluchadlo. Dítě zůstává v péči foniatra.
Klinický nález svědčí pro branchio-otický syndrom dítěte.
DISKUSE
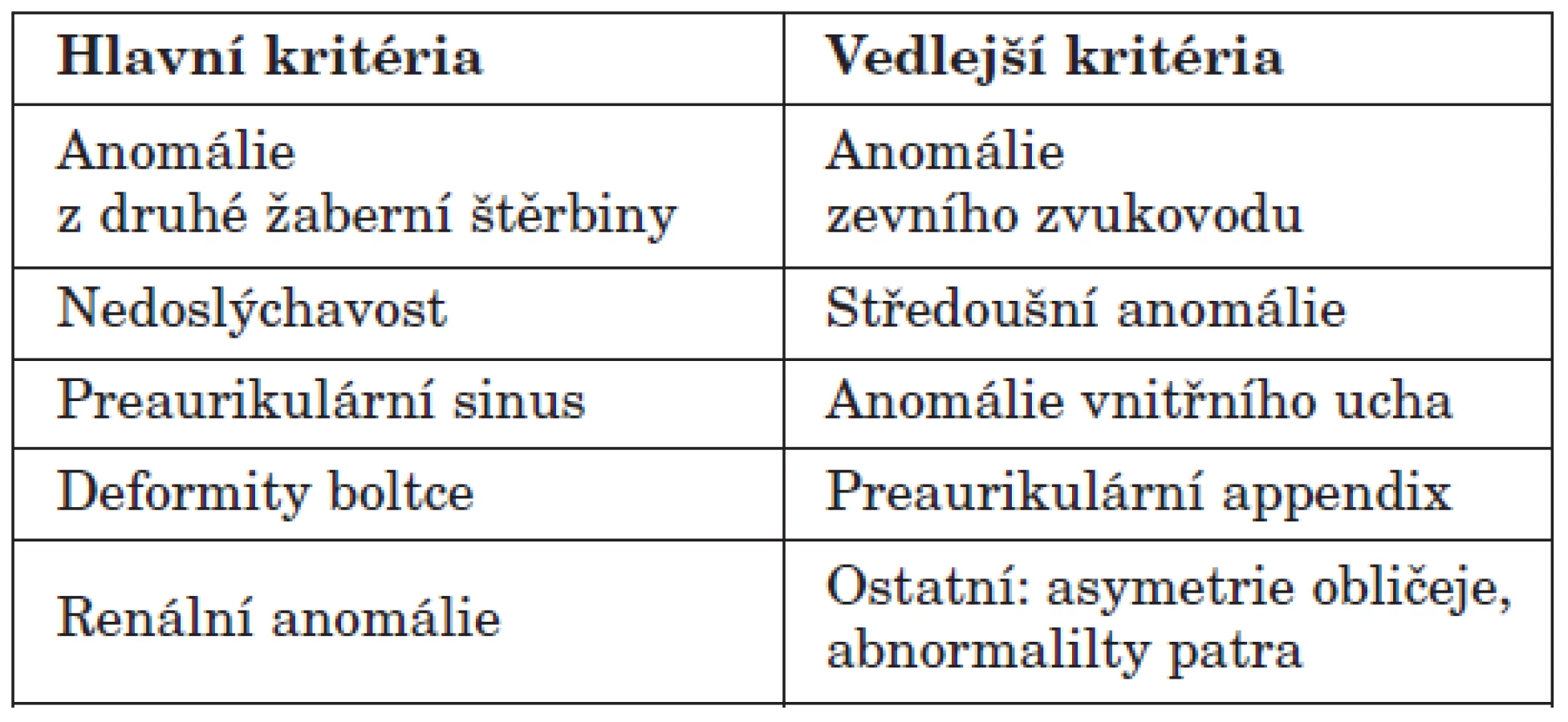
Poprvé byl popsán společný výskyt sluchového postižení a branchálních píštělí Heusingerem již v roce 1864. Pojem branchio-oto-renální syndrom s popisem specifického fenotypu tohoto syndromu byl zaveden Melnickem v roce 1975 (8). Incidence onemocnění se pohybuje kolem 1 případu na 40 000 porodů (2, 10). Diagnóza onemocnění je obvykle stanovena na základě klinických kritérií postižení ucha, branchiální malformace a ledvinného postižení (2, 4, 9, 10). Branchio-oto-renální syndrom je onemocnění s velmi variabilní expresivitou klinického obrazu i v rámci jedné rodiny. V případě absence renálního postižení je syndrom nazýván branchio-otický a patří do stejné skupiny jako syndrom branchio-oto-renální, liší se pouze nižší expresivitou klinického obrazu. Chen a kol. (4) prokazují v souboru 45 nemocných s branchio-oto-renálním syndromem postižení sluchu v 93 %, preaurikulární píštěle v 82 %, laterální krční cysty nebo píštěle v 50 %, ledvinné abnormality v 67 % případů. Mezi méně časté malformace patří preaurikulární apendix (13 %), aplazie slzných cest (11 %) nebo zkrácení patra (7 %). Chang a kol. (3) definovali klinická kritéria branchio-oto-renálního syndrom výskytem nejméně 3 z hlavních kritérií nebo dvou hlavních a dvou vedlejších kritérií dle tabulky 1.
Branchiogenní malformace u tohoto syndromu jsou v oblasti 2. žaberní štěrbiny. Jedná se o branchiogenní cysty a cervikální píštěle. Zevní ústí těchto píštělí se nachází laterálně na krku v průběhu m. sternocleidomastoideus, vnitřní ústí bývá obvykle na předním povrchu zadního patrového oblouku a m. palatoglossus tvoří jeho svalový kryt. Při chirurgickém ošetření těchto píštělí je proto doporučována exstirpace v celém průběhu píštěle až k jejímu vnitřnímu ústní spolu s tonzilektomií. Velmi často se u branchi-oto-renálního syndromu vyskytují abnormality boltce, především preaurikulární píštěle a apendixy.
V naší kazuistice byla matka dítěte operována na chirurgickém pracovišti, kde byla provedena exstirpace pouze zevní části laterální krční píštěle bez preparace jejího celého průběhu, přesto do dospělosti již nedošlo k recidivě píštěle či vzniku laterální krční cysty. Dítě operované na našem pracovišti podstoupilo exstirpaci celého průběhu píštěle, včetně ipsilaterální tonzilektomie. Byla indikovaná pouze jednostranná operace – na straně obstrukce ústí píštěle s vytvořením cysty a sekundární zánětlivou komplikací. Chirurgické řešení druhostranné píštěle bylo ponecháno do vyššího věku dítěte z důvodu zachování patrové tonzily.
Ušní postižení bývá charakterizováno malformacemi zevního, středního a vnitřního ucha. Projevuje se percepční, smíšenou nebo jen převodní nedoslýchavostí. Je-li indikováno chirurgické řešení převodní nedoslýchavosti, častým nálezem je fixace třmínku v oválném okénku a ageneze šlachy třmínkového svalu, ale jsou popsány i jiné malformace a defekty středoušních kůstek. Častější jsou ageneze oválného než okrouhlého okénka. Byly publikovány také nálezy dehiscence kanálu n.VII, rozšíření vestibulárního aqueduktu a Mondiniho anomálie vnitřního ucha (3, 4, 6, 13). Úspěchy rekonstrukční chirurgie nejsou zdaleka tak jednoznačné jako u nemocných s otosklerózou.
Matka dítěte měla oboustranně symetrickou percepční nedoslýchavost apikochleárního typu. U dítěte byla diagnostikovaná chronická sekretorická otitida superponovaná na pravděpodobně vrozenou percepční vadu sluchu. Po 3 měsících prokázané sekretorické otitidy bez spontánní úpravy byla indikována adenotomie a paracentéza, při nálezu mukotympana, vzhledem k preexistující vrozené sluchové vadě, byly v jedné době oboustranně zavedeny ventilační trubičky. Následné vyšetření frekvenčně specifickým vyšetřením evokovaných kmenových potenciálů Notched-Noise-BERA prokázalo středně těžkou nedoslýchavost vlevo a těžkou nedoslýchavost vpravo (klasifikace dle WHO) a umožnilo nastavení sluchadla ke kompenzaci sluchové vady. Dítě nadále zůstává v péči naší foniatrické ambulance.
Renální malformace mohou být jednostranné nebo oboustranné. Postižení ledvin může být velmi variabilní, od mírné hypoplazie bez funkčního postižení, až po oboustrannou renální agenezi (5,10,11). Na souboru 21 postižených (10) byly prokázány renální ageneze ve 29 %, hypoplazie v 19 %, dysplazie ve 14 %, obstrukce ureteropelvické junkce v 10 %, pelviektazie, hydronefróza a vesiko-ureterální reflux v 5 %. Na základě renální biopsie byla popsána oligomeganefrotická hypoplazie a multicystická dysplazie (10).
Matka dítěte má refluxní nefropatii s hypoplastickou, téměř afunkční levou ledvinou. U dítěte nebylo zjištěno ledvinné postižení.
Onemocnění je autosomálně dominantně dědičné, riziko postižení dětí nemocného je 50%. Geneticky je onemocnění heterogenní. Převážná většina případů je podmíněná mutacemi v EYA1 genu, ale byly také popsány mutace v SIX5 a SIX1 genech (3, 5, 9, 10, 12). V současné době není DNA analýza těchto genů v ČR dostupná.
V rodině dítěte byl diagnostikován branchio-oto-renální syndrom u matky. Ze dvou dětí je postiženo pouze jedno, druhé dítě je zdravé. Riziko pro děti postiženého dítěte je v případě zdravého partnera 50%.
ZÁVĚR
U pacientů s branchiální cystou či píštělí je nutno myslet na možnost branchio-oto-renálního syndromu. Doporučujeme u těchto pacientů vyšetření sluchu a při kombinaci sluchové vady s branchiogenní malformací by mělo být pátráno po možném ledvinném postižení.
MUDr. Lukáš Školoudík
Klinika ušní, nosní a krční LF UK a FN
Sokolská 581
500 05 Hradec Králové
Sources
1. De Caluwé, D., Hayes, R., McDermott, M., Corbally, M. T.: Complex Branchial Fistula: A variant arch anomaly. J. Pediatr. Surg., 36, 2001, s. 1087-1088.
2. Fraser, F. C., Sproule, j.r., Halal, F.: Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am. J. Med. Genet., 7, 1980, s. 341-349.
3. Chang, E. H., Menezes, J. R., Meyer, N. C. et al.: Branchio-oto.renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum. Mutat., 23, 2004, s. 582-589.
4. Chen, A., Francis, M., Ni, L. et al.: Phenotypic manifestation of branchio-oto-renal syndrome. Am. J. Med. Genet., 58, 1995, s. 365-377.
5. Kochhar, A., Orten, D. J., Sorensen J. L. et al.: SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurent missense mutation associated with BOR. Hum. Mutat., 29, 2008, s. 565.
6. Kemperman, M. H., Koch, S. M., Joosten, F. B., Kumar, S., Hyugen, P. L., Cremers C. W.: Inner ear anomalies are frequent but nonobligatory features of the branchio-oto-renal syndrome. Arch. Otolaryngol. Head Neck Surg., 128, 2002, s.1033-1038.
7. Keyvan, N., Ginger, R., Jaecklin, T., Pope, H. G., Dulguerov, P.: Management of congenital third branchial arch anomalies: A systematic review. Otolaryngol. Head Neck Surg., 142, 2010, s. 21-28.
8. Melnick, M., Bixler, D., Nance, W., Dilk, K., Yune, H.: Familiar branchio-oto-renal dysplasia: a new addition to the branchial arch syndromes. Clin. Genet., 9, 1976: s. 25-34.
9. Morisada, N., Rendtorff, N. D., Nozu, K. at al.: Branchio-oto-renal syndrome caused by partial EYA1 deletion due to LINE-1 insertion. Pediatr. Nephrol., 25, 2010, s. 1343-1348.
10. Okada, M., Fujimaru, R., Morimoto, N., Satomura, K., Kaku, Y, Tsuzuki, K., Nozu, K., Okuyama, T., Iijima, K.: EYA1 and SIX1 gene mutations in Japanede patients with branchio-oto-renal syndrome and related conditions. Pediatr. Nephrol., 21, 2006, s. 475-481.
11. Pellant , A., Pellantová, Z., Frank, M.: Příspěvek k hereditárním nefropatiím II. Nedoslýchavost v dětském věku. Českosl. Otolaryngol., 36, 1987, 6, s. 335-339.
12. Ruf, R. G., Xu, P. X. Silvius, D. et al.: SIX1 mutations cause branchio-oto-renal syndrome by dysruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. USA, 101, 2004, s. 8090-8095.
Labels
Audiology Paediatric ENT ENT (Otorhinolaryngology)Article was published in
Otorhinolaryngology and Phoniatrics

2011 Issue 2
Most read in this issue
- Změna komunikace je u pacientů s trvalou tracheostomií největším hendikepem
- Extraezofageální refluxní choroba - mezioborový konsenzus
- Pendredův syndrom v České republice
-
Nádorová trombóza v. jugularis interna
Kazuistika a přehled jejích epidemiologických, patogenetických a klinických aspektů