
Primární vaskulitidy v otorinolaryngologii: přehled literatury a kazuistické případy
Primary Vasculitis in Otorhinolaryngology: Review of the Literature and Case Reports
Primary vasculitis is a rare group of diseases with diverse local and systemic symptoms. The disease can affect any organ of the human body including head and neck region. Moreover, manifestation in head and neck is not rare and for some pathologic units it is an elemental part of the established diagnostic criteria. In view of that fact, ENT specialist can be the first one who comes to a contact with a patient affected by a vasculitis and be the one who hastens the diagnostic and therapeutic process. However, because of the unspecific symptomatology and a very low incidence of the diseases, the determination of the diagnosis can be quite a difficult quest. Even when a clinician is suspicious about systemic vasculitis and other cooperative specialists are involved into a diagnostic process, it can be difficult to precisely categorize the pathologic unit. In this article we review vasculitis most commonly occurring in ENT, including their various symptomatology. Complexity of the diagnostic process is demonstrated on two case reports: a patient with granulomatosis with polyangiitis and a patient with unclassified systemic vasculitis.
Keywords:
primary vasculitis – ANCA – head and neck
Authors:
V. Koucky; J. Bouček; D. Kalfeřt; J. Plzák
Authors‘ workplace:
Klinika otorinolaryngologie a chirurgie hlavy a krku, 1. lékařská fakulta, Univerzita Karlova a Fakultní, nemocnice v Motole, Praha
Published in:
Otorinolaryngol Foniatr, 68, 2019, No. 4, pp. 237-246.
Category:
Review Article
Overview
Primární vaskulitidy jsou vzácnou skupinou onemocnění s rozmanitými lokálními a systémovými projevy. Onemocnění může zasáhnout jakýkoli orgán, včetně oblastí spadajících do problematiky otorinolaryngologie. Projevy na hlavě a krku pak nejsou u řady patologických jednotek vzácnými a u některých vaskulitid jsou přímou součástí diagnostických kritérií. Otorinolaryngolog se tak může stát jedním z prvních lékařů, který může přijít s pacientem s vaskulitidou do kontaktu a být tím, kdo uspíší diagnosticko-terapeutický proces. Vzhledem k nespecifické symptomatologii a velmi nízké incidenci těchto chorob je však určení diagnózy na základě prvotních symptomů značně obtížné. Navíc, i po vyslovení podezření na systémovou vaskulitidu a zapojení dalších odborníků do diagnostického procesu, může být přesné zařazení diagnózy komplikovaným úkolem. V tomto přehledu uvádíme popis vaskulitid nejčastěji se vyskytujících ve vztahu k ORL, včetně jejich různorodých klinických manifestací. Složitost diagnostiky těchto onemocnění je demonstrována na kazuistickém případu pacienta postiženého granulomatózou s polyangiitidou a pacienta s dosud nejasně klasifikovanou systémovou vaskulitidou.
Klíčová slova:
primární vaskulitidy – ANCA – hlava a krk
ÚVOD
Vaskulitidy jsou heterogenní skupinou onemocnění, které mohou postihnout jakýkoli orgán lidského těla. Nejčastěji zmiňované primární vaskulitidy ve vztahu k otorinolaryngologii (ORL) jsou ANCA (anti-neutrophilic cytoplasmic antibody) asociované vaskulitidy, které mají postižení horních dýchacích cest v rámci svých diagnostických kritérií, avšak i ostatní patologické jednotky mohou postihnout oblast hlavy a krku. Společnou charakteristikou je zánětlivé postižení stěny cév, které může být způsobeno několika různými mechanismy. Popsána byla cytotoxická reakce T lymfocytů, ukládání imunokomplexů do stěny cév, přímé poškození protilátkami a protilátkami zprostředkovaná buněčná cytotoxicita. Mnohdy se tyto patologické reakce účastní v jedné době. Přes rozdílný mechanismus poškození jsou klinické symptomy často velmi podobné, mezi jednotlivými diagnostickými jednotkami zaměnitelné a ani následná imunologická a histologická vyšetření nemusí přinést přesné stanovení diagnózy. Onemocnění může začít jako izolované postižení jednoho orgánu a postupně se šířit či se již v počátečních fázích projevit jako systémová choroba. Prvotní manifestace v oblasti hlavy a krku pak není vzácností. Klinický obraz je však zaměnitelný i s mnoha dalšími, mnohem častějšími onemocněními, které nespadají do skupiny vaskulitid. Jsou jimi různé infekční choroby, kožní a autoimunitní onemocnění. Přesné určení diagnózy a nastavení nejlepšího terapeutického postupu tak může být v počátcích diagnostického procesu velmi obtížné. Nejucelenější nomenklatura vaskulitid vychází z druhé “International Chapel Hill Consensus Conference“ konané roku 2012 (50). Základní diagnostické jednotky jsou zde rozděleny dle asociace s jiným onemocněním, dle predilekčně postiženého orgánu či dle velikosti cév, které jsou v rámci daného onemocnění typicky zasaženy. Kompletní rozdělení je uvedeno v tab. 1.

V tomto přehledu uvádíme popis vaskulitid nejčastěji se vyskytujících ve vztahu k ORL, včetně jejich rozmanitých klinických manifestací. Všechna uváděná onemocnění mohou mít i nespecifické systémové příznaky v podobě febrilií, úbytku na váze, nechutenství, únavy, bolesti kloubů a svalů, a tím upozornit na závažnější diagnózu než by naznačoval samotný lokální nález. Rozmanitost příznaků a diagnostických postupů dokládáme dvěma kazuistickými případy.
MATERIÁL A METODY
Přehled literatury za využití databází Medline a Scopus. K vyhledávání bylo použito klíčových identifikátorů – ENT, otolaryngology, otorhinolaryngology, ear, nose, throat, hearing loss, vertigo v kombinacích s identifikátory jednotlivých diagnostických jednotek. Dále jsou prezentovány dva kazuistické případy pacientů diagnostikovaných, léčených a dispenzarizovaných na Klinice otorinolaryngologie a chirurgie hlavy a krku 1. LF UK a FN v Motole.
KLINICKO-PATOLOGICKÉ JEDNOTKY
Vaskulitidy postihující velké cévy
Do této podskupiny spadají obrovskobuněčná temporální arteritida (OTA) a Takayasuova arteritida.
V případě obrovskobuněčné temporální arteritidy se jedná o chronické systémové onemocnění postihující primárně větve zevní karotidy (arteria carotis externa). V literatuře ji můžeme najít i pod názvy jako velkobuněčná nebo Hortonova arteritida. Je to nejčastější primární vaskulitida v severoamerické a evropské populaci nad 50 let věku s incidencí mezi 12-17 případy na 100 tisíc obyvatel a vyšším rizikem rozvoje u ženského pohlaví (30, 32, 93). Dominujícím příznakem je náhle vzniklá bolest hlavy, která se vyskytuje přibližně u 2/3 postižených (69). Může se vykytovat jednostranně i oboustranně a častěji bývá lokalizována do temporální a parietální krajiny. Dalším typickým příznakem jsou abnormality postihující temporální arterie, které se vyznačují prominující, bolestivou cévou se sníženou pulzací. Obávanou ischemickou komplikací onemocnění je okluze očních arterií (větví arteria ophtalmica) s rozvojem náhlé částečné či úplné ztráty zraku. Tento stav vyžaduje promptní zhodnocení a případnou intenzivní kortikoterapii (31). Z dalších očních projevů se může objevit ptóza, mióza či oftalmoplegie (71). Z méně častých ischemických projevů jsou popsány případy cévních mozkových příhod s parézami hlavových nervů a okrskové kožní a slizniční infarzace. Ty mohou postihnout rty i jazyk (6, 77). Často pozorovaným příznakem u pacientů jsou čelistní klaudikace při delším mluvení a žvýkání (69). Nicméně, vzhledem k možnému postižení různých cév v oblasti hlavy a krku jsou popsány další rozmanité příznaky zasahující do problematiky ORL lékaře, jako je např.: periferní vestibulární syndrom (4), náhlá percepční ztráta sluchu (40) či dysartrie (60). Onemocnění se v asi 50 % případů vyskytuje zároveň s revmatickou polymyalgií, příbuzným systémovým zánětlivým onemocnění projevujícím se bolestí v oblasti krku, ramen a ranní ztuhlostí svalů a kloubů (8). Diagnostická kritéria pro OTA vyžadují pozitivitu 3 z následujících 5 symptomů: věk nad 50 let; nově vzniklá bolest hlavy; abnormality temporální arterie; zvýšená sedimentace; abnormální biopsie temporální arterie (45). Zlatým standardem pro potvrzení diagnózy je histologické vyšetření biopsie z temporální arterie, která se provádí v temporální nebo preaurikulární krajině (66).
Takayasuova arteritida, známá těž jako bezpulzová nemoc, postihuje především větve aortálního oblouku. Jedná se o velmi vzácné onemocnění nejčastěji se vyskytující mezi 10. a 30. rokem věku (62). Incidence v Severní Americe byla vyčíslena 2,6 případu na 1 milion obyvatel (39). U více jak 50 % pacientů je možné pozorovat vymizení pulzací spojené s rozdílnostmi v krevním tlaku, poslechovými šelesty a dalšími závažnými hemodynamickými a ischemickými symptomy, spojenými se zánětlivým postižením a stenózou velkých cév (62). Projevy v oblasti hlavy a krku jsou velmi raritní. Popsány byly jednotlivé případy pacientů s Takayasuovou arteritidou asociovanou s perforací nosního septa (1), sedlovitou deformací nosu (79) či percepční ztrátou sluchu (81). Základem léčby jsou vysoké dávky kortikoidů.
Vaskulitidy postihující cévy o středním průměru
Polyarteritis nodosa je vzácná nekrotizující systémová vaskulitida postihující primárně cévy středního kalibru, především v místech jejich větvení. Incidence v Evropě dosahuje 1,6 případu na 1 milion obyvatel (41). Stran etiopatogeneze se může jednat buď o primární, idiopatickou formu, či o nemoc asociovanou s infekcí virem hepatitidy B (28). Byly však popsány případy spojené s řadou jiných virových onemocnění (16). Příznaky jsou značně rozmanité, od izolovaných kožních forem, přes průběh dominantně postihující jeden orgán, až po rychle progredující multisystémové onemocnění. Nejčastěji se jedná o postižení kůže v podobě purpury, vředů a podkožních uzlíků spojené dále s postižením periferních nervů. Často popisovaným příznakem je i náhlá percepční či kombinovaná ztráta sluchu s přítomností serózního středoušního výpotku s příměsí krve (49, 88). Postižení bývá oboustranné, symetrické, může rychle progredovat a v některých případech se jedná o primární projev onemocnění (76, 95). Dále byly popsány i případy spojené s obrnou lícního nervu či s oboustrannou parézou zvratných nervů (23, 98). Základem diagnostiky je odebrání biopsie k histologickému vyšetření s následným typickým mikroskopickým obrazem. Diferenciálně diagnosticky je třeba vyloučit ANCA-asociované vaskulititidy. Při typické polyarteritis nodosa jsou ANCA negativní.
Druhým zástupcem této podskupiny vaskulitid je Kawasakiho syndrom, vaskulitida primárně postihující dětské pacienty (20). Onemocnění má tendenci ke spontánní regresi, pokud však není včas léčeno, může vést k závažným komplikacím. Až ve 25 % neléčených případů totiž dochází k tichému rozvoji koronárních aneurysmat, které mohou i mnoho let po prodělané nemoci vést k náhlému úmrtí (52). 85 % pacientů se pohybuje v rozmezí 6 měsíců a 5 let věku. Incidence je značně rozmanitá, kdy v České republice je udávána 1,6 případu na 100 tisíc dětí do 5 let věku (18), v USA 17 případů na 100 tisíc dětí do 5 let věku (44) a nejvyšší výskyt je zaznamenán v Japonsku s více jak 100 případy na 100 tisíc dětí do věku 5 let (70). Typický je pro tento syndrom sezonní výskyt v zimních a jarních měsících. Diagnóza je stanovena na základě přítomnosti horečky a 4 z následující sestavy příznaků: oboustranná konjunktivitida, krční lymfadenopatie, polymorfní exantém, změny na sliznicích horních dýchacích cest a edematózní změny na horních či dolních končetinách. Zvětšení mízních uzlin na krku je většinou jednostranné a s výskytem u 25–75 % pacientů nejméně častým projevem zahrnutým do diagnostických kritérií (20, 56, 97). Postižení horních dýchacích cest se vyskytuje téměř u všech zasažených, a to ve formě erozivních lézí na rtech, faryngitidy a jahodového jazyka, obrazu vzniklého vymizením filiformních papil a vystoupením papil fungiformních. Exantém se může vyskytovat na kterékoliv části těla, ale často postihuje kůži tváří a má skarlatiformní či morbiliformní charakter. Základní, zpravidla účinnou, terapií není klasická imunosupresivní léčba, ale intravenózně podávané imunoglobuliny a kyselina acetylsalicylová (24). I z toho důvodu je zásadní časné stanovení správné diagnózy.
ANCA-asociované vaskulitidy
Tato skupina nekrotizujících vaskulitid, postihujících cévy malého kalibru, je charakteristická sérologickým nálezem protilátek specifických vůči neutrofilním cytoplazmatickým antigenům, které se však mohou vyskytovat i u řady jiných onemocnění. Zároveň nález ANCA není pro rozvoj vaskulitid ani pro diagnózu nezbytný a jejich přesný patogenetický vliv ještě není zcela objasněn. Na základě klasických imunofluorescenčních testů rozeznáváme tři možné obrazy, vzniklé jako artefakt laboratorního zpracování: cytoplazmatické (cANCA) a perinukleární (pANCA). Ve vztahu ke konkrétním antigenů můžeme rozlišit protilátky proti proteináze 3 (PR3-ANCA), neutrofilní myeloperoxidáze (MPO-ANCA) a atypické ANCA proti elastáze, laktoferrinu či katepsinu G. Nejčastěji pak koreluje nález pANCA s MPO-ANCA a cANCA s PR3-ANCA (11). U daného pacienta zpravidla nacházíme pouze jeden sérotyp ANCA a v současnosti je známa řada rozdílů v klinickém průběhu onemocnění i odpovědí na léčbu mezi pacienty s pozitivitou PR3-ANCA nebo MPO-ANCA. Ve vztahu k postižení v ORL oblasti mají pacienti s nálezem PR3-ANCA častěji nekrotizující léze v horních dýchacích cestách, naproti tomu MPO-ANCA pozitivní jedinci jsou typičtější výskytem nosní polypózy (42). V léčbě této skupiny onemocnění jsou využívány kortikoidy a cyklofosfamid.
Mikroskopická polyangiitida (MPA). Toto onemocnění je nejméně často asociováno s projevy v ORL oblasti. U více jak 50 % případů prokazujeme MPO-ANCA, jsou však i případy ANCA negativní. Dominantně postiženými orgány jsou ledviny, v podobě rychle progredující glomerulonefritidy, a plíce. Jak bylo zmíněno, postižení v ORL oblasti není pro tuto diagnózu typické, avšak dle dostupných prací se může vyskytovat v 9-30 % ze všech případů MPA (35). Nejčastěji se jedná o epistaxi, sinusitidy a nespecifické rinitidy (22, 65). Přesto, že v histologickém nálezu nedominuje granulomatózní zánět, tak odlišení od granulomatózy s polyangiitidou může být vzhledem k podobnému klinickému průběhu velmi obtížné.
Granulomatóza s polyangiitidou (GPA). Nejvíce dohledatelných publikací o projevech vaskulitidy v ORL je ve vztahu ke GPA, dříve známé jako Wegenerova granulomatóza. Jedná se o systémové onemocnění s typickou tvorbou nekrotizujících granulomů v postižených orgánech. Incidence onemocnění v Evropě je 5-10 případů na 1 milion obyvatel a mezi postiženými mírně převládají muži (91). Diagnostická kritéria stanovila už v roce 1990 American College of Rheumatology (59), s potřebou pozitivity alespoň 2 z následujících 4 příznaků: postižení nosu nebo nosní dutiny (slizniční ulcerace, hnisavý či krvavý výtok), nemigrující nodulární infiltráty na plicích, abnormální výsledky při vyšetření moči (hematurie) a arteriální či periarteriální granulomy při histologickém vyšetření. Nicméně tato kritéria byla stanovena ještě před odlišením mikroskopické polyangiitidy jako samostatné diagnostické jednoty a zároveň před objevením významu ANCA. Následně tedy byla mezi diagnostická kritéria přidána pozitivita ANCA (90). V případě nálezu výše určených symptomů a pozitivity ANCA, typicky anti-PR3, tak není potřeba biopsie ke stanovení diagnózy GPA. Ta může být navíc u lokalizovaných mimoplicních forem málo výtěžná, kdy nemusí být klasický obraz granulomů zachycen (78). Senzitivita a specificita vyšetření ANCA během akutních vzplanutí onemocnění se pohybuje okolo 90 % (72).
Dle klinického průběhu můžeme onemocnění rozdělit na 3 různé formy – lokalizovanou, časně systémovou a generalizovanou. Až v 95 % je iniciální manifestací ORL symptomatologie, nejčastěji jako ulcerativní rinosinusitidy s případným otologickým postižením (38, 51, 61, 85, 86). Plně rozvinuté generalizované onemocnění má, pokud není adekvátně léčeno, rychlý průběh s vysokou mortalitou. Celkové symptomy v podobě úbytku na váze, nechutenství a horeček jsou doprovázeny bolestmi na hrudi, kašlem, dušností, hemoptýzou a/nebo akutním renální selháním v důsledku rychle progredující glomerulonefritidy s obrazem nefritického syndromu.
V rámci postižení nosu a vedlejších nosních dutin je typicky zasažena přední část nosního septa. Častými příznaky je nosní obstrukce, anosmie, epistaxe a bolest. V důsledku zánětlivého procesu mohou být poškozeny kostěné a chrupavčité struktury a vést tak ke vzniku perforací nosního septa či deformitě v podobě sedlovitého nosu (73). Jako jeden z prvotních symptomů se může objevit i epifora při poškození ústí nasolakrimálního duktu (95). Eroze kostěných struktur se netýká pouze okolí nosu a vedlejších nosních dutin, ale může zasáhnout i další oblasti lební baze, vzácně může vést k paréze hlavových nervů (52, 54). V důsledku bakteriálního osídlení poškozené sliznice můžeme také nacházet četné krusty a hnisavou sekreci.
Otologické projevy jsou popisovány u 19–61 % nemocných (87). Nejčastěji je zánětlivým procesem postižena středoušní dutina, a to sekundárně na podkladě obstrukce Eustachovy trubice (33). Samotné přímé postižení středoušní dutiny zánětem není příliš častým nálezem a spíše se jedná o bakteriální superinfekci. Stejně tak postižení vnějšího zvukovodu není pro onemocnění typické. Pokud se vyskytne, může imitovat obraz klasické perichondritidy (17). Nejčastější sluchovou vadou je nález kombinované poruchy, některé zdroje ale udávají čistě percepční vadu, která se může objevit až u 1/3 pacientů. Ta má tendenci progredovat a je doprovázena tinnitem (73). Zasažení vestibulárního systému a projevy v podobě akutní závratě jsou spíše vzácné. Stejně tak jsou popsány pouze kazuistické případy s periferním postižením lícního nervu (51, 92).
Zasažení sliznice ústní dutiny, faryngu a laryngu jsou rovněž relativně častým nálezem. Ulcerace v ústní dutině jsou popisovány u 5–10 % pacientů (94). Postiženy mohou být i dásně, které jsou edematózní, zarudlé, s petechiálním krvácením a dávají tak obraz tzv. jahodové hyperplazie gingivy (54). Postižení laryngu se projevuje chrapotem, dušností, hemoptýzou či kašlem. Relativně typickým nálezem jsou stenózy subglotické oblasti (27). Jejich incidence je udávaná až u 20 % pacientů s GPA (2).
Prognóza pacientů závisí na formě onemocnění a rozsahu postižní, kdy lokalizované formy mají přirozeně méně závažný průběh. Se současnými imunosupresivními přípravky je popisovaný medián přežití 21,7 let (74). V indukční terapii se dominantně uplatňují cyklofosfamid a glukokortikoidy. V indikovaných případech je možné podat rituximab či metotrexát. Dlouhodobá udržovací terapie je důležitou součástí léčby, neboť onemocnění má vysokou tendenci k relapsům. Nejvíce se v léčbě uplatňuje kombinace perorálních glukokortikoidů a azathioprinu.
Chirurgická terapie projevů diagnostikované GPA není primárně indikovaná, neboť při včasně zavedené imunosupresivní terapii dojde zpravidla k úpravě stavu. U otologických projevů však někteří autoři nacházejí místo pro chirurgickou sanaci v případě purulentní sekrece, špatné odpovědi na antibiotickou terapii a při projevech mastoiditidy (33, 63). Pokud dojde i k projevům v podobě zasažení lícního nervu, pak chirurgický zásah nevede ke zlepšení stavu a může poškození nervu ještě prohloubit (75). Chirurgické řešení nosních projevů se nabízí v případě rekonstrukcí typických sedlovitých deformací. Dle systematického přehledu Ezzata a kol. je chirurgie v podobě rinoseptoplastiky bezpečnou metodou s úspěšností až 84 % (19). Chirurgická terapie se dále nabízí u perzistujících symptomatických subglotických stenóz. Dle dostupné literatury je metodou s dobrými výsledky intralesionální aplikace kortikoidů a balónková dilatace (43, 58). K dispozici jsou i studie zkoumající efekt laserové terapie, eventuálně kombinované s dilatací a lokální aplikací mitomycinu C, které v krátkodobém horizontu ukazují vysokou úspěšnost, ale zároveň častý výskyt pozdních restenóz (80, 84). Obecně však výsledky intervenčních studií trpí malými počty pacientů a mnohdy krátkou dobou sledování.
Diferenciální diagnostika GPA je široká. Patří do ní další ANCA-asociované vaskulitidy, granulomatózní procesy jako sarkoidóza, mykobakteriální či mykotické infekce a pochopitelně i nádorová onemocnění, především lymfomy.
Eosinofilní granulomatóza s polyangiitidou (EGPA). EGPA je primární systémová vaskulitida, do roku 2012 známa pod názvem syndrom Churga-Straussové, který byl poprvé popsán v roce 1951 (5, 46). Nyní je charakterizována jako eosinofilní a nekrotizující granulomatózní zánět často postihující dýchací cesty, který je asociován s astmatem a eosinofílii. Incidence v evropské populaci je 0,5–6,8 případů na 1 milion obyvatel a nejvyšší výskyt je zaznamenám u věkových skupin mezi 40 a 60 lety (68). Používaná diagnostická kritéria American College of Rheumatology z roku 1990 udávají potřebu nálezu 4 z následujících 6 symptomů (64): astma, periferní eosinofilie >10 % WBC, migrující plicní infiltráty, anomálie vedlejších nosních dutin a biopsie dokumentující akumulaci eosinofilů v perivaskulární tkáni. Přítomnost ANCA, především pANCA, je popisován u 25-75 % pacientů (82, 83). U řady pacientů klasifikovaných jako EGPA pak dle současných studií ani není nález typické vaskulitidy. Probíhají tedy diskuse o možném rozdělení EGPA ve 2 odlišné jednotky – typickou vaskulitidu a hypereosinofilní astma se systémovými projevy (13). Případný klinický dopad však musí být ještě objasněn.
U onemocnění je popisován průběh ve 3 fázích (5, 25). První je fáze prodromální, kdy je typická symptomatologie v horních a dolních dýchacích cestách. Jedná se o astmatické projevy a alergickou rinitidu či rinosinusitidu s nosní polypózou. Ve druhé fázi dochází k rozvoji eosinofilie a infiltraci periferních tkání, především plic a gastrointestinálního traktu, eosinofilními granulocyty. V závěrečné, třetí fázi onemocnění, dochází k rozvoji nekrotizující vaskulitidy typicky se projevující polyneuropatií, avšak případná symptomatologie se odvíjí od dominantně zasaženého orgánu (5, 25).
Dle systematického přehledu Goldfarba a kol. z roku 2016, zahrnujícího 1175 pacientů, byly projevy v oblasti hlavy a krku zřejmé u 48 až 96 % pacientů (29). Kromě rinosinusitidy s nosními polypy je také častá i ušní manifestace v podobě lehké až středně těžké ztráty sluchu, případně spojená s otalgii a otoreou. Nejčastěji je však popsán percepční typ poruchy.
Základem léčby jsou glukokortikoidy, eventuálně v kombinaci s dalšími imunosupresivy jako cyklofosfamid, methothrexát či azathioprin. Dle výsledků French Vasculitis Study Group cohort bylo po primární léčbě a použití následné udržovací terapie 64,8 % pacientů bez známek onemocnění v 5 letech a 54,4 % pacientů po 10 letech od stanovení diagnózy (12).
Behcetova choroba
Behcetova choroba je systémová vaskulitida projevující se predominantně postižením ústní dutiny a orofaryngu, ulcerujícími lézemi na genitálu a postižením struktur oka, především ve formě uveitidy. Onemocnění má největší prevalenci v Turecku, dále je zaznamenán vyšší výskyt v oblasti Středozemního moře (3). Prevalence v euroamerické populaci je kolem 0,5 případů na 100 tisíc obyvatel (9, 15). Postihuje především mladší pacienty ve věku 20–30 let (3) a distribuce mezi pohlavími se značně liší mezi jednotlivými regiony. Většina pacientů je postižena aftózními ulceracemi v ústní dutině. Ty se mohou vyskytnout na jazyku, bukální sliznici, orofaryngu či gingivě. Svým vzhledem jsou snadno zaměnitelně s prostou aftózní stomatitidou. Jejich počet se pohybuje v průměru kolem 5 lézí při rozvoji onemocnění a velikostí do 10 mm (67). Ulceracemi mohu být postiženy i struktury laryngu a hypofaryngu, vzácně pak s časovým odstupem od propuknutí onemocnění může dojít k rozvoji stenóz či tvorbě píštělí v těchto lokalitách (7, 21, 37). Audiovestibulární postižení je rovněž jako u jiných vaskulitid relativně častým nálezem (57). Až třetina pacientů s diagnostikovanou Behcetovou chorobou má popsánu percepční ztrátu sluchu a/nebo vestibulární obtíže. Postižení sluchu je typicky oboustranné a ve vyšších frekvencích (14).
Coganův syndrom
Toto onemocnění bylo původně popsané jako nonsyfilitická intersticiální keratitida s audiovestibulárními symptomy podobnými Meniérovu syndromu (10, 48). Kromě této symptomatologie však více jak 70 % pacientů trpí i systémovými projevy onemocnění v podobě postižení gastrointestinálního traktu, centrálního nervového a kardiovaskulárního systému (36). Jedná se obecně o velmi vzácný stav, kdy celosvětově bylo dosud popsáno okolo 250 případů, převážně v bělošské populaci (47). Ve většině případů se onemocnění projeví nejprve samostatnými očními či audiovestibulárními příznaky. V případě typické formy onemocnění se jedná o intersticiální keratitidu, v případě atypické formy se oční poškození může projevit i jako episkleritida či konjunktivitida. Další symptomy se zpravidla přidávají v průběhu několika měsíců až let. Postižení sluchu bývá u většiny postižených náhlé, oboustranné, postihující všechny frekvence a během několika měsíců může progredovat k praktické hluchotě (26). Naproti tomu oční postižení málokdy vede ke slepotě a hlavní morbidita pacientů spočívá v postižení sluchu. Percepční sluchová ztráta je pak doprovázena závratí, nauzeou a zvracením (34). Časná terapie intravenózními glukokortikoidy může vést k významnému zlepšení sluchu (89). V případě, že dojde k systémové postižení, především ve formě aortitidy, je pak celková prognóza pacientů horší.
KAZUISTIKY
Kazuistika č. 1
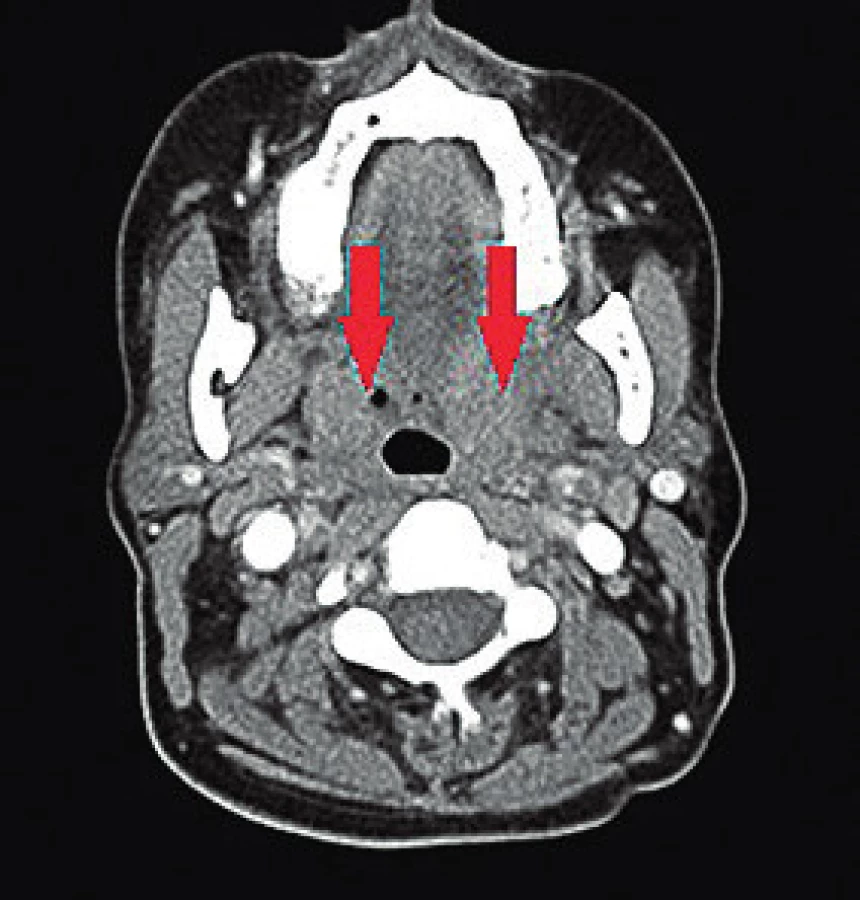
38letá žena, pocházející z Ruska, byla přijata na lůžkové oddělení Kliniky ORL a chirurgie hlavy a krku 1. LF UK a FN Motol. Důvodem hospitalizace byla 2 týdny trvající, postupně progredující, silná bolest v hrdle a z toho vyplývající polykací obtíže. Stav byl dále doprovázen rekurentními febríliemi pohybujícími se mezi 38°C a 39°C. Během klinického vyšetření byl dominující nález v orofaryngu, který imponoval jako peritonzilární flegmóna s mírnou krční lymfadenopatií v submandibulární oblasti oboustranně. Laryngoskopické vyšetření neprokázalo žádnou další patologii. Pro pravděpodobný infekt orofaryngu byla pacientka již 3 týdny ambulantně léčena praktickým lékařem a ORL specialistou, bez efektu antibiotické terapie (makrolidy, penicilin, cefalosporiny II. generace). Dále byly při vstupním vyšetřením identifikovány palpačně bolestivé kožní eflorescence růžovo-červeného zabarvení, mírně vystupující nad niveau, na vnitřní straně stehen, nártech a ojediněle na trupu. V laboratorním vyšetření byla patrna leukocytóza 16,5x109/l a elevace CRP na 35,5 mg/l. Pacientce byla nasazena intravenózní antibiotika, nejdříve cefalosporin II. generace, posléze pro nelepšící se stav kombinace klindamycinu s gentamycinem. Stav pacientky se však nadále klinicky i laboratorně horšil s potřebou zavést nasogastrickou sondu pro absolutní nemožnost polykat tekutiny a stravu. Došlo k vzestupu leukocytózy (32,9x109/l) s neutrofilií, elevaci CRP na hodnoty 300 mg/l a trombocytóze (742x109/l). Pacientka byla indikována ke kompletnímu vyšetření pro vyloučení možné infekční etiologie, včetně screeningu virových a mykotických agens. Vzhledem k progresi stavu a kožním příznakům bylo doporučeno imunologické vyšetření, v rámci kterého bylo vysloveno podezření na blíže neurčenou vaskulitidu. Sérové hladiny ENA, ANA, ANCA a RF však byly negativní. Byly zaznamenány pouze mírně zvýšené hladiny cirkulujících imunokomplexů. V diferenciální diagnostice byla zvažována neutrofilní dermatóza či vaskulitida v rámci systémového autoimunitního postižení pojiva. Pro progredující nález v krevním obrazu byla dále provedena i punkční biopsie kostní dřeně, která neprokázala možnou onko-hematologickou etiologii. Zobrazovací metody v podobě RTG hrudníku, sonografie břicha a echokardiografie neprokázaly postižení vnitřních orgánů. Na základě dominujících obtíží v oblasti krku bylo provedeno CT vyšetření, na kterém byly patrné infiltráty při dolním pólu tonzil oboustranně, nemající charakter abscesu (obr. 1). Na doporučení imunologa a revmatologa byla provedena biopsie z kožních lézí a dále i hypofaryngoskopie v celkové anestezii s odběrem tkáně z lividních, tuhých ložisek ložisek v oro-hypofaryngu. Vzhledem k rychle progredujícímu septickému stavu byla pacientka k další péči přeložena na jednotku intenzivní péče Interní kliniky 2. LF UK FN v Motole. Pacientce byla nasazena pulzní terapie metylprednisolonem, která vedla k částečnému zlepšení stavu. Z histologických preparátů pak byl odečten obraz leukocytoklastické vaskulitidy malých cév a pacientka byla přeložena na specializované revmatologické pracoviště. V dalším průběhu pak pacientka přes dispenzarizaci a imunosupresivní léčbu protrpěla několik recidiv v podobě kožních výsevů a dysfagických obtíží. Na základě celotělového PET/CT nebyla prokázána aktivita onemocnění v jiných orgánových systémech. Relativně uspokojivého stavu bylo dosaženo až po pulzní intenzivní terapii cyklofosfamidem v kombinaci s kortikosteroidy. Vzhledem k atypickému klinickému a laboratornímu nálezu je pacientka vedena pouze pod histologickým nálezem leukocytoklastické vaskulitidy kůže a sliznic bez jasně zařaditelné klinické jednotky.
Kazuistika č. 2
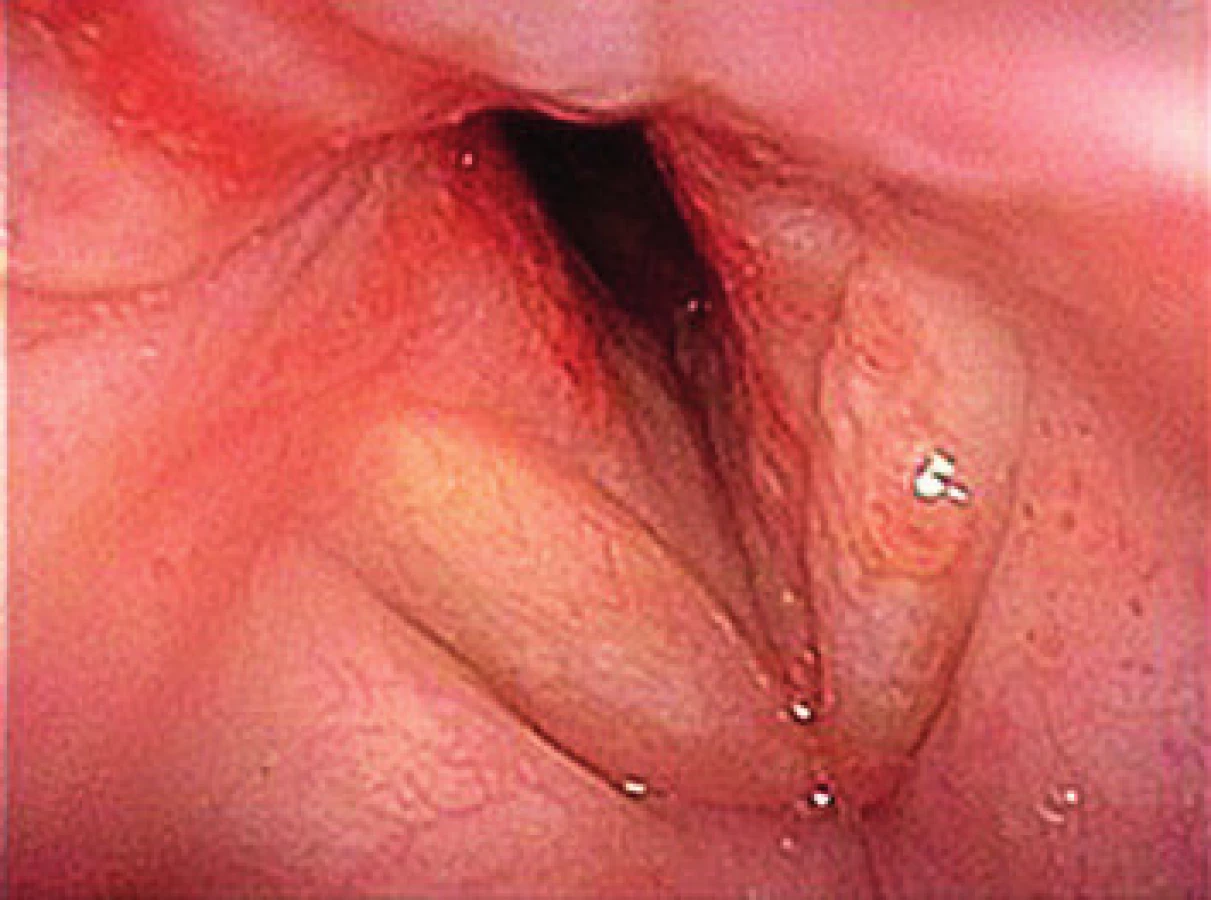
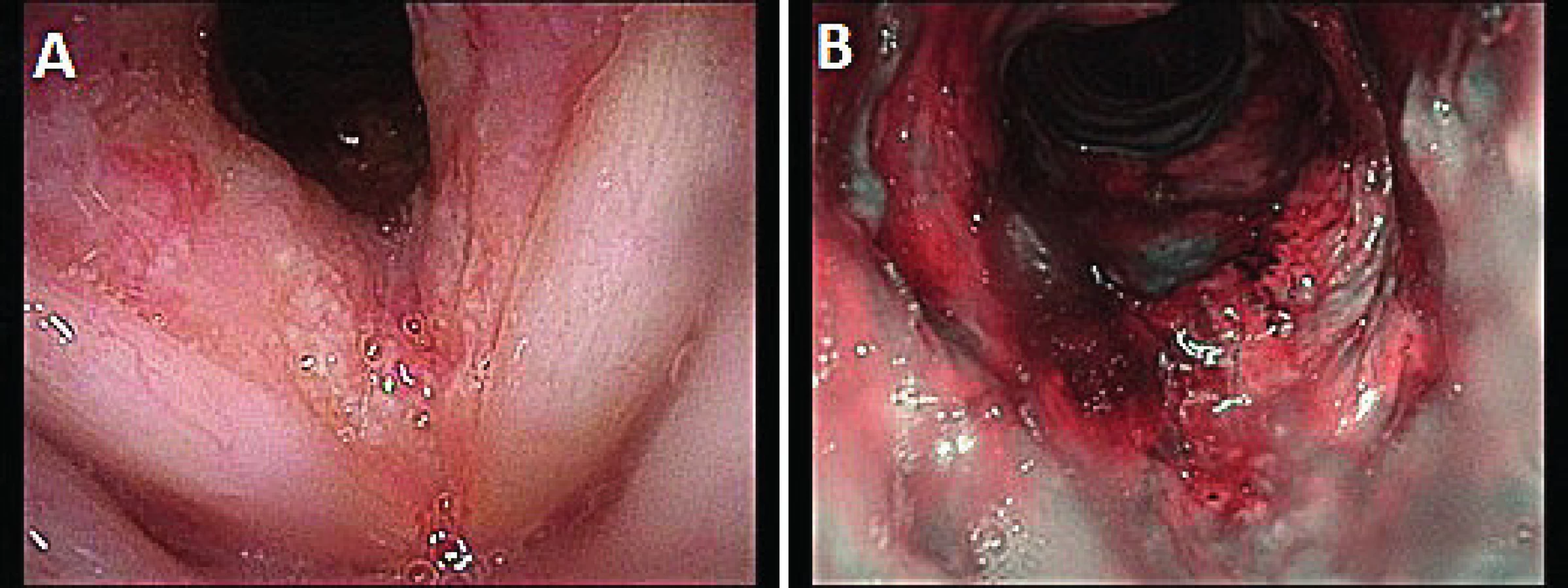
66letá pacientka byla hospitalizována na Pneumologické klinice 2. LF UK FN v Motole pro stridor, dušnost a hemoptýzu. V počátku byl stav hodnocen jako akutní tracheobronchitida. V rámci konziliárního ORL vyšetření však byla popsána i subglotická laryngitida (obr. 2). Po zlepšení stavu byla pacientka s perorálními antibiotiky propuštěna do domácí péče. Za týden od propuštění došlo ke zhoršení dechových obtíží s výrazným stridorem. V klinickém nálezu dominovalo zarudnutí sliznic a stenóza subglotické krajiny (obr. 3). Pacientka byla hospitalizována na JIP ORL kliniky. Pacientka měla při příjmu elevované zánětlivé parametry - CRP 184 mg/l, RTG plic bylo bez nálezu ložiskových změn či jiné patologie. Po zavedení intravenózních antibiotik a kortikoterapie došlo k částečnému zlepšení klinických obtíží, avšak lokálně přetrvával obraz subglotické stenózy. Z laboratorních nálezů se po podané kortikoterapii manifestoval diabetes mellitus typu II a došlo k záchytu dosud neléčené arteriální hypertenze. Klinický stav a laboratorní výsledky však nesvědčily pro probíhající systémové onemocnění. Na základě konziliárního vyšetření byl doplněn panel imunologických parametrů, kde dominoval silně pozitivní nález PR3-ANCA. Pacientce byla nasazena pulzní terapie metylprednidolonem, po kterém došlo k rychlému ústupu obtíží, poklesu zánětlivých parametrů (CRP 6,9 mg/l) a hladiny ANCA. Pacientka byla pracovně vedena jako lokalizovaná forma granulomatózy s polyangiitididou. V rámci screeningového vyšetření bylo provedeno HRCT plic, USG břicha a opakované vyšetření moči, které neprokázaly patologické změny na jiných orgánech. Po zlepšení stavu pacientka podstoupila endoskopické vyšetření v celkové anestezii s odběrem vzorků k potvrzení diagnózy. V histologickém obrazu však nebyl potvrzen nález granulomů a nebyl tak zcela v souladu s diagnózou granulomatózy s polyangiitidou.
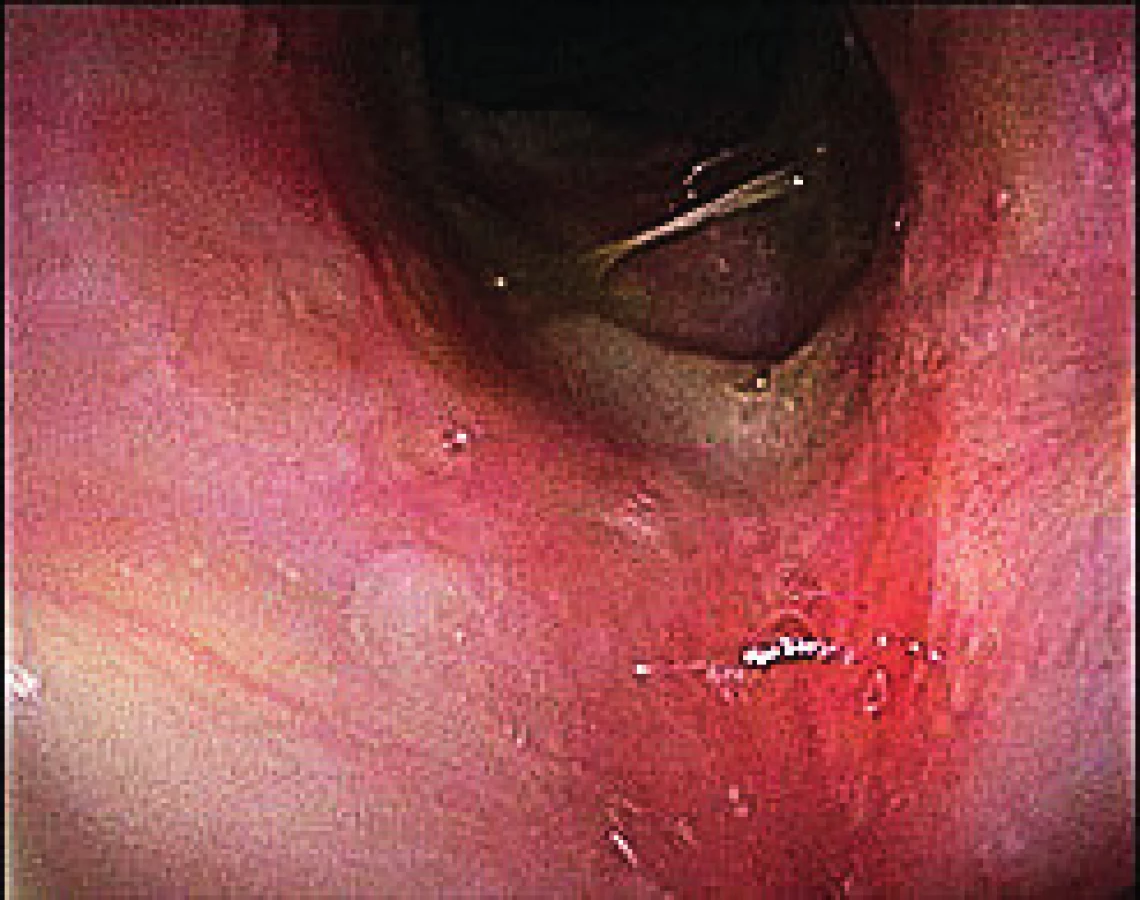
Tři měsíce od propuštění do ambulantní péče na postupně se snižující perorální kortikoteraii došlo opětovně ke zhoršení stavu ve smyslu dysfonie a poruše sluchu. Při kontrolním vyšetření dominoval nález zarudlé, oteklé sliznice v nosohltanu a nález sekretorické otitidy, pro kterou byla zavedena grometa do levého ucha. Byla opětovně navýšena imunosupresivní terapie s dočasným zlepšením stavu. V dalším průběhu se objevily dechové obtíže s videoendoskopickým nálezem subglotické stenózy s ulceracemi sahajícími až k prvnímu tracheálnímu prstenci. Ty se po dalším navýšení imunosupresivní terapie zhojily (obr. 4). Pacientka nadále podléhá pečlivé dispenzarizaci, je na udržovací terapii prednisonem a je vedena jako blíže nespecifikovaná ANCA-asociovaná vaskulitida.
DISKUSE
V tomto článku jsme představili problematiku primárních vaskulitid. Hlavním cílem bylo zpřístupnění tohoto tématu lékařům otorinolaryngologům, kteří se mohou stát významnou součástí diagnosticko-terapeutického týmu pečujícího o pacienta s vaskulitidou.
Primární vaskulitidy jsou vzácná onemocnění, avšak jejich projevy v oblasti hlavy a krku jsou relativně častými. Většina těchto symptomů není specifických pro žádné konkrétní onemocnění a podezření na vaskulitidu tak není mezi prvními diferenciálně-diagnostickými jednotkami. Vzhledem ke značné specializaci zdravotnictví v jednotlivé obory pak není vzácností ignorování případných projevů nespadajících pod odbornost vyšetřujícího lékaře. Vysoce suspektními jsou právě pacienti s abnormálně kumulovanými příznaky nasvědčujícími pro postižení více orgánových systému či případy s atypickými lokálními nálezy, u kterých se nepotvrdí infekční či nádorová etiologie a které nereagují na empirickou antimikrobiální léčbu.
Jak ukazují námi představené kazuistiky, tak i po vyslovení podezření na vaskulitidu a zapojení specialistů do diagnostického procesu může být přesné stanovení diagnózy obtížné až nemožné. Nevýhodou pro pacienta je pak problematické nastavení terapie, která se nemůže opírat o data medicíny založené na důkazech či velké empirické zkušenosti. Značně ztížena je samozřejmě i možnost predikce vývoje daného onemocnění. Snaha o přesné stanovení diagnózy je tak zásadní. Navíc, tak jako v případě naší pacientky č. 1, i časový faktor může být důležitou proměnnou. Vaskulitická onemocnění mohou vyústit v závažné stavy s život ohrožujícími systémovými reakcemi a rizikem trvalého postižení důležitých orgánů. I „naslepo“ podaná imunosupresivní terapie tak může alespoň pomoci zvládnout nejakutnější fáze onemocnění a poskytnout čas k dokončení diagnostického procesu.
V rámci článku jsme se zaměřili na primární vaskulitidy. Avšak zánětlivé postižení cév se může rozvinout i na podkladě již probíhajícího systémového onemocnění. Typickými spouštěči může být reakce proti vlastním antigenům v rámci autoimunitních onemocnění, jako je systémový lupus erythematodes, nebo v rámci paraneoplastického syndromu při maligním tumoru. Dalšími etiologickými faktory mohou být virové infekce, typicky viry hepatitidy B a C, nebo zánětlivé reakce vyvolané aktivací imunitního systému v rámci alergické reakce. I tyto faktory je třeba zohlednit v diferenciálně-diagnostických úvahách a neopomenout je při odběru anamnézy.
ZÁVĚR
Primární systémové vaskulitidy jsou vzácná onemocnění s možností manifestace v oblasti hlavy a krku a otorinolaryngolog se tak může stát jedním z lékařů stojícím na počátku diagnostického procesu. Měl by si být vědom možných projevů vaskulitid s typickou manifestací v ORL oblasti a v případě atypického klinického průběhu onemocnění zahrnout vaskulitidy do diferenciální diagnostiky.
Poděkování
Podpořeno grantem Progres Q28 1. LF UK.
Prohlášení o střetu zájmu
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa ke korespondenci:
MUDr. Vladimír Koucký
Klinika ORL a chirurgie hlavy a krku
1. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: vladimir.koucky@fnmotol.cz
Sources
1. Akar, S., Dogan, E., Goktay, Y. et al.: Nasal septal perforation in a patient with Takayasu‘s arteritis; a rare association. Intern Med, 48, 2009, 17, s. 1551-1554.
2. Alam, D. S., Seth, R., Sindwani, R. et al.: Upper airway manifestations of granulomatosis with polyangiitis. Cleve Clin J Med, 79, Suppl 3, 2012, s. S16-21.
3. Altenburg, A., Papoutsis, N., Orawa, H. et al.: Epidemiology and clinical manifestations of Adamantiades-Behcet disease in Germany -- current pathogenetic concepts and therapeutic possibilities. J Dtsch Dermatol Ges, 4, 2006, 1, s. 49-64; quiz 65-46.
4. Amor-Dorado, J. C., Llorca, J., Garcia-Porrua, C. et al.: Audiovestibular manifestations in giant cell arteritis: a prospective study. Medicine (Baltimore), 82, 2003, 1, s. 13-26.
5. Baráth, L., Liška, M., Mézesová, C. et al.: Churg-Straussov syndróm. (Kazuistika). Otorinolaryng a Foniat /Prague/, 52, 2003, 1, s. 42-44.
6. Biebl, M. O., Hugl, B., Posch, L. et al.: Subtotal tongue necrosis in delayed diagnosed giant-cell arteritis: a case report. Am J Otolaryngol, 25, 2004, 6, s. 438-441.
7. Brookes, G. B.: Pharyngeal stenosis in Behcet‘s syndrome. The first reported case. Arch Otolaryngol, 109, 1983, 5, s. 338-340.
8. Buttgereit, F., Dejaco, C., Matteson, E. L. et al.: Polymyalgia rheumatica and giant cell arteritis: A systematic review. JAMA, 315, 2016, 22, s. 2442-2458.
9. Calamia, K. T., Wilson, F. C., Icen, M. et al.: Epidemiology and clinical characteristics of Behcet‘s disease in the US: a population-based study. Arthritis Rheum, 61, 2009, 5, s. 600-604.
10. Cogan, D.: Syndrome of non-syphilitic interstitial keratitis and vestibuloauditorysymptoms. Arch Ophthalmol, 1945, 33, s. 144-149.
11. Cohen Tervaert, J. W., Damoiseaux, J.: Antineutrophil cytoplasmic autoantibodies: how are they detected and what is their use for diagnosis, classification and follow-up? Clin Rev Allergy Immunol, 43, 2012, 3, s. 211-219.
12. Comarmond, C., Pagnoux, C., Khellaf, M. et al.: Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum, 65, 2013, 1, s. 270-281.
13. Cottin, V., Bel, E., Bottero, P. et al.: Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): A study of 157 patients by the Groupe d‘Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev, 16, 2017, 1, s. 1-9.
14. Dagli, M., Eryilmaz, A., Tanrikulu, S. et al.: Evaluation of cochlear involvement by distortion product otoacoustic emission in Behcet‘s disease. Auris Nasus Larynx, 35, 2008, 3, s. 333-337.
15. Davatchi, F., Chams-Davatchi, C., Shams, H. et al.: Behcet‘s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol, 13, 2017, 1, s. 57-65.
16. De Virgilio, A., Greco, A., Magliulo, G. et al.: Polyarteritis nodosa: A contemporary overview. Autoimmun Rev, 15, 2016, 6, s. 564-570.
17. Diaz-Jouanen, E. D., Alarcon-Segovia, D.: Chondritis of the ear in Wegener‘s granulomatosis. Arthritis Rheum, 20, 1977, 6, s. 1286-1288.
18. Dolezalova, P., Telekesova, P., Nemcova, D. et al.: Incidence of vasculitis in children in the Czech Republic: 2-year prospective epidemiology survey. J Rheumatol, 31, 2004, 11, s. 2295-2299.
19. Ezzat, W. H., Compton, R. A., Basa, K. C. et al.: Reconstructive techniques for the saddle nose deformity in granulomatosis with polyangiitis: A systematic review. JAMA Otolaryngol Head Neck Surg, 143, 2017, 5, s. 507-512.
20. Faitlová, H., Bártová, I., Němec, V.: Kawasakiho syndrom - možná příčina krční lymfonoditidy u dětí. Otorinolaryng a Foniatr /Prague/, 59, 2010, 4, s. 202-208.
21. Fitzgerald, C. W., Adeeb, F., Timon, C. V. et al.: Significant laryngeal destruction in a northern European cohort of Behcet‘s disease patients. Clin Exp Rheumatol, 33, 2015, 6 Suppl 94, s. S123-128.
22. Flores-Suarez, L. F., Alba, M.A., Tona, G.: Severe microscopic polyangiitis with unilateral vocal cord paralysis as initial manifestation. Colomb Med (Cali), 48, 2017, 1, s. 32-34.
23. Fujiki, N., Nakamura, H., Nonomura, M. et al.: Bilateral vocal fold paralysis caused by polyarteritis nodosa. Am J Otolaryngol, 20, 1999, 6, s. 412-414.
24. Furusho, K., Kamiya, T., Nakano, H. et al.: High-dose intravenous gammaglobulin for Kawasaki disease. Lancet, 2, 1984, 8411, s. 1055-1058.
25. Gioffredi, A., Maritati, F., Oliva, E. et al.: Eosinophilic granulomatosis with polyangiitis: an overview. Front Immunol, 5, 2014, s. 549.
26. Gluth, M. B., Baratz, K. H., Matteson, E. L. et al.: Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc, 81, 2006, 4, s. 483-488.
27. Gluth, M. B., Shinners, P. A., Kasperbauer, J. L.: Subglottic stenosis associated with Wegener‘s granulomatosis. Laryngoscope, 113, 2003, 8, s. 1304-1307.
28. Gocke, D. J., Hsu, K., Morgan, C. et al.: Association between polyarteritis and Australia antigen. Lancet, 2, 1970, 7684, s. 1149-1153.
29. Goldfarb, J. M., Rabinowitz, M. R., Basnyat, S. et al.: Head and neck Manifestations of Eosinophilic Granulomatosis with Polyangiitis: A Systematic Review. Otolaryngol Head Neck Surg, 155, 2016, 5, s. 771-778.
30. Gonzalez-Gay, M. A., Garcia-Porrua, C.: Epidemiology of the vasculitides. Rheum Dis Clin North Am, 27, 2001, 4, s. 729-749.
31. Gonzalez-Gay, M. A., Garcia-Porrua, C., Llorca, J. et al.: Visual manifestations of giant cell arteritis. Trends and clinical spectrum in 161 patients. Medicine (Baltimore), 79, 2000, 5, s. 283-292.
32. Gonzalez-Gay, M. A., Vazquez-Rodriguez, T. R., Lopez-Diaz, M. J. et al.: Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum, 61, 2009, 10, s. 1454-1461.
33. Gottschlich, S., Ambrosch, P., Kramkowski, D. et al.: Head and neck manifestations of Wegener‘s granulomatosis. Rhinology, 44, 2006, 4, s. 227-233.
34. Grasland, A., Pouchot, J., Hachulla, E. et al.: Typical and atypical Cogan‘s syndrome: 32 cases and review of the literature. Rheumatology (Oxford), 43, 2004, 8, s. 1007-1015.
35. Greco, A., De Virgilio, A., Rizzo, M. I. et al.: Microscopic polyangiitis: Advances in diagnostic and therapeutic approaches. Autoimmun Rev, 14, 2015, 9, s. 837-844.
36. Greco, A., Gallo, A., Fusconi, M. et al.: Cogan‘s syndrome: an autoimmune inner ear disease. Autoimmun Rev, 12, 2013, 3, s. 396-400.
37. Gross, M., Ben-Chetrit, E.: Laryngeal involvement in Behcet‘s disease--a challenge for treatment. Clin Rheumatol, 32 Suppl 1, 2013, s. S75-77.
38. Gubbels, S. P., Barkhuizen, A., Hwang, P. H.: Head and neck manifestations of Wegener‘s granulomatosis. Otolaryngol Clin North Am, 36, 2003, 4, s. 685-705.
39. Hall, S., Barr, W., Lie, J. T. et al.: Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore), 64, 1985, 2, s. 89-99.
40. Hausch, R. C., Harrington, T.: Temporal arteritis and sensorineural hearing loss. Semin Arthritis Rheum, 28, 1998, 3, s. 206-209.
41. Hernandez-Rodriguez, J., Alba, M. A., Prieto-Gonzalez, S. et al.: Diagnosis and classification of polyarteritis nodosa. J Autoimmun, 48-49, 2014, s. 84-89.
42. Hilhorst, M., van Paassen, P., Tervaert, J. W. et al.: Proteinase 3-ANCA vasculitis versus myeloperoxidase-ANCA vasculitis. J Am Soc Nephrol, 26, 2015, 10, s. 2314-2327.
43. Hoffman, G. S., Thomas-Golbanov, C. K., Chan, J. et al.: Treatment of subglottic stenosis, due to Wegener‘s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol, 30, 2003, 5, s. 1017-1021.
44. Holman, R. C., Curns, A. T., Belay, E. D. et al.: Kawasaki syndrome hospitalizations in the United States, 1997 and 2000. Pediatrics, 112, 2003, 3 Pt 1, s. 495-501.
45. Hunder, G. G., Bloch, D. A., Michel, B. A. et al.: The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum, 33, 1990, 8, s. 1122-1128.
46. Churg, J., Strauss, L.: Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol, 27, 1951, 2, s. 277-301.
47. Iliescu, D. A., Timaru, C. M., Batras, M. et al.: Cogan‘s Syndrome. Rom J Ophthalmol, 59, 2015, 1, s. 6-13.
48. Jančatová, D., Komínek, P., Zeleník, K.: Coganův syndrom. Otorinolaryng a Foniat /Prague/, 64, 2015, 1, s. 42-45.
49. Jenkins, A. D., Mintz, P. D.: Optimal blood use in genitourinary surgery. J Urol, 126, 1981, 4, s. 497-499.
50. Jennette, J. C., Falk, R. J., Bacon, P. A. et al.: 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum, 65, 2013, 1, s. 1-11.
51. Kalfeřt, D., Čelakovský, P., Školoudík, L. et al.: Periferní paréza lícního nervu při středoušní manifestaci Wegenerovy granulomatózy. Otorinolaryng a Foniat /Prague/, 58, 2009, 2, s. 117-120.
52. Kato, H., Inoue, O., Kawasaki, T. et al.: Adult coronary artery disease probably due to childhood Kawasaki disease. Lancet, 340, 1992, 8828, s. 1127-1129.
53. Keni, S. P., Wiley, E. L., Dutra, J. C. et al.: Skull base Wegener‘s granulomatosis resulting in multiple cranial neuropathies. Am J Otolaryngol, 26, 2005, 2, s. 146-149.
54. Knight, J. M., Hayduk, M. J., Summerlin, D. J. et al.: „Strawberry“ gingival hyperplasia: a pathognomonic mucocutaneous finding in Wegener granulomatosis. Arch Dermatol, 136, 2000, 2, s. 171-173.
55. Kristin, J., Beutner, C., Klenzner, T. et al.: Granulomatosis with polyangiitis and multiple bilateral cranial nerve palsies: a diagnostic challenge. B-ENT, 10, 2014, 3, s. 231-235.
56. Kubota, M., Usami, I., Yamakawa, M. et al.: Kawasaki disease with lymphadenopathy and fever as sole initial manifestations. J Paediatr Child Health, 44, 2008, 6, s. 359-362.
57. Kulahli, I., Balci, K., Koseoglu, E. et al.: Audio-vestibular disturbances in Behcet‘s patients: report of 62 cases. Hear Res, 203, 2005, 1-2, s. 28-31.
58. Langford, C. A., Sneller, M. C., Hallahan, C. W. et al.: Clinical features and therapeutic management of subglottic stenosis in patients with Wegener‘s granulomatosis. Arthritis Rheum, 39, 1996, 10, s. 1754-1760.
59. Leavitt, R. Y., Fauci, A. S., Bloch, D. A. et al.: The American College of Rheumatology 1990 criteria for the classification of Wegener‘s granulomatosis. Arthritis Rheum, 33, 1990, 8, s. 1101-1107.
60. Lee, C. C., Su, W. W., Hunder, G. G.: Dysarthria associated with giant cell arteritis. J Rheumatol, 26, 1999, 4, s. 931-932.
61. Lukáš, J., Říhová, Z., Skalická, P. et al.: Nosní a krční manifestace Wegenerovy granulomatózy při multiorgánovém postižení. Otorinolaryng a Foniat /Prague/, 50, 2001, 4, s. 229-232.
62. Lupi-Herrera, E., Sanchez-Torres, G., Marcushamer, J. et al.: Takayasu‘s arteritis. Clinical study of 107 cases. Am Heart J, 93, 1977, 1, s. 94-103.
63. Magliulo, G., Parrotto, D., Alla, F. R. et al.: Acute bilateral facial palsy and Wegener‘s disease. Otolaryngol Head Neck Surg, 139, 2008, 3, s. 476-477.
64. Masi, A. T., Hunder, G. G., Lie, J. T. et al.: The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum, 33, 1990, 8, s. 1094-1100.
65. Metaxaris, G., Prokopakis, E. P., Karatzanis, A. D. et al.: Otolaryngologic manifestations of small vessel vasculitis. Auris Nasus Larynx, 29, 2002, 4, s. 353-356.
66. Meyers, A. D., Said, S.: Temporal artery biopsy: concise guidelines for otolaryngologists. Laryngoscope, 114, 2004, 11, s. 2056-2059.
67. Morales-Angulo, C., Vergara Pastrana, S., Obeso-Aguera, S. et al.: Otorhinolaryngological manifestations in patients with Behcet disease. Acta Otorrinolaringol Esp, 65, 2014, 1, s. 15-21.
68. Mouthon, L., Dunogue, B., Guillevin, L.: Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun, 48-49, 2014, s. 99-103.
69. Myklebust, G., Gran, J. T.: A prospective study of 287 patients with polymyalgia rheumatica and temporal arteritis: clinical and laboratory manifestations at onset of disease and at the time of diagnosis. Br J Rheumatol, 35, 1996, 11, s. 1161-1168.
70. Newburger, J. W., Taubert, K. A., Shulman, S. T. et al.: Summary and abstracts of the Seventh International Kawasaki Disease Symposium: December 4-7, 2001, Hakone, Japan. Pediatr Res, 53, 2003, 1, s. 153-157.
71. Paraskevas, K. I., Boumpas, D. T., Vrentzos, G. E. et al.: Oral and ocular/orbital manifestations of temporal arteritis: a disease with deceptive clinical symptoms and devastating consequences. Clin Rheumatol, 26, 2007, 7, s. 1044-1048.
72. Rao, J. K., Weinberger, M., Oddone, E. Z. et al.: The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis. A literature review and meta-analysis. Ann Intern Med, 123, 1995, 12, s. 925-932.
73. Rasmussen, N.: Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist‘s perspective. Curr Opin Rheumatol, 13, 2001, 1, s. 3-11.
74. Reinhold-Keller, E., Beuge, N., Latza, U. et al.: An interdisciplinary approach to the care of patients with Wegener‘s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum, 43, 2000, 5, s. 1021-1032.
75. Roszkowska, A., Morawska-Kochman, M., Temporale, H. et al.: Bilateral facial palsy in rapidly progressive course of Wegener‘s granulomatosis: a case report. Case Reports in Otolaryngology, 2013, 2013, s. 875108.
76. Rubin, F., Tran Khai Hoan, N., Bonfils, P.: Sudden bilateral hearing loss revealing polyarteritis nodosa. Eur Ann Otorhinolaryngol Head Neck Dis, 131, 2014, 4, s. 265-266.
77. Scully, C., Eveson, J. W., Barrett, A. W. et al.: Necrosis of the lip in giant cell arteritis: report of a case. J Oral Maxillofac Surg, 51, 1993, 5, s. 581-583.
78. Seo, P., Stone, J. H.: The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med, 117, 2004, 1, s. 39-50.
79. Shine, N. P., Hamilton, S., McShane, D. P.: Takayasu‘s arteritis and saddle nose deformity: a new association. J Laryngol Otol, 120, 2006, 1, s. 59-62.
80. Shvero, J., Shitrit, D., Koren, R. et al.: Endoscopic laser surgery for subglottic stenosis in Wegener‘s granulomatosis. Yonsei Med J, 48, 2007, 5, s. 748-753.
81. Siglock, T. J., Brookler, K. H.: Sensorineural hearing loss associated with Takayasu‘s disease. Laryngoscope, 97, 1987, 7 Pt 1, s. 797-800.
82. Sinico, R. A., Di Toma, L., Maggiore, U. et al.: Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum, 52, 2005, 9, s. 2926-2935.
83. Sinico, R.A., Di Toma, L., Maggiore, U. et al.: Renal involvement in Churg-Strauss syndrome. Am J Kidney Dis, 47, 2006, 5, s. 770-779.
84. Smith, M.E., Elstad, M.: Mitomycin C and the endoscopic treatment of laryngotracheal stenosis: are two applications better than one? Laryngoscope, 119, 2009, 2, s. 272-283.
85. Srouji, I.A., Andrews, P., Edwards, C. et al.: Patterns of presentation and diagnosis of patients with Wegener‘s granulomatosis: ENT aspects. J Laryngol Otol, 121, 2007, 7, s. 653-658.
86. Srouji, S., Kizhner, T., Suss-Tobi, E. et al.: 3-D Nanofibrous electrospun multilayered construct is an alternative ECM mimicking scaffold. J Mater Sci Mater Med, 19, 2008, 3, s. 1249-1255.
87. Trimarchi, M., Sinico, R. A., Teggi, R. et al.: Otorhinolaryngological manifestations in granulomatosis with polyangiitis (Wegener‘s). Autoimmun Rev, 12, 2013, 4, s. 501-505.
88. Tsunoda, K., Akaogi, J., Ohya, N. et al.: Sensorineural hearing loss as the initial manifestation of polyarteritis nodosa. J Laryngol Otol, 115, 2001, 4, s. 311-312.
89. Van Doornum, S., McColl, G., Walter, M. et al.: Prolonged prodrome, systemic vasculitis, and deafness in Cogan‘s syndrome. Ann Rheum Dis, 60, 2001, 1, s. 69-71.
90. Watts, R., Lane, S., Hanslik, T. et al.: Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis, 66, 2007, 2, s. 222-227.
91. Watts, R. A., Lane, S. E., Scott, D. G. et al.: Epidemiology of vasculitis in Europe. Ann Rheum Dis, 60, 2001, 12, s. 1156-1157.
92. Wawrzecka, A., Szymanska, A., Jeleniewicz, R. et al.: Granulomatosis with polyangiitis with bilateral facial palsy and severe mixed hearing loss. Case Reports in Otolaryngology, 2016, 2016, s. 5206170.
93. Weyand, C. M., Goronzy, J. J.: Medium- and large-vessel vasculitis. N Engl J Med, 349, 2003, 2, s. 160-169.
94. Wojciechowska, J., Krajewski, W., Krajewski, P. et al.: Granulomatosis with polyangiitis in otolaryngologist practice: A Review of Current Knowledge. Clin Exp Otorhinolaryngol, 9, 2016, 1, s. 8-13.
95. Wolf, M., Kronenberg, J., Engelberg, S. et al.: Rapidly progressive hearing loss as a symptom of polyarteritis nodosa. Am J Otolaryngol, 8, 1987, 2, s. 105-108.
96. Wong, R. J., Gliklich, R. E., Rubin, P. A. et al.: Bilateral nasolacrimal duct obstruction managed with endoscopic techniques. Arch Otolaryngol Head Neck Surg, 124, 1998, 6, s. 703-706.
97. Yoskovitch, A., Tewfik, T. L., Duffy, C. M. et al.: Head and neck manifestations of Kawasaki disease. Int J Pediatr Otorhinolaryngol, 52, 2000, 2, s. 123-129.
98. Yuminaga, Y., Richards, B., Rasiah, K. et al.: Polyarteritis nodosa presenting with bilateral testicular swelling and complicated by unilateral facial nerve palsy. Korean J Urol, 52, 2011, 5, s. 364-367.
Labels
Audiology Paediatric ENT ENT (Otorhinolaryngology)Article was published in
Otorhinolaryngology and Phoniatrics

2019 Issue 4
Most read in this issue
- První zkušenosti s vyšetřením závrativých stavů video Head Impulse Testem
- Obojstranný peritonzilárny absces
- Primární vaskulitidy v otorinolaryngologii: přehled literatury a kazuistické případy
- Terapie chronického sekretorického středoušního zánětu balónkovou dilatací Eustachovy tuby