
Postižení kardiovaskulárního systému u žen s Turnerovým syndromem, kardiovaskulární rizika spojená s těhotenstvím
Cardiovascular disease in Turner syndrome, cardiovascular risks associated with pregnancy
Turner syndrome (TS), or monosomy X, is characterised by short stature, premature ovarian failure and congenital cardiovascular defects in phenotypic females. Congenital cardiovascular disease affects approximately 50% of individuals and is the major cause of premature mortality in adults with TS. Dissection of the aorta occurs in TS patients at a remarkably young age (mean 36 years). Aortic dissection is usually associated with additional risk factors including bicuspid aortic valve or other abnormalities of the aortic valve, coarctation or dilatation of the aorta, and systemic arterial hypertension. In recent years, increasing numbers of women with TS have become pregnant via assisted reproduction. The risk for aortic dissection or rupture during pregnancy is 2% or higher and the risk of death is increased as much as 100-fold. Because of this fact, the Practice Committee of the American Society for Reproductive Medicine recommended in 2006 a careful screening for cardiovascular abnormalities including echocardiography and cardiovascular magnetic resonance imaging in all TS patients before any planned attempt at pregnancy. Women with any risk factors for aortic dissection should not undertake pregnancy.
Key words:
Turner syndrome – congenital heart disease – aortic dissection – systemic arterial hypertension – pregnancy
Authors:
E. Klásková; J. Zapletalová; A. Sobek; D. Horák; J. Wiedermann; A. Sobek jr.
Authors‘ workplace:
Dětská klinika FN a LF UP, Olomouc
1; FERTIMED, Centrum pro léčbu neplodnosti, Olomouc
2; Radiologická klinika FN a LF UP, Olomouc
3
Published in:
Prakt Gyn 2010; 14(3): 142-146
Category:
Review Article
Overview
Turnerův syndrom (TS), neboli monozomie X (karyotyp 45,X), je charakterizována malým vzrůstem, předčasným ovariálním selháním a přítomností vrozených srdečních vad. TS je vždy spojen s ženským fenotypem. Vrozené srdeční vady postihují přibližně 50 % nositelek TS a jsou hlavní příčinou předčasné mortality u dospělých žen s TS. Disekce aorty postihuje pacientky s TS v nápadně nižším věku (průměr 36 let). Aortální disekce je obvykle spojena s rizikovými faktory, jako jsou bikuspidální aortální chlopeň nebo jiné abnormality aortální chlopně, koarktace nebo dilatace aorty a systémová arteriální hypertenze. V posledních letech otěhotní narůstající počet žen s TS díky metodám arteriální hypertenze. Riziko disekce nebo ruptury aorty během gravidity je více než 2 % a riziko úmrtí těhotné ženy je zvýšeno více než stokrát. Z tohoto důvodu Practice Committee of the American Society for Reproductive Medicine vydal v roce 2006 doporučení provést u všech žen s TS, které plánují těhotenství, podrobné screeningové kardiologické vyšetření včetně echokardiografického vyšetření a magnetické rezonance srdce a velkých cév. U žen s jakýmkoli rizikovým faktorem pro aortální disekci je gravidita kontraindikována.
Klíčová slova:
Turnerův syndrom – vrozená srdeční vada – aortální disekce – systémová arteriální hypertenze – těhotenství
Úvod
Turnerův syndrom (TS) je jednou z nejčastějších chromozomálních aberací. Postnatální incidence je udávána přibližně 1 na 2 000 živě narozených děvčátek. V České republice žije v současné době více než 2 000 dívek a žen s TS a každoročně se narodí dalších 20–25 novorozenců s TS.
Za nejčastější chromozomální odchylku u TS bývá považována monozomie X (45,X), kdy zcela chybí druhý pohlavní chromozom. Strukturální abnormity chromozomu X jsou méně časté. Patří k nim izochromozom z dlouhých ramének (iXq), ring chromozom (rX), případně může chybět krátké (Xp-) nebo dlouhé (Xp-) raménko X chromozomu. Tyto abnormity mohou postihovat všechny buňky nebo jen jejich část ve formě chromozomální mozaiky. Výjimečně bývá přítomen Y chromozom (45,X/46,XY) nebo jeho část.
Díky rozvoji nových cytogenetických a molekulárních metod jsou původní genetické nálezy přehodnocovány a častěji jsou diagnostikovány i minoritní mozaikové formy TS. Abnormity X chromozomu, typické pro TS, se vyskytují u 1–3% všech koncepcí, ale 99% fétů s karyotypem 45,X je spontánně potraceno již v rané fázi gravidity v důsledku srdečního selhání [1]. Toto zjištění vedlo k předpokladu, že větší „životaschopnost“ mají plody s chromozomální mozaikou, jejichž druhý pohlavní chromozom je v některých tkáních alespoň ve zbytku zachován. Někteří autoři se domnívají, že čistá monozomie (45,X) není slučitelná se životem, a že se tedy u TS vždy jedná o skrytou, nerozpoznanou mozaiku.
K příznakům, které vedou k podezření na TS, patří malá postava, dysgeneze gonád a pozůstatky fetálního lymfedému, jako jsou kožní duplikatury po stranách krku (pterygium colli), nízká vlasová hranice, široký hrudník s oddálenými bradavkami a postnatální lymfedém na dorzech rukou a na nártech (obr. 1, 2). U TS je vysoká incidence renálních a kardiovaskulárních vad. Nositelky TS nemají mentální retardaci s výjimkou pacientek s ring chromozomem.

Stanovení diagnózy TS nečiní potíže u pacientek, které mají zřejmé klinické projevy (zejména dívky s karyotypem 45,X). Existují ale dívky a ženy s TS, které mají minimální symptomy, a jediným projevem TS může být kromě nevýrazné růstové retardace porucha menstruačního cyklu, předčasné ovariální vyhasnutí nebo poruchy plodnosti. Jsou to především pacientky s minoritní chromozomální mozaikou (za TS je považován karyotyp s více než 5% patologických buněčných linií) nebo s delecí dlouhého raménka chromozomu X [2]. Tyto ženy mohou být klientkami IVF center a pokud nemají vyšetřen karyotyp, nemusí být rozpoznány jako nositelky TS.
Sdělení je věnováno kardiovaskulárním projevům TS a jeho cílem je upozornit na vysokou prevalenci a široké spektrum kardiovaskulárních anomálií, které jsou často klinicky zcela němé. První manifestací může být fatálně probíhající disekce a ruptura aorty u mladé ženy.
Vrozené srdeční vady u TS
Spektrum vrozených i získaných srdečních anomálií je široké a postihuje zejména levostranné srdeční oddíly, tj. aortální chlopeň a ascendentní aortu. Nejčastějšími srdečními vadami jsou dvojcípá aortální chlopeň, koarktace aorty a dilatace ascendentní aorty [3–5]. Méně známé je, že pacientky s TS mají nápadně vyšší výskyt parciálního anomálního návratu plicních žil (13%) a perzistující levostranné horní duté žíly (13%). Z recentních studií vyplývá, že až 50% žen s TS má nějakou vrozenou srdeční anomálii.
Příčina vzniku vrozených srdečních vad není doposud jednoznačně objasněna. Nejvíce je akceptována teorie, že jsou důsledkem haploinsuficience lymfogenního genu v pseudoautozomální oblasti 1 chromozomu X (PAR1). Kritické místo zodpovědné za vývoj lymfatického systému leží v lokusu Xp11.4 [6].
V roce 1984 vyslovil Clark [7] hypotézu, že vznik vrozených srdečních vad u TS je způsoben pozdním vývojem nebo úplně chybějící drenáží mezi ductus thoracicus a jugulárními a podkličkovými vénami. Hromadící se lymfa rozšiřuje mízní systém v zadních a bočních oblastech krku. Důsledkem je vznik tzv. hygroma colli cysticum, které je vedoucím symptomem pro prenatální ultrazvukovou diagnostiku TS. Generalizovaný lymfedém je příčinou spontánních potratů u více než 99,9% plodů s karyotypem 45,X. Hypotetickým důvodem potratu je porušený venózní návrat v důsledku tlaku nadměrně dilatovaných hrudních lymfatických cév, což následně vede k srdečnímu selhání.
Intrauterinně vzniklá dilatace mízního systému způsobuje i řadu klinicky nápadných postnatálních projevů TS (pterygium colli, nízká vlasová hranice, abnormální tvar a posazení ušních boltců, „štítovitý“ tvar hrudníku a lymfedémy nártů, hřbetů rukou a zevního genitálu). Opakovaně bylo potvrzeno, že jedinci s těmito typickým stigmaty mají signifikantně vyšší výskyt vrozených srdečních vad [8].
Možnost detekce kardiovaskulárních abnormalit se výrazně zvýšila se zavedením nových vyšetřovacích metod, zejména magnetické rezonance (MRI) srdce a velkých cév. Echokardiografické vyšetření u dívek a žen s TS bývá komplikováno zhoršenou viditelností při transtorakálním vyšetření. Příčinou je sklon k obezitě a neobvyklá architektura hrudního koše, která je pozůstatkem fetálního lymfedému. MRI naopak umožňuje spolehlivě zobrazit hrudní aortu a stanovit morfologii aortální chlopně u žen, které nejsou echokardiograficky vyšetřitelné [5,9,10].
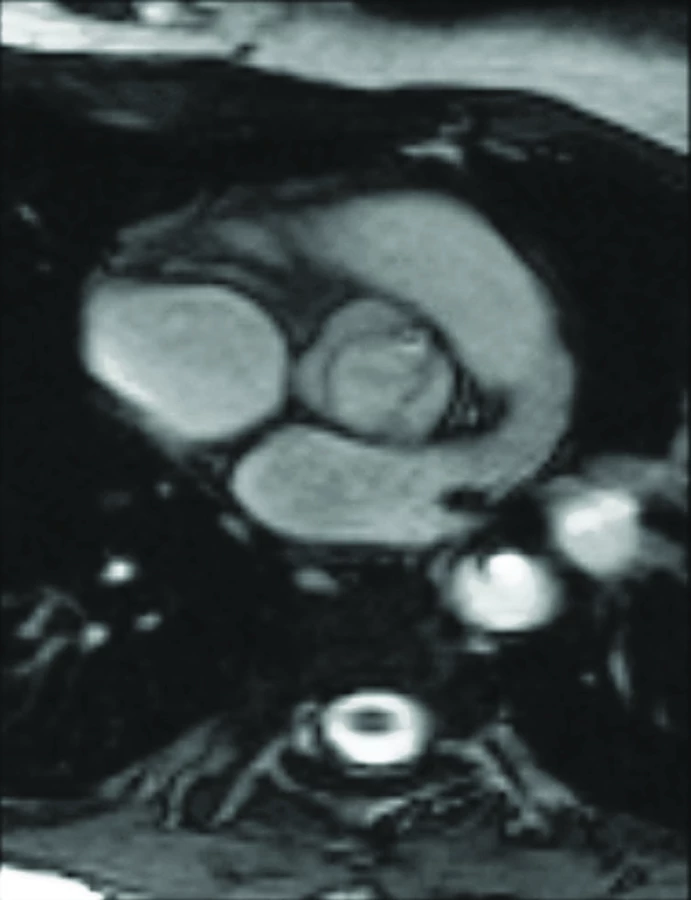
Nejčastější vrozenou anomálií u TS je bikuspidální aortální chlopeň. Její prevalence u TS byla v minulosti podhodnocována [3,4,11–14]. V poslední publikované studii, v níž byla vyšetřena neselektovaná populace 250 žen s TS, byla diagnostikována bikuspidální aortální chlopeň u 30% pacientek [5]. U 6% nebylo možné echokardiograficky vizualizovat aortální chlopeň pro špatnou vyšetřitelnost. Při kombinaci echokardiografie a MRI se podařilo stanovit morfologii 99% aortálních chlopní (obr. 3). Bylo zjištěno, že ženy s bikuspidální aortální chlopní mají signifikantně větší rozměr aorty na úrovni aortálního anulu, sinů, sinotubulární junkce a větší průměr ascendentní aorty. Ve skupině 51 žen, které prodělaly disekci aorty a zároveň u nich bylo provedeno kardiologické vyšetření před touto komplikací, mělo 27% z nich izolovanou bikuspidální chlopeň a 18% kombinaci bikuspidální aortální chlopně a koarktace aorty [15].
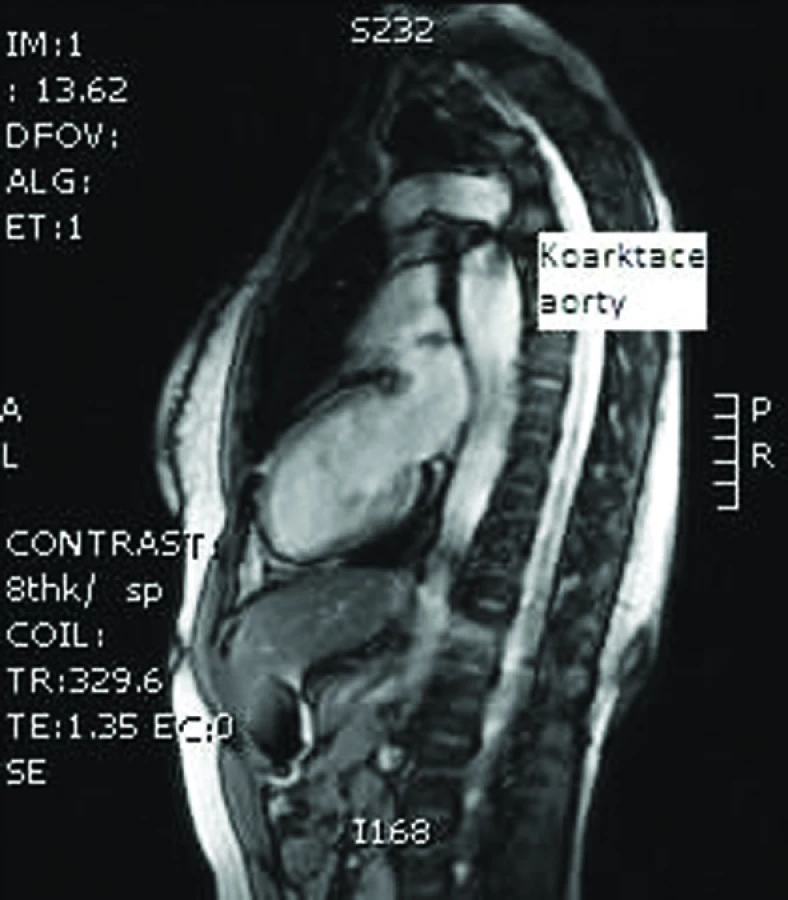
Diagnóza koarktace aorty je na rozdíl od bikuspidální aortální chlopně obvykle stanovena na podkladě klinických příznaků v kojeneckém a batolecím věku, kdy vyžaduje chirurgické řešení. Prevalence koarktace aorty je udávána u dívek a žen s TS mezi 7–18% [3,4,12,13,16]. Se zavedením kardiovaskulárního screeningu u všech pacientek s TS a se zvýšenou dostupností MRI se setkáváme s nově diagnostikovanými případy klinicky němých koarktací aorty i po 10. roce života (obr. 4). I tyto nálezy jsou vysoce rizikové pro disekci aorty.
MRI angiografie umožňuje až u 50% nositelek s TS detekovat anomálie aortálního oblouku obtížně rozpoznatelné echokardiograficky, a to zejména elongaci transverzálního oblouku (ETA) s prohnutím oblouku v místě aortálního istmu [16]. V budoucnu bude nutné objasnit, zda přítomnost ETA predikuje aortální komplikace ve smyslu dilatace a disekce aorty, nebo zda mají tendenci k progresi do koarktace.
Dilatace aorty a disekce aorty
Nejméně 1,4% dívek a žen s TS prodělá v průběhu života aortální disekci [17,18]. Většina z nich má prokazatelný alespoň jeden ze známých rizikových faktorů, k nimž patří koarktace aorty, bikuspidální aortální chlopeň, dilatace ascendentní aorty a arteriální hypertenze [19,20]. U 10–25% žen s disekcí ale nebyl žádný z rizikových faktorů nalezen [8,15]. Z tohoto důvodu je TS považován za samostatný rizikový faktor pro disekci i bez přítomnosti jiných rizikových faktorů. Nejvyšší riziko disekce mají ženy s typickými stigmaty TS.
S aortální disekcí se u pacientek s TS setkáváme ve výrazně nižším věku než v běžné populaci, a to ve druhém a třetím decenniu (medián věku je 36 let oproti 68 letům v běžné populaci). Tento nízký věk vede k podcenění prvních symptomů, jako jsou bolest na hrudi, bolest zad, pocení, tachykardie. Důsledkem je vysoká mortalita, která přesahuje 50% [15]. Při histologickém vyšetření bývá zjištěna cystická nekróza medie aorty.
Jedním z nejzávažnějších rizikových faktorů pro disekci aorty je dilatace ascendentní aorty. Její prevalence je udávána 15–30% dívek a žen s TS [10,21,22]. Zásadním problémem je, jak definovat dilataci aorty u této specifické populace. Rozměry aorty jsou určeny věkem a tělesným povrchem. Populace dívek a žen s TS je malého vzrůstu a má sklon k obezitě. Používání absolutních hodnot naměřených rozměrů aorty bez vztažení k tělesnému povrchu vede u pacientek s TS k poddiagnostikování dilatace aorty a může mít závažné důsledky.
V současné době je zlatým standardem k detekci dilatace ascendentní aorty MRI (obr. 5) [9,22,23]. Jedním z prvních používaných markerů dilatace ascendentní aorty bylo stanovení poměru ascendentní a descendentní aorty (Asc.Ao/Desc.Ao ratio) na úrovni pravé větve plicnice. Aplikace tohoto poměru vychází z předpokladu, že dilatace postihuje výhradně ascendentní aortu a rozměr descendentní aorty je normální. Za signifikantní dilataci je považován poměr ≥ 1,5 [21].
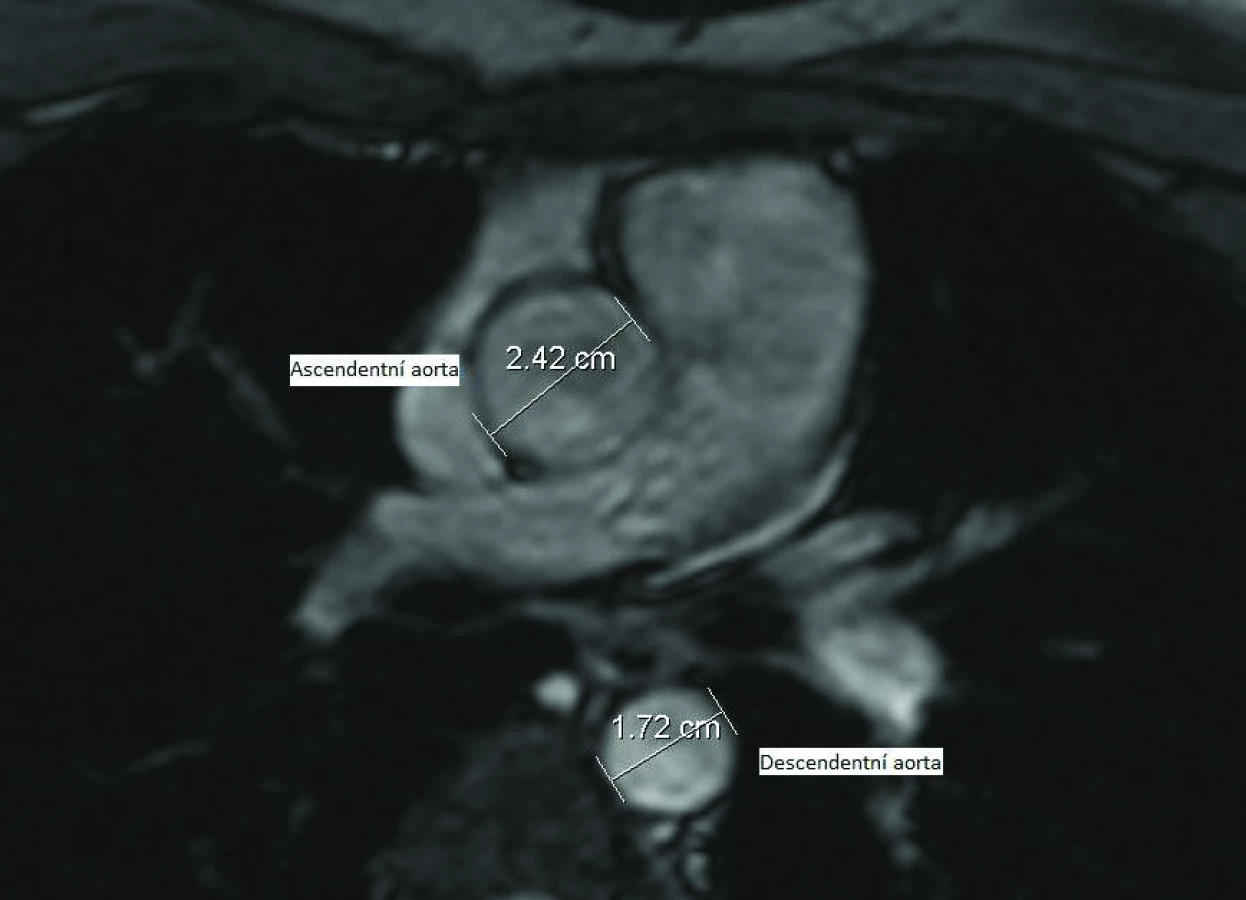
Dle poslední studie z roku 2007 je doporučován pro screening dilatace aorty u TS indexovaný aortální rozměr (ASI – aortic size index), což je průměr ascendentní aorty vztažený k tělesnému povrchu. Tato metoda má nejvyšší senzitivitu i specificitu při predikci dalších komplikací. Za 95. percentil byla stanovena hodnota 2,0cm/m2 a 99. percentil 2,5cm/m2 [24]. Ženy s ASI > 2,5cm/m2 měly v této studii riziko disekce v následujících třech letech 33%. V současnosti se jedná o nejvíce akceptovaný ukazatel klinicky závažné dilatace.
Dosud není vyřešena podstatná otázka, zda má dilatace ascendentní aorty u konkrétní pacientky progresivní charakter. Lanzarini [25] prokázal při tříletém echokardiografickém sledování 78 dívek a žen s TS pomalou progresi dilatace aorty na všech úrovních. I když nebyla klinicky závažná, pacientky musí být celoživotně sledovány kardiologem. Zároveň chybí jednoznačný algoritmus, kdy by žena s TS a dilatací aorty měla podstoupit preventivní chirurgický zákrok ke stabilizaci ascendentní aorty. Rovněž není vyřešeno, zda je možno progresi aortální dilatace ovlivnit farmakologicky (b-blokátory nebo ACE inhibitory).
K dalším závažným rizikovým faktorům patří arteriální hypertenze [10]. Obecně je známo, že více než 50% žen s TS má při 24hodinovém monitorování (ABPM) arteriální hypertenzi. Typické je narušení diurnálního rytmu arteriálního tlaku, kdy je redukován nebo zcela chybí noční pokles. U nositelek TS je nutná přísná farmakologická a režimová kontrola hypertenze. V graviditě platí toto pravidlo dvojnásobně.
Kardiovaskulární rizika v graviditě
V posledních dvou desetiletích se pro ženy s TS otevřela reálná možnost otěhotnění s využitím metod asistované reprodukce z dárcovského oocytu. Narůstající počet těhotenství a živě narozených dětí je vykoupen vysokou frekvencí mateřských komplikací, jako jsou arteriální hypertenze, eklampsie a aortální disekce [26].
V práci španělských autorů byla přítomna arteriální hypertenze u pěti z osmi gravidních žen s TS, z toho se u tří pacientek vyvinula preeklampsie [27]. Důsledkem vysokého výskytu arteriální hypertenze byla intrauterinní růstová retardace a předčasný porod ve více než 50% případů. Další autoři sledovali arteriální hypertenzi u 6 z 18 těhotných [28]. Všeobecně je doporučeno snížit riziko hypertenze transferem jediného embrya ve snaze vyhnout se mnohočetným graviditám.
Mateřská mortalita u TS v důsledku aortální disekce je odhadována na více než 2% [15]. V nejrozsáhlejší metaanalýze 85 případů žen s TS a aortální disekcí bylo sedm případů v souvislosti s graviditou a jen jedna ze sedmi žen přežila chirurgickou intervenci. V další sérii pacientek s TS a aortální disekcí byly dvě těhotné ženy z 16 případů [17]. Zvýšené riziko disekce během těhotenství se týká i vzácných případů žen s TS, které spontánně otěhotní. Nejvyšší riziko disekce je na začátku gravidity a v průběhu třetího trimestru v důsledku hyperkinetické cirkulace.
Na podkladě narůstajících dat o riziku aortální disekce v graviditě vydal The Practice Committee of the American Society for Reproductive Medicine [29] v roce 2006 tato doporučení, jak přistupovat k těhotenství u žen s TS.
- signifikantní srdeční vady (dilatace ascendentní aorty, bikuspidální aortální chlopeň a koarktace aorty), jsou kontraindikací těhotenství
- u žen s normálním kardiologickým nálezem je nutno zajistit důslednou léčbu arteriální hypertenze a opakované echokardiografické vyšetření v graviditě
- ženy ve stabilizovaném stavu s průměrem aortálního kořene < 4cm mohou rodit spontánně v epidurální anestezii, u žen s dilatací aortálního kořene je doporučen porod císařským řezem
Výše zmíněná doporučení jsou v současné době předmětem diskuze [30]. Jedna z největších autorit v kardiovaskulární problematice TS Carolyn Bondy [31] doporučuje na podkladě nových poznatků upravit doporučení následujícím způsobem:
- u žen se srdeční vadou (bikuspidální aortální chlopeň nebo koarktace aorty) nebo s hypertenzí je gravidita kontraindikována
- ženy bez známé srdeční vady mají být podrobně kardiologicky vyšetřeny před plánovanou graviditou včetně magnetické rezonance srdce a hrudní aorty
- ženy s indexovaným rozměrem ascendentní aorty nad 2,0cm/m2 by neměly otěhotnět
Autorka rovněž zdůrazňuje, že i ženy bez výše zmíněných rizikových faktorů mohou být nositelkami latentní vaskulopatie, která se může manifestovat až v graviditě disekcí aorty.
Závěr
Nositelky TS mohou v případě včasné diagnózy a při adekvátní medikamentózní léčbě dosáhnout plné feminizace a skoro normální dospělé výšky. Kvalita jejich života se téměř neliší od běžné populace.
Výhlídky na normální délku života však bývají limitovány rizikem předčasné kardiovaskulární morbidity a mortality, zejména v důsledku disekce ascendentní aorty. Gravidita představuje pro tyto ženy vysoce rizikové období s nejméně dvouprocentní mateřskou úmrtností. Ženy, které se rozhodnou využít metod umělého oplodnění, musí být podrobně informovány o kardiovaskulárních rizicích a projít podrobným kardiologickým vyšetřením včetně MRI srdce a hrudní aorty. Při zjištění signifikantní srdeční vady, dilatace ascendentní aorty a nekontrolované arteriální hypertenze je těhotenství kontraindikováno.
Ženy menšího vzrůstu, které se léčí pro neplodnost, by měly podstoupit genetické vyšetření včetně karyotypu. Pokud se u nich prokáže TS, je u nich nutné provést před plánovanou graviditou kardiologické vyšetření.
Doručeno do redakce: 19. 9. 2010
Přijato po recenzi: 11. 10. 2010
MUDr. Eva Klásková1,2
doc. MUDr. Jiřina Zapletalová, Ph.D.1
doc. MUDr. Aleš Sobek, CSc.2
MUDr. David Horák, Ph.D.3
MUDr. Jaroslav Wiedermann, CSc.1
MUDr. Aleš Sobek Jr.2
1Dětská klinika FN a LF UP, Olomouc
2Fertimed, centrum pro léčbu neplodnosti, Olomouc
3Radiologická klinika FN a LF UP, Olomouc
E.Klaskova@seznam.cz
Sources
1. Surerus E, Huggon IC, Allan LD. Turner‘s syndrome in fetal life. Ultrasound Obstet Gynecol 2003; 22(3): 264–267.
2. Bondy CA, Turner Syndrome Study Group. Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metabo 2007; 92(1): 10–25.
3. Gøtzsche CO, Krag-Olsen B, Nielsen J et al. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child 1994; 71(5): 433-436.
4. Sybert VP. Cardiovascular malformations and complications in Turner syndrome. Pediatrics 1998; 101(1): E11.
5. Sachdev V, Matura LA, Sidenko S et al. Aortic valve disease in Turner syndrome. JACC 2008; 51(19): 1904–1909.
6. Boucher CA, Sargent CA, Ogata T et al. Breakpoint analysis of Turner patients with partial Xp deletions: implications for the lymphoedema gene location. J Med Genet 2001; 38(9): 591–598.
7. Clark EB. Neck Web and Congenital Heart Defects: A Pathogenetic Association in 45 X-0 Turner Syndrome. Teratology 1984; 29 : 355–361.
8. Bondy CA. Congenital cardiovascular disease in Turner syndrome. Congenit Heart Dis 2008; 3 : 2–15.
9. Cleemann L, Mortensen KH, Holm K et al. Aortic Dimensions in Girls and Young Women with Turner Syndrome: A Magnetic Resonance Imaging Study. Pediatr Cardiol 2010; 31(4): 497–504.
10. Hjerrild BE, Mortensen KH, Sørensen KE et al. Thoracic aortopathy in Turner syndrome and the influence of bicuspid aortic valves and blood pressure: a CMR study. J Cardiovasc Magn Reson 2010; 12(3): 12.
11. Price WH, Clayton JF, Collyer S et al. Mortality ratios, life expectancy, and causes of death in patients with Turner‘s syndrome. J Epidemiol Community Health 1986; 40(2): 97–102.
12. Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner‘s syndrome. J Pediatr 1998; 133(5): 688–692.
13. Völkl TM, Degenhardt K, Koch A et al. Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome the Erlagen experience. Clin Cardiol 2005; 28(2): 88–92.
14. Loscalzo M, Van Phillip L, Ho VB et al. Association Between Fetal Lymfedema and Congenital Cardiovascular Defects in Turner Syndrome. Pediatrics 2005; 115(3): 732–735.
15. Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. J Med Genet 2007; 44(2): 745–749.
16. Ho VB, Bakalov VK, Cooley M et al. Major vascular abnormalities in Turner syndrome: Prevalence and magnetic resonance angiographic features. Circulation 2004; 110(12): 1694–1700.
17. Gravholt CH, Landin-Wilhelsemsen K, Stochholm K et al. Clinical and epidemiological description of aortic dissection in Turner’s syndrome. Cardiol Young 2006; 16(5): 430–436.
18. Lopez L, Arheart KL, Colan SD et al. Turner Syndrome Is an Independent Risk Factor for Aortic Dilatation in the Young. Pediatrics 2008; 121(6): e1622–e1627.
19. Lin Angela E, Lippe B, Rosenfeld RG. Further Delineation of Aortic Dilatation, Dissection, an Rupture in Patients With Turner syndrome. Pediatrics 1998; 102(1): E12.
20. Bondy CA. Aortic dissection in Turner syndrome. Curr Opin Cardiol 2008; 23(6): 519–526.
21. Ostberg JE, Brookes JAS, McCarthy C et al. A Comparison of Echocardiography and Magnetic Resonance Imaging in Cardiovascular Screening of Adults with Turner Syndrome. J Clin Endocrinol Metabolism. J Clin Endocrinol Metabolism 2004; 80(12): 5966–5971.
22. Chalard F, Ferey S, Teinturier C et al. Aortic dilatation in Turner syndrome: the role of MRI in early recognition. Pediatr Radiol 2005; 35(3): 323–326.
23. Mortensen KH, Hjerrild BE, Andersen NH. Abnormalities of the major intrathoracic arteries in Turner syndrome as revealed by magnetic resonance imaging. Cardiol Young 2010; 20(2): 191–200.
24. Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation 2007; 116(15): 1663–1670.
25. Lanzarini L, Larizza D, Prete Prete G et al. Prospective evaluation of aortic dimensions in Turner syndrome: A 2-dimensional echocardiographic study. J Am Soc Echocardiogr 2007; 20(3): 307–313.
26. Hovatta O, Hreinsson J, Fridström M et al. Fertility and pregnancy aspects in Turner syndrome. International Congress Series 1298 2006; 185–189.
27. Bodri D, Vernaeve V, Figueras F et al. Oocyte donation in patients with Turner‘s syndrome: a successful technique but with an accompanying high risk of hypersensitive disorders during pregnancy. Hum Reprod 2006; 21(3): 829–832.
28. Foudila T, Söderström-Anttila V, Hovatta O. Turner‘s syndrome and pregnancies after oocyte donation. Hum Reprod 1999; 14(2): 532–535.
29. The Practice Comimittee of the American Society for Reproductive Medicine. Increa - sed maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome. Fertil Steril 2008; 90 (Suppl 3): 185–186.
30. Šnajderová M, Heresová J, Mardešič T et al. Turnerův syndrom: přehled problematiky, současný stav, návrh koncepce péče a protokol sledování v dětství, adolescenci a dospělosti. Lék. čes 2001; 140 : 533–537.
31. Bondy CA, Rosing D, Reindollar R. Cardiovascular risks of pregnancy in women with Turner syndrome. Letter to the editor. Fertil Steril 2009; 91(5): e31–32.
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Practical Gynecology

2010 Issue 3
Most read in this issue
- Borderline ovariální tumory
- Monozygotní dvojčata v asistované reprodukci
- Elektivní single embryo transfer
- Postižení kardiovaskulárního systému u žen s Turnerovým syndromem, kardiovaskulární rizika spojená s těhotenstvím