
Diagnostika komplexu tuberózní sklerózy se zaměřením na prenatální období
Diagnosis of tuberous sclerosis complex focusing on prenatal period
Tuberous sclerosis is a disease with an autosomal dominant pattern of inheritance which is characterized by the development of benign tumours in many tissues and organs. Clinical signs are extremely variable, causing mutations in the gene TSC1 or TSC2. Complex formed by the products of the TSC genes regulates cell growth and proliferation by inhibition of mTORC1 signalling. Early diagnosis of TSC is very important to plan appropriate perinatal care. Using ultrasound and eventually MRI it is possible in the prenatal period to capture the following major features of tuberous sclerosis: cardiac rhabdomyoma, subependymal nodules, cortical tubers and renal angiomyolipomas. In connection with the syndrome of contiguous genes TSC2 / PKD1 can also be detect foetal renal cysts. Often these TSC-associated lesions represent an incidental finding during a routine ultrasound. In the period from the 20th week of pregnancy it is most often found cardiac rhabdomyoma/s as the first marker suggestive of tuberous sclerosis. In the case, where one of the parents is a carrier of already identified mutation in the TSC gene, it is possible to carry out targeted genetic testing of a sample of DNA isolated from cells of chorionic villi, amniocytes or tissue from aborted foetuses. Significantly more time consuming is to perform molecular analysis of the TSC genes in foetuses with suspected tuberous sclerosis without the occurrence of illness in the family. After finding a causal mutation and its confirmation, it is possible to offer genetic testing for other persons at risk, prenatal (eventually preimplantation) diagnosis for future pregnancies. It is also necessary to consider the possibility of gonadal mosaicism.
Design:
Review of the literature.
Keywords:
tuberous sclerosis complex, TSC1, TSC2, foetal rhabdomyoma, sequencing, mosaicism
Autori:
H. Filipová; M. Procházka; R. Vrtěl
Pôsobisko autorov:
Ústav lékařské genetiky FN a LF UP Olomouc
přednosta doc. MUDr. M. Procházka, Ph. D.
Vyšlo v časopise:
Ceska Gynekol 2016; 81(2): 147-154
Súhrn
Tuberózní skleróza je onemocnění s autosomálně dominantním typem dědičnosti, pro které je charakteristický vývoj benigních tumorů v mnoha tkáních a orgánech. Klinické příznaky jsou extrémně variabilní. Příčinou jsou mutace v genu TSC1 nebo genu TSC2. Komplex vytvořený produkty těchto TSC genů reguluje buněčný růst a proliferaci inhibicí mTORC1 signalizace. Včasně stanovená diagnóza TSC je velmi důležitá pro naplánování příslušné perinatální péče. Použitím ultrazvuku a případně MRI je možné v prenatálním období zachytit následující majoritní znaky tuberózní sklerózy: srdeční rhabdomyomy, subependymální noduly, kortikální tubery a renální angiomyolipomy. V souvislosti se syndromem přilehlých genů TSC2/PKD1 lze u plodu detekovat i renální cysty. Často tyto TSC asociované léze představují náhodný nález během rutinního ultrazvuku. V období od 20. týdne gravidity je nejčastějším markerem vyvolávajícím podezření na tuberózní sklerózu srdeční rhabdomyom(y). V případě, kdy jeden z rodičů je nositelem již identifikované mutace v TSC genu, je možné provést cílené genetické testování vzorku DNA izolovaného z buněk choriových klků, amniocytů, případně z tkáně potracených plodů. Výrazně časově náročnější je provedení molekulární analýzy TSC genů u plodů se suspektní tuberózní sklerózou bez výskytu onemocnění v rodině. Po nálezu příčinné mutace a její konfirmace je možné nabídnout genetické testování dalších osob v riziku, prenatální (eventuálně preimplantační) diagnostiku pro další těhotenství. Rovněž je nutné zvážit možnost výskytu gonadálního mozaicismu.
Typ studie:
Literární přehled.
Klíčová slova:
komplex tuberózní sklerózy, TSC1, TSC2, fetální rhabdomyom, sekvenování, mozaicismus
ÚVOD
Komplex tuberózní sklerózy (TSC) je onemocnění s autosomálně dominantním typem dědičnosti, pro které je charakteristický vývoj hamartomů (benigních tumorů) v mnoha tkáních a orgánech. Nejčastěji bývá zasažena pokožka, mozek, srdce, oči, ledviny a plíce. Frekvence výskytu TSC v populaci je odhadována na 1/6000 – 1/10 000 [28]. Za příčinu vzniku hamartomů byly označeny inaktivující mutace v tumor supresorových genech TSC1 a TSC2 [7]. TSC se vyznačuje lokusovou heterogenitou. Gen TSC1 byl identifikován v roce 1997 na 9. chromozomu (9q34) [25]. V roce 1993 byl objeven na 16 chromozomu (16p13.3) gen TSC2 [4].
Produkt genu TSC1 (hamartin) vytváří s produktem genu TSC2 (tuberinem) komplex, který prostřednictvím signalizace mTOR (mammalian target of rapamycin) funguje jako negativní regulátor buněčného růstu a proliferace [22]. Právě zprostředkování inhibice mTOR je středem zájmu při vývoji strategie léčby TSC asociovaných lézí. Použití everolimu, inhibitoru mTOR, se ukazuje jako nadějná farmakologická léčba u pacientů se subependymálními obrovskobuněčnými astrocytomy a s renálními angiomyolipomy [5, 14].
Vzhledem k tomu, že se symptomy TSC objevují se vzrůstajícím věkem pacienta [27] (viz tab. 1) a jsou extrémně variabilní, byla v roce 1998 zavedena diagnostická kritéria. Podle míry specifičnosti byly klinické symptomy rozděleny do dvou kategorií, kdy jedna zahrnovala majoritní znaky a druhá minoritní znaky TSC [20].
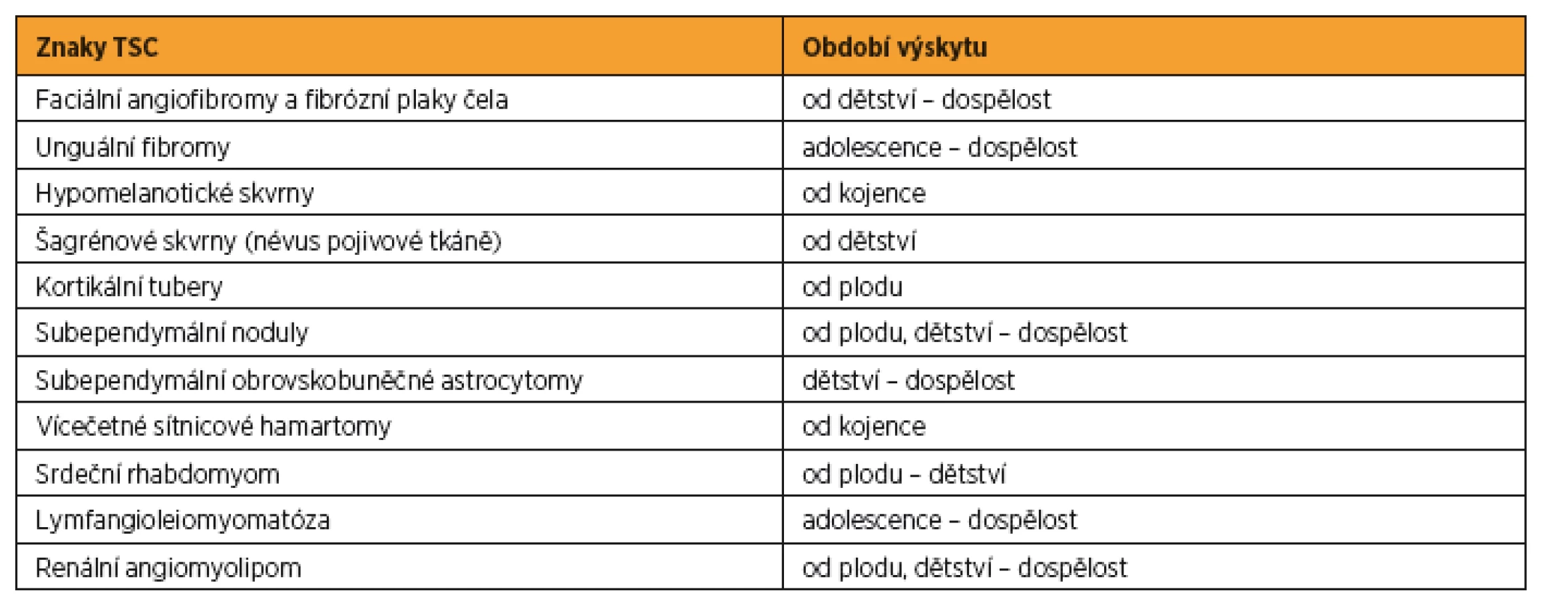
Na základě narůstajícího množství poznatků o TSC byla v roce 2012 ve Washingtonu uspořádána mezinárodní konsenzuální konference, jejímž cílem bylo aktualizovat diagnostická kritéria tuberózní sklerózy stanovená v roce 1998. Byla přehodnocena prevalence a specifičnost dosud známých markerů asociovaných s TSC [17] a zároveň formulována doporučení pro klinickou péči o pacienty s TSC [18]. Výsledná diagnostická kritéria TSC jsou shrnuta v tabulce 2 a doporučení pro klinickou péči pro nově diagnostikovanou a suspektní TSC jsou popsána v tabulce 3, pro již diagnostikovanou TSC v tabulce 4.

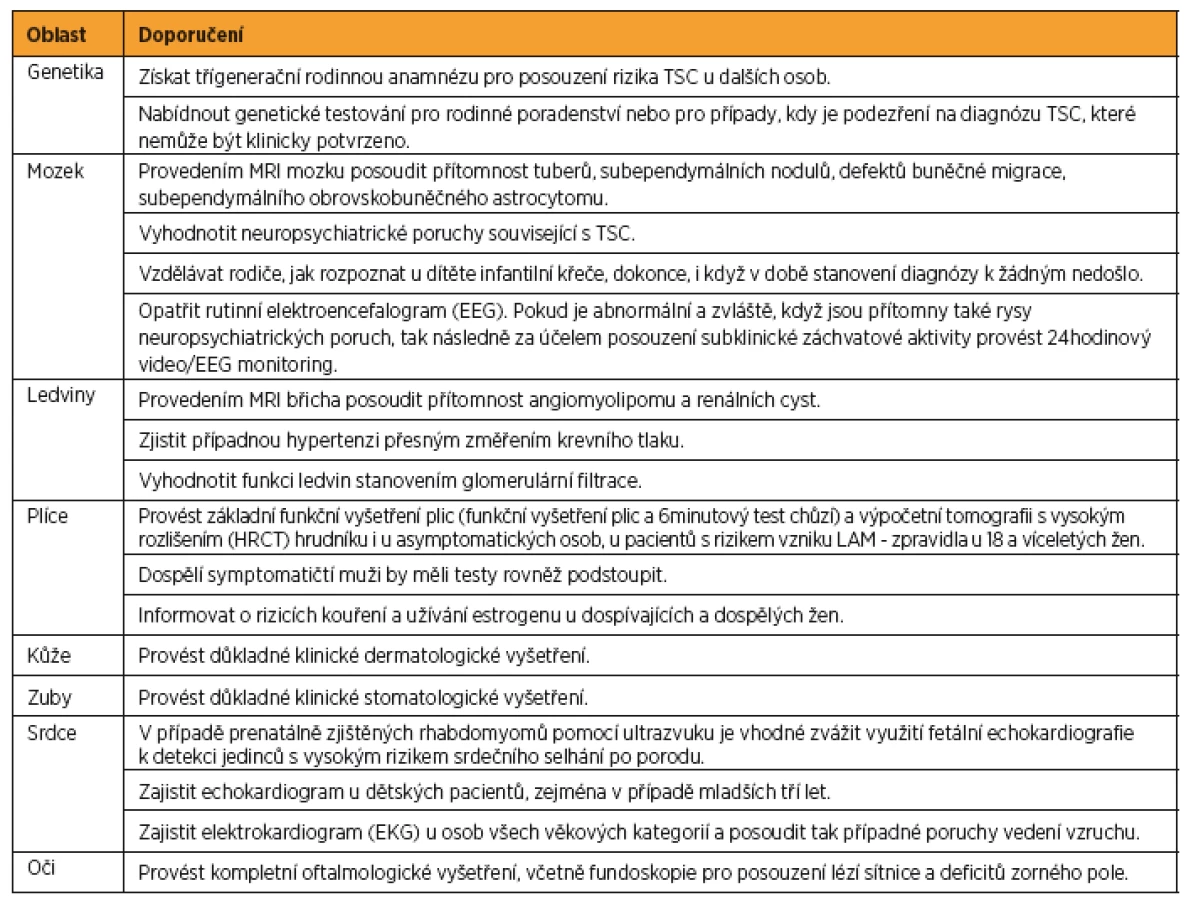
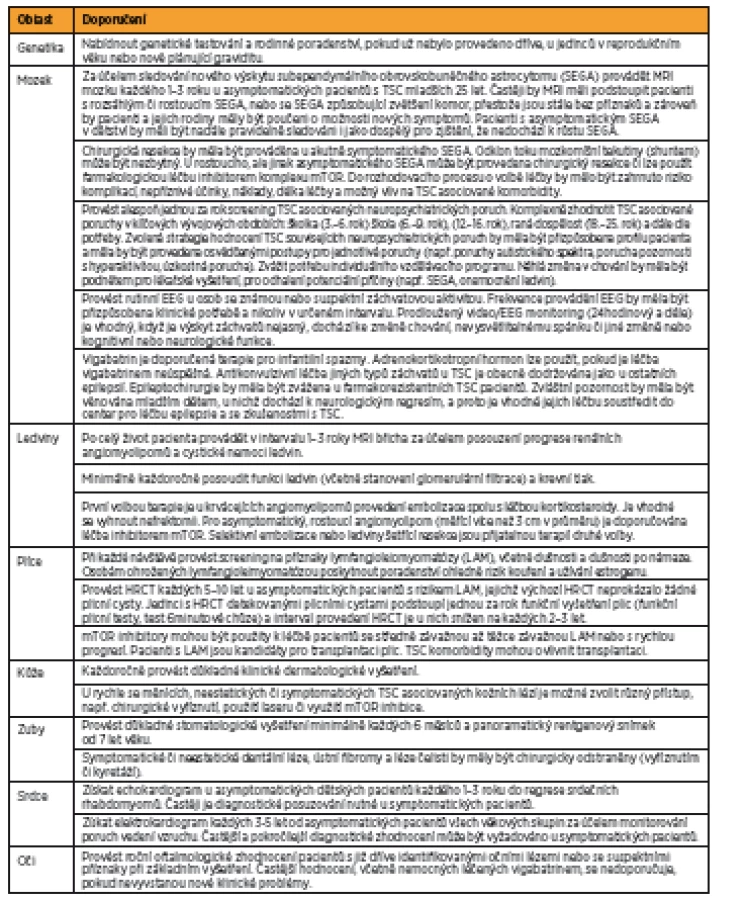
V důsledku technického pokroku a progresivního vývoje nových strategií je molekulární analýza genů dnes široce dostupná, a proto analýza TSC1 a TSC2 genu byla začleněna do nových diagnostických kritérií TSC. Další zásadní změnou oproti původním kritériím je redukce kategorií TSC diagnóz. Z dřívějších tří (definitivní, pravděpodobná a možná diagnóza TSC) zůstaly zachovány pouze dvě (definitivní a možná). Definitivní diagnóza TSC je dána přítomností dvou majoritních znaků nebo jednoho majoritního znaku v kombinaci s dvěma a více znaky minoritními. O možné diagnóze TCS lze uvažovat v případě výskytu jednoho majoritního znaku či dvou a více minoritních znaků. Rovněž došlo k úpravě ve výčtu minoritních znaků TSC. Hamartomatózní rektální polypy a cysty kostí byly pro svoji nedostatečnou specifičnost k TSC odebrány z tohoto seznamu. Cerebrální radiální migrační dráhy bílé hmoty byly přesunuty z minoritních znaků do majoritních, kde společně s kortikálními tubery reprezentují položku „kortikální dysplazie“.
MOLEKULÁRNÍ ANALÝZA TSC GENŮ
Vzhledem k tomu, že do dnešního dne nebylo objeveno žádné „horké“ mutační místo v TSC genech, nelze molekulární analýzu DNA vzorku probanda primárně zaměřit na určitou oblast. V TSC asociovaných genech dochází ve velké míře k de novo mutacím, neboť přibližně 2/3 TSC případů představují sporadické případy [23]. Podle záznamů v databázi LOVD TSC1/TSC2 bylo již popsáno více než 840 unikátních sekvenčních variant v genu TSC1 a přes 2390 různých variant v genu TSC2.
Bodové mutace a posunové mutace (způsobené inzercemi a delecemi malého rozsahu) tvoří většinu ze spektra mutací identifikovaných v TSC1 a TSC2 genu. Rozsáhlé delece/duplikace jsou výrazněji zastoupeny v genu TSC2. Zkoumáním 261 DNA vzorků TSC pacientů metodou MLPA (multiplex ligation-dependent probe amplification) byla zachycena delece/duplikace u 50 z nich v genu TSC2 a pouze čtyři rozsáhlé mutace postihující gen TSC1 [15].
Analýzou velkých skupin TSC pacientů se prokázalo, že u sporadických případů jsou mutace častější v genu TSC2 než v TSC1 genu. Dokladem je např. mutační analýza 224 vzorků TSC osob, kdy se u 186 (u 83 %) podařilo identifikovat příčinnou mutaci, která se ve 12 % případů nalézala v genu TSC1, u 62 % ji tvořily mutace malého rozsahu TSC2 genu a 9 % zaujímaly rozsáhlé delece TSC2 genu [3]. V souboru čítajícím 490 TSC jedinců byla odhalena kauzální mutace u 362 pacientů, přičemž poměr nalezených mutací byl rovněž ve prospěch TSC2 genu, konkrétně 77 % ku 23 %. Provedením korelace fenotypu s genotypem bylo prokázáno v souladu s již dříve zdokumentovanými studiemi, že mutace v genu TSC2 jsou spjaty se závažnější mírou klinických znaků tuberózní sklerózy než mutace v genu TSC1 [23].
V závislosti na technologickém pokroku se postupně měnil způsob analýzy těchto genů. Nepřímé metody (např. vazebná analýza) ustoupily technikám přímé analýzy. Pro svůj rozsah (23 exonů genu TSC1 a 42 exonů genu TSC2) byly v dřívějších letech nejprve používány tzv. skenovací metody (např. denaturační gradientová gelová elektroforéza, vysokotlaká kapalinová chromatografie či metoda jednořetězcového konformačního polymorfismu) sloužící k vytipování oblastí nesoucích sekvenční změny. Následně za účelem identifikace sekvenční změny (zjištění její přesné polohy a posouzení, zda se jedná o patologickou změnu) bylo nutné příslušnou oblast podrobit sekvenování (nejčastěji Sangerovou metodou). Pracné i časově náročné skenovací techniky byly nahrazeny metodou přímého sekvenování všech kódujících oblastí TSC genů a oblastí zahrnující exon/intronové rozhraní.
Pro detekci rozsáhlých změn (delecí/duplikací) v TSC genech se s úspěchem používá metoda MLPA. Nález je vhodné konfirmovat i jinou metodou (např. long-range PCR), popřípadě prostřednictvím MLPA provedené s kitem obsahujícím odlišné sondy. Přestože je metoda MLPA přednostně určená k záchytu velkých přestaveb genů, někdy lze s její pomocí nalézt i sekvenční změny malého rozsahu (např. bodové), ovšem pouze tehdy, když dojde k mutaci/polymorfismu v místě nasedání sondy pro určitý exon. Pro konkretizaci sekvenční změny je nutno tento exon sekvenovat. Pro identifikaci rozsáhlé změny sekvence je sekvenování vhodné, jestliže se začátek nebo konec změny nachází v oblasti mezi použitými primery. Pokud mix sond v MLPA kitu obsahuje kromě sond pro jednotlivé exony TSC2 také sondu pro terminální část genu PKD1, existuje možnost zachycení i tzv. syndromu přilehlých genů – stavu, kdy delece je natolik rozsáhlá, že zasáhne nejen gen TSC2, ale i gen PKD1, jenž svým koncem navazuje na konec genu TSC2.
Nástup masivního paralelního sekvenování, metod představujících novou generaci sekvenování (NGS) za použití nejrůznějších platforem (např. Illumina, IonTorent, 454, SOLiD aj.) s sebou přinesl výhody v podobě mnohonásobného „čtení“ jednotlivých amplikonů kódujících i nekódujících oblastí (v závislosti na navržení knihovny) současně u celé skupiny pacientů. Mnohdy jsou TSC geny součástí již komerčně dostupných panelů zahrnujících i desítky různých genů. Do popředí se dostává i přístup celogenomového sekvenování.
Masivní paralelní sekvenování se jeví jako velmi užitečné i při odhalování mutací ve formě mozaik, což dokládá studie Tyburczy a kol., kteří metodou NGS vyšetřovali DNA vzorky izolované z krve, slin či angiofibromů u 53 TSC pacientů, u nichž konvenčními technikami nebyla identifikována žádná mutace. Kromě genu TSC1 a TSC2 zaměřili analýzu i na kódující regiony potenciálních kandidátních genů pro gen TSC3, tedy na geny DEPTOR, PRAS40, TBC1D7, DEPDC5, NPRL2 a NPRL3. Zatímco v kandidátních genech nebyla zaznamenána žádná mutace, u 45 (85 %) jedinců byla zjištěna mutace v TSC1 nebo TSC2 genu. Značná část detekovaných mutací (26 ze 45; 58 %) bylo ve formě mozaiky, přičemž frekvence mutované alely se pohybovala v rozsahu 0,21 % až 34 %. U 17 osob byla frekvence mutované alely pod 5 %, v pěti případech dokonce pod 1 %. U dvou jedinců byla mutace prokazatelná pouze ve vzorku z kožního tumoru. Významný podíl z celkově nalezených mutací (18 ze 45; 40 %) se nacházel v intronových oblastech. Na základě těchto výsledků se autoři domnívají, že gen TSC3 pravděpodobně neexistuje, že zastoupení mutací ve formě mozaik je vyšší, než se usuzuje, zdůrazňují významnost plného pokrytí lokusu genu TSC1 a TSC2 a zároveň doporučují NGS i pro analýzu DNA z tumorů asociovaných s TSC [24]. Některé typy TSC lézí (nejčastěji AML) vykazují ztrátu heterozygozity (LOH), kterou je možno odhalit molekulární analýzou.
Obrovské množství výstupních dat získané prostřednictvím NGS je sice na jednu stranu přínosné, na druhou stranu však klade zvýšené nároky na jejich třídění, vyhodnocování a interpretaci možného klinického dopadu identifikovaných sekvenčních změn, obzvláště změn nejasného významu.
Při vyhodnocování sekvenčních dat jsou hojně využívány následující NCBI referenční sekvence. Pro gen TSC1 genomická referenční sekvence NG_012386.1 a referenční sekvence pro transkript NM_000368.4, pro gen TSC2 genomická sekvence NG_005895.1 a pro jeho transkript NM_000548.3 (v lednu 2016 aktualizováno na verzi NM_000548.4). Na základě povahy sekvenční změny, její lokalizace (např. v místě sestřihu) a údajů uvedených v databázi LOVD TSC1/TSC2 (pokud byla varianta již popsána) jsou mutace odlišeny od polymorfismů. U variant nejasného významu lze posoudit jejich vliv na funkci proteinu využitím predikčních programů (např. SIFT, PolyPhen), výsledky tohoto „virtuálního“ testování však nemají takovou výpovědní hodnotu jako skutečně provedená funkční analýza. K výraznému usnadnění v procesu rozhodování o případné patogenicitě varianty nejasného významu dochází v situaci, kdy výskyt varianty u probanda koreluje s výskytem i u dalších TSC postižených členů rodiny a u zdravých příbuzných je výsledek genetického testování negativní.
Mutace zachycené prostřednictvím NGS jsou obvykle potvrzovány Sangerovým sekvenováním. Rovněž je vhodné, aby nález kauzální mutace byl konfirmován i analýzou dalšího vzorku pocházejícího z nezávislého odběru. Posléze je možné poskytnout cílené testování pro další osoby v riziku, případně nabídnout možnost prenatální diagnostiky, eventuálně preimplantační diagnostiky.
PRENATÁLNÍ DIAGNOSTIKA TSC
Včasně stanovená diagnóza TSC má významný vliv na osud gravidity a novorozence, neboť v případě pokračování těhotenství umožňuje naplánovat perinatální a následnou péči. Prostřednictvím vhodných zobrazovacích technik lze prenatálně u plodu zachytit srdeční rhabdomyomy, kortikální tubery, subependymální noduly a renální angiomyolipomy. Při přítomnosti dvou z výše uvedených majoritních znaků TSC jsou splněna kritéria pro stanovení definitivní diagnózy TSC [9].
Nejčastěji nalézanými typy kongenitálních srdečních nádorů jsou rhabdomyomy, které tvoří 60–86 % případů. Výrazně méně jsou zastoupeny např. teratomy či fibromy. Při ultrazvuku srdce plodu se fibrom i rhabdomyom jeví jako homogenní, kompaktní, echogenní masa [29]. Rhabdomyomy představují intramurální i intrakavitární tumory, častěji postihující srdeční komory nežli síně, přičemž preferují levou stranu srdce před pravou [7]. Rozlišení rhabdomyomu od jiných srdečních nádorů může být obtížné v situaci, kdy je na srdci nalezen pouze jeden útvar, či se nádory objevují na neobvyklých místech. Srdeční rhabdomyomy jsou hormon-dependentní tumory, jejichž velikost se pohybuje od několika milimetrů po několik centimetrů. Přibližně v 90 % případů se vyskytují jako vícečetné a právě většina z těchto případů je asociovaná s TSC [10]. Důkazem vysokého výskytu TSC v souvislosti se srdečními rhabdomyomy je i studie 40 plodů, které vykazovaly jeden či více srdečních tumorů. Z nich 33 (75 %) mělo rhabdomyomy, tři gravidity byly ukončeny, ve čtyřech případech došlo k úmrtí při porodu a 26 (79 %) přežilo. Celkem u 95 % z živě narozených s rhabdomyomy nebyly přítomny žádné srdeční symptomy a u 88% z nich byla stanovena diagnóza TSC [29].
Srdeční rhabdomyom(y) lze ultrasonograficky prokázat od 20. týdne gravidity [10]. Obvykle jsou tyto intrakardiální útvary zjištěné při rutinním ultrazvuku plodu a jejich objev vyvolává první podezření na TSC. Pro upřesnění nálezu a pro odhalení případných komplikací je velmi užitečná fetální echokardiografie, neboť umožňuje posoudit vliv hmoty rhabdomyomu(ů) na funkci srdečních komor, na průtok krve či na vznik arytmií [9].
Kromě gestačního věku v době stanovení diagnózy má právě umístění a velikost rhabdomyomu, počet útvarů a progrese tumoru zásadní vliv na perinatální výsledky. V některých případech může být výskyt rhabdomyomu spojen s hydropsem plodu, intrauterinní smrtí plodu či s náhlým úmrtím kojenců [2].
Většina rhabdomyomů je asymptomatická a spontánně regreduje během prvních let života. K chirurgickému řešení se přistupuje u hemodynamicky významných rhabdomyomů. U novorozence s vícečetnými rhabdomyomy, u něhož z důvodu nezralosti a nízké porodní váhy nebylo možné provést operaci [16] a u tří novorozenců (dva s hemodynamicky významnými rhabdomyomy a jeden s rozsáhlým SEGA) byla s úspěchem popsána farmakologická strategie léčby, kdy k zmenšení tumorů došlo po aplikacích everolimu [8].
Při nálezu fetálního srdečního rhabdomyomu(ů) lze během těhotenství provést MRI plodu a zobrazením případných intrakraniálních TSC asociovaných lézí či renálních angiomyolipomů, tak potvrdit diagnózu TSC.
Prabowo s kolegy zdokumentoval výskyt prenatálních TSC znaků u monozygotních dvojčat. Ve 22. týdnu těhotenství při rutinním ultrasonografickém vyšetření zaznamenal útvar v oblasti pravého čelního laloku u jednoho z plodů. MRI mozku plodu provedená rovněž ve 22. týdnu gravidity nález léze potvrdila a zároveň odhalila u druhého plodu vícečetné subependymální léze připomínající SEGA. Gravidita byla ukončena ve 23. týdnu [19].
U plodu z 33. týdne gravidity se pomocí ultrazvuku zjistily mnohočetné rhabdomyomy a renální angiomyolipomy a následným využitím MRI byly objeveny i léze na mozku [6].
Byl zaznamenán i případ plodu, u něhož ve 20. týdnu těhotenství byla ultrasonograficky zachycena renální cysta a útvar na srdci. Pitva plodu prokázala srdeční rhabdomyom a polycystickou nemoc ledvin. Vzhledem k tomu, že i matka plodu vykazovala jak znaky TSC, tak znaky autosomálně dominantního onemocnění polycystických ledvin bylo vysloveno podezření na syndrom přilehlých genů TSC2/PKD1. Molekulární analýza genů, která tento mechanismus mohla potvrdit, ovšem nebyla provedena [30].
Pokud matka či otec plodu má stanovenu diagnózu TSC a příčinná mutace je již známa, je možné, v případě zájmu rodičů, provést cílené genetické testování vzorku DNA izolované z buněk choriových klků nebo z amniocytů (obvykle zcela postačuje DNA z nekultivovaného biologického materiálu) a tímto způsobem během několika dní vyloučit, či potvrdit diagnózu TSC. Častěji však dochází k situaci, kdy v rodině nikdo není tuberózní sklerózou postižen a nálezy související s TSC představují náhodný záchyt při běžném ultrazvuku plodu. Jestliže jsou tyto markery zjištěny před 24. týdnem gravidity a je požadována molekulární analýza TSC genů, je k dispozici pouze malý časový interval pro provedení příslušných molekulárních metod, neboť pro identifikaci případné kauzální mutace je zapotřebí vyšetřit oba celé TSC geny, což je výrazně časově náročnější než cílené testování na známou mutaci. Poměrně rychle lze zjistit, zda příčinou onemocnění je rozsáhlá delece/amplifikace genu metodou MLPA, jejíž provedení trvá přibližně dva dny. Pakliže je toto vyšetření negativní, je nutné TSC geny sekvenovat. Při zvolení konvenčního přístupu (nejběžněji používané Sangerovo sekvenování), lze očekávat výsledek analýzy spíše v horizontu několika týdnů než dnů. V některých případech nelze provést kompletní analýzu genů v důsledku nedostatečného množství vzorku DNA pocházejícího z nekultivovaných amniocytů, takže je nutné vyčkat na DNA z kultivovaného biologického materiálu, čímž se doba dodání výsledku prodlouží. Naopak pro analýzu TSC genů metodou NGS je zapotřebí pouze malé množství DNA o nízké koncentraci. Při masivním paralelním sekvenování je vyšetřována současně celá skupina pacientů, proto záleží na režimu daného pracoviště, jak často toto vyšetření provádí.
Z hlediska plánování další gravidity je vhodné podrobit molekulární analýze TSC genů i DNA vzorky pocházející z tkání plodů z ukončených gravidit či z tkání mrtvorozenců, u nichž byla na základě prenatálně zachycených markerů či TSC znaků odhalených při pitvě stanovena diagnóza TSC nebo bylo vyjádřeno podezření na toto onemocnění.
Jestliže je jeden z rodičů postižen TSC, je vzhledem k autosomálně dominantnímu typu dědičnosti onemocnění 50% riziko pro potomky, a to jak pro případy, kdy se příčinná mutace vyskytuje v ne-mozaikovém heterozygotním stavu, tak i pro případy somatických mozaik, u nichž je mutovaná alela s různou frekvencí zastoupena v DNA vzorcích pocházejících z rozličných biologických materiálů (např. krev, močový sediment, bukální stěr, vlasové kořínky, ejakulát).
Kromě toho jedinci reprezentující případy somatického mozaicismu s nízkou frekvencí výskytu mutované alely mohou mít pouze mírný TSC fenotyp [26] nebo, bez detailního klinického vyšetření, mohou být mylně považováni za zdravé, či skutečně nevykazují obtíže související s TSC.
Vyjádření míry rizika TSC pro dalšího potomka může tedy zkomplikovat fakt, že za zdánlivě sporadickým případem TSC se skrývá případ familiární, v podobě nerozpoznané (např. z důvodu technických limitů sekvenačních metod) mutace ve formě somatické mozaiky s nízkou hladinou výskytu u jednoho z rodičů. Rovněž nelze vyloučit ani možnost gonadálního mozaicismu, a to zejména v situaci, kdy se rodičům, kteří nevykazují symptomy TSC, narodí dítě s tuberózní sklerózou a následně i další potomek nebo některý z dalších potomků je postižen TSC. Výskyt gonadálního mozaicismu je podle literatury vzácný, přibližně 2% [26].
Vyšší záchyt (5%) gonadálního mozaicismu popisuje studie 120 TSC rodin, kde se na podkladě klinického zhodnocení rodinných příslušníků gonadální mozaicismus očekával u sedmi rodin (7/120; 6 %). Z těchto sedmi rodin se podařilo detekovat příčinnou mutaci u šesti rodin, přičemž mozaika zárodečné linie byla u pěti rodin zastoupena mutací v genu TSC2 a v jedné rodině mutací genu TSC1 [21].
Vzhledem k výše zmíněným úskalím je vždy vhodné, pokud je u probanda-dítěte již identifikovaná mutace a rodiče jsou asymptomatičtí a cílené genetické testování jejich vzorku DNA izolované z krve bylo negativní, nabídnout této rodině pro další těhotenství možnost prenatální diagnostiky.
Vedle dědičných forem TSC existuje i nedědičná frustní forma TSC. Například u pacientů s izolovaným SEGA a bez jakýchkoli jiných příznaků TSC je tato forma obvykle považována za frustní formu [13]. Sporadická LAM, ať už s přítomností renálních angiomyolipomů, či nikoliv [1], či výskyt pouze jednoho angiomyolipomu je některými autory řazen rovněž do této kategorie TSC.
Doc. RNDr. Radek Vrtěl, Ph.D.
ÚLG FN a LF UP
I. P. Pavlova 6
779 00 Olomouc,
e-mail: vrtel@fnol.cz
Zdroje
1. Bisceglia, M., D'Alessandro, V., Simeone, A., et al. Selected case from the Arkadi M. Rywlin International Pathology Slide Seminar: Sporadic lymphangioleiomyomatosis. Adv Anat Pathol, 2010, 17(6), p. 445–452.
2. Colosi, E., Russo, C., Macaluso, G., et al. Sonographic diagnosis of fetal cardiac rhabdomyomas and cerebral tubers: a case report of prenatal tuberous sclerosis. J Prenat Med, 2013, 7(4), p. 51–55.
3. Dabora, SL., Jozwiak, S., Franz, DN., et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet, 2001, 68, p. 64–80.
4. European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell, 1993, 31, 75(7), p. 1305–1315.
5. Franz, D., Belousova, E., Sparagana, S., et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet, 2012, 381, p. 125–132.
6. Gedikbasi, A., Oztarhan, K., Ulker, V., et al. Pre-natal sonographic diagnosis of tuberous sclerosis complex. J Clin Ultrasound, 2011, 39(7), p. 427–430.
7. Gómez, MR., Sampson, JR., Whittemore, VH. Tuberous sclerosis complex, Developmental perspectives in psychiatry. Oxford University Press, New York 1999, 3rd ed., p. 313–316, 195.
8. Goyer, I., Dahdah, N., Major, P. Use of mTOR inhibitor everolimus in three neonates for treatment of tumors associated with tuberous sclerosis complex. Pediatr Neurol, 2015, 52(4), p. 450–453.
9. Gusman, M., Servaes, S., Feygin, T., et al. Multimodal imaging in the prenatal diagnosis of tuberous sclerosis complex. Case Rep Pediatr, 2012, 2012, p. 925646.
10. Hinton, RB., Prakash, A., Romp, RL., Krueger, DA., Knilans, TK., International Tuberous Sclerosis Consensus Group. Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. Am Heart Assoc. 2014, 25; 3(6): e001493 doi 10.1161/JAHA.114.001493
11. Hoogeveen-Westerveld, M., Ekong, R., Povey, S., et al. Functional assessment of TSC1 missense variants identified in individuals with tuberous sclerosis complex. Hum Mutat, 2012, 33, p. 476–479.
12. Hoogeveen-Westerveld, M., Ekong, R., Povey, S., et al. Functional assessment of TSC2 variants identified in individuals with tuberous sclerosis complex. Hum Mutat, 2013, 34, p. 167–175.
13. Ichikawa, T., Wakisaka, A., Daido, S., et al. A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J Mol Diagn, 2005, 7(4), p. 544–549.
14. Kingswood, JC., Jozwiak, S., Belousova, ED., et al. The effect of everolimus on renal angiomyolipoma in patients with tuberous sclerosis complex being treated for subependymal giant cell astrocytoma: subgroup results from the randomized, placebo-controlled, Phase 3 trial EXIST-1. Nephrol Dial Transplant, 2014, 29(6), p. 1203–1210.
15. Kozlowski, P., Roberts, P., Dabora, S., et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet, 2007, 121(3–4), p. 389–400.
16. Mohamed, I., Ethier, G., Goyer, I., et al. Oral everolimus treatment in a preterm infant with multifocal inoperable cardiac rhabdomyoma associated with tuberous sclerosis complex and a structural heart defect. BMJ Case Rep, 2014, 26. doi: 10.1136/bcr-2014-205138.
17. Northrup, H., Krueger, D. The International Tuberous Sclerosis Complex Consensus Group. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol, 2013, 49, p. 243–254.
18. Northrup, H., Krueger, D. Tuberous Sclerosis Complex Surveillance and Management: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol, 2013, 49(4), p. 255–265.
19. Prabowo, AS., Anink, JJ., Lammens, M., et al. Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol, 2013, 23(1), p. 45–59.
20. Roach, E., Gomez, M., Northrup, H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol, 1998, 13, p. 624–628.
21. Rose, VM., Au, KS., Pollom, G., et al. Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet, 1999, 64(4), p. 986–992.
22. Roux, PP., Ballif, BA., Anjum, R., et al. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribo-somal S6 kinase. Proc Natl Acad Sci USA, 2004, 101(37), p. 13489–13494.
23. Sancak, O., Nellist, M., Goedbloed, M., et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet, 2005, 13(6), p. 731–741.
24. Tyburczy, ME., Dies, KA., Glass, J., et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet, 2015, 11(11), p. e1005637.
25. van Slegtenhorst, M., deHoogt, R., Hermans, C., et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science, 1997, 277, p. 805–808.
26. Verhoef, S., Bakker, L., Tempelaars, AM., et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet, 1999, 64(6), p. 1632–1637.
27. Vrtěl, R., Filipová, H., Vodička, R., et al. Tuberózní skleróza. Klin Onkol, 2009, 22 (Suppl. 1), p. 50-53.
28. Vrtel, R., Verhoef, S., Bouman, K., et al. Identification of a nonsense mutation at the 5' end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J Med Genet, 1996, 33(1), p. 47–51.
29. Yinon, Y., Chitayat, D., Blaser, S., et al. Fetal cardiac tumors: a single-center experience of 40 cases. Prenat Diagn, 2010, 30(10), p. 941–949.
30. Zhang, YX., Meng, H., Zhong, DR., et al. Cardiac rhabdomyoma and renal cyst in a fetus: early onset of tuberous sclerosis with renal cystic disease. J Ultrasound Med, 2008, 27(6), p. 979–982.
Štítky
Detská gynekológia Gynekológia a pôrodníctvo Reprodukčná medicínaČlánok vyšiel v časopise
Česká gynekologie

2016 Číslo 2
- Ne každé mimoděložní těhotenství musí končit salpingektomií
- Mýty a fakta ohledně doporučení v těhotenství
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
- Gynekologické potíže pomáhá účinně zvládat benzydamin
- Jak podpořit využití železa organismem bez nežádoucích účinků
Najčítanejšie v tomto čísle
- Současné možnosti a doporučení pro intrapartální monitorování ozev plodu
- Indukce porodu
- Potermínové těhotenství
- Porod velkého plodu