
Adultní forma Pompeho nemoci
Adult Form of Pompe Disease
Pompe disease (glycogen storage disease type 2, acid maltase deficiency) is inherited autosomal recessive metabolic disorder caused by deficiency of acid alpha-glucosidase and resulting in lysosomal glycogen storage in various tissues, mainly heart and skeletal muscle. Continuous spectrum of phenotypes from the rapidly progressive infantile form to the slowly progressive late onset form of the disease can be observed. Classical infantile form of the disease manifests soon after birth due to absent or nearly absent activity of the key enzyme. Typical manifestations include failure to thrive, muscle weakness, cardiomegaly, and respiratory failure. Before the era of substitution therapy, the majority of children died within the first year of life. Partial enzyme deficiency (severe mutation on one allele and milder on the second) leads to the less severe phenotype with manifestation in child- or adulthood. Time span is from the first to the sixth decade of life. Leading symptoms include slowly progressing limb girdle and trunk muscle weakness with significant involvement of respiratory muscles. There is no cardiomegaly. Suspicion of Pompe disease is confirmed in three steps. The first involves screening with the Dried Blood Spot test. Testing of the activity of alfa glucosidase in leukocytes is used to confirm the disease. Mutation analysis is important to assess the correlation between genotype and phenotype and to identify familial carriers.
Key words:
Pompe disease – alpha glucosidase – lysosomal storage diseases – limb-girdle muscle weakness
The author declares he has no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autori:
S. Voháňka
Pôsobisko autorov:
Neurologická klinika LF MU a FN Brno
Vyšlo v časopise:
Cesk Slov Neurol N 2014; 77/110(6): 667-676
Kategória:
Minimonografie
doi:
https://doi.org/10.14735/amcsnn2014667
Súhrn
Pompeho nemoc (glykogenóza typ 2, deficit alfa glukosidázy) je autozomálně recesivní dědičné metabolické onemocnění, jehož podkladem je defekt lysozomální kyselé alfa glukosidázy vedoucí k hromadění- střádání lysozomálního glykogenu v buňkách a tkáních s následnou dysfunkcí především ve svalové tkáni srdce a kosterních svalech. Průběh je velmi variabilní: od těžkého rychle progredujícího postižení novorozenců (klasická infantilní forma) po postupné postižení s manifestací v dětství či pozdní dospělosti. Je-li alfa glukosidáza zcela nebo téměř nefunkční, vzniká tzv. klasická infantilní forma choroby. Potíže se rozvíjí v prvních měsících života a mají charakter neprospívání, svalové slabosti a potíží s dýcháním. Dochází k výraznému zvětšení srdce, většina dětí se před zavedením substituční terapie nedožila prvních narozenin. Je-li enzym štěpící glykogen alespoň částečně funkční, vzniká tzv. pozdní (juvenilní či adultní) forma onemocnění. Tato situace nastává, pokud vznikne závažná mutace jedné alely a lehčí na druhé. To vede k méně progresivnímu fenotypu. Rozpětí manifestace nemoci je u této formy od první do šesté věkové dekády. Hlavním projevem je svalová slabost, která postupuje a významně zasahuje dýchací svalstvo. U těchto nemocných nebývá srdce zvětšeno. Při podezření na PN máme tři diagnostické úrovně. První je skríningové vyšetření pomocí testu suché kapky (DBS, Dried Blood Spot). Potvrzení diagnózy se provádí vyšetřením aktivity GAA v leukocytech. DNA vyšetření je důležité pro stanovení korelace genotyp- fenotyp a detekci přenašečů v rodině.
Klíčová slova:
Pompeho nemoc – deficit alfa glukosidázy – lysozomální střádavé nemoci – pletencová svalová slabost
Použité zkratky
CK kreatinkináza
DBS Dried Blood Spot (suchá krevní kapka)
ERT Enzyme Replacement Therapy (enzymová substituční terapie)
FVC Forced Vital Capacity (usilovná vitální kapacita)
GAA kyselá alfa-1,4- glukosidáza
GSD Glykogen Storage Disease (onemocnění se střádáním glykogenu)
LGMD Limb Girdle Muscular Dystrophy (pletencová svalová dystrofie)
OMIM On line Mendelian Inheritance in Man
PN Pompeho nemoc
rhGAA rekombinanatní lidská kyselá alfa-1,4-glukosidáza
Úvod
Pompeho nemoc byla pojmenována po svém objeviteli, nizozemském patologovi Joannesu Cassianu Pompem (1901– 1945), který první informoval o dívce, jež zemřela v sedmi měsících na selhání srdce. Doktor Pompe nebyl jen vynikající patolog, ale i skutečný nizozemský vlastenec. Byl členem ilegálního hnutí odporu a pomáhal zajišťovat bezpečné úkryty pro židovské spoluobčany. Ve zvířetníku vedle své laboratoře skrýval ilegální vysílačku, která byla v únoru 1945 okupační správou zaměřena a lokalizována. Doktor Pompe byl za dramatických okolností 25. 2. 1945 před zraky své ženy a dětí zatčen a poté uvězněn. Když byl v dubnu toho roku při blížící se frontě vyhozen do povětří hnutím odporu železniční most, byl s dalšími devíti vězni pro výstrahu popraven a uložen do hromadného hrobu.
Biologická podstata choroby
Příčinou Pompeho nemoci (OMIM 232300) neboli glykogenózy II. typu (Glykogen Storage Disease type II, GSD II) je mutace genu pro lysozomální kyselou alfa-1,4- glukosidázu (GAA). Tento glykoprotein o 952 aminokyselinách fyziologicky štěpí alfa-1,4 vazby glykogenu. Dochází k intralysozomální akumulaci glykogenu. Jde především o svalovou tkáň (kosterní svaly, myokard i hladké svaly), ale hromadí se také v játrech, endotelu, nervové tkáni (buňky předních rohů míšních, motorická jádra v kmeni, spinální ganglia, astrocyty), ledvinách a jinde. Taktéž ve Schwannových buňkách periferních nervů zemřelých pacientů byla nalezena depozita glykogenu [1].
Dochází k zvětšování svalových vláken, rupturám lysozomů s únikem glykogenu do cytosolu. Aktivace hydrolytických enzymů poškozuje sousední myofibrily. Spekuluje se o tom, že svalové buňky svými kontrakcemi potencují poškozování naplněných lysozomů, čímž se vysvětluje relativně větší poškození svalů oproti ostatním tkáním [2]. Nakonec dochází k lipofuscinem zprostředkované apoptóze.
Lysozom je subcelulární organela obsahující katabolické enzymy podílející se na fyziologickém obratu buněčných látek. Lysozomální střádavé nemoci jsou heterogenní skupina dědičných onemocnění charakterizovaná akumulací nestrávených molekul intralysozomálně. Klasifikují se obvykle podle akumulovaného substrátu (např. sfingolipidóza, mukopolysacharidóza apod.). Patří sem téměř 50 nemocí, jako třeba Krabbeho nemoc, metachromatická leukodystrofie, ceroidní lipofuscinóza a další.
Gen pro GAA byl lokalizován na dlouhém raménku 17. chromozomu (17q25.2– 25.3), má 20 exonů, z toho 19 kódujících, a obsahuje 18,5 kb genomové DNA. Je známo kolem 300 mutací tohoto genu, jen 200 je však relevantních (viz www.pompecenter.nl). Většinou jde o substituce, malé delece a inzerce či sestřihové mutace.
Pro vznik poruchy musí být přítomny relevantní mutace na obou alelách významně snižující aktivitu enzymu (autozomálně recesivní typ dědičnosti). Pro bezproblémovou funkci stačí jedna plně funkční alela, kritická hodnota aktivity enzymu je asi 25 %.
Je-li kyselá alfa glukosidáza zcela nebo téměř zcela nefunkční, vzniká tzv. klasická infantilní forma choroby. Potíže se začínají projevovat v prvních měsících života a mají charakter neprospívání, svalové slabosti a potíží s dýcháním. Dochází k výraznému zvětšení srdce, většina dětí se před zavedením substituční terapie nedožila prvních narozenin.
Je-li enzym štěpící glykogen alespoň částečně funkční, vzniká tzv. pozdní (juvenilní či adultní) forma onemocnění. Tato situace nastává, když vznikne závažná mutace jedné alely a lehčí na druhé. To vede k méně progresivnímu fenotypu s reziduální aktivitou do 23 %. Rozpětí manifestace nemoci je u této formy od první do šesté věkové dekády. Hlavní projev je svalová slabost, která postupuje a významně zasahuje dýchací svalstvo. U těchto nemocných nebývá srdce zvětšeno.
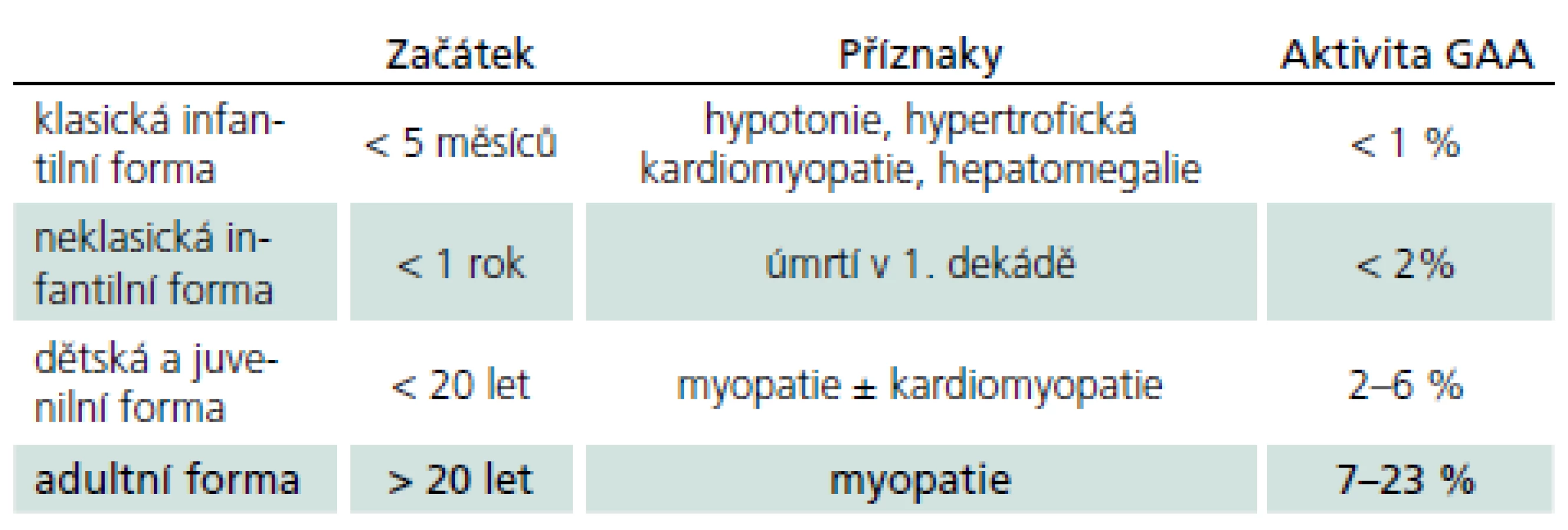
Vymezení je ovšem arbitrární a existuje kontinuum fenotypů od narození do pozdní dospělosti. Obecně ale platí, že u klasické infantilní formy není aktivita GAA nikdy více než 2 %. Naopak u starších dětí a dospělých s mírnější formou PN není genotyp a enzymová aktivita lineárně prediktivní a mohou mít jak velmi nízkou, tak i hraniční či téměř normální hodnotu aktivity GAA. Chorobu zpravidla rozdělujeme do čtyř typů (tab. 1). Toto rozdělení lze racionálně redukovat na dvě hlavní skupiny: infantilní formu se začátkem v kojeneckém věku a postižením srdce a na kultní formu s rozvojem v dospělosti bez postižení srdce. Zbylé dvě formy tvoří přechodné varianty, tedy kontinuum fenotypů.
Epidemiologie
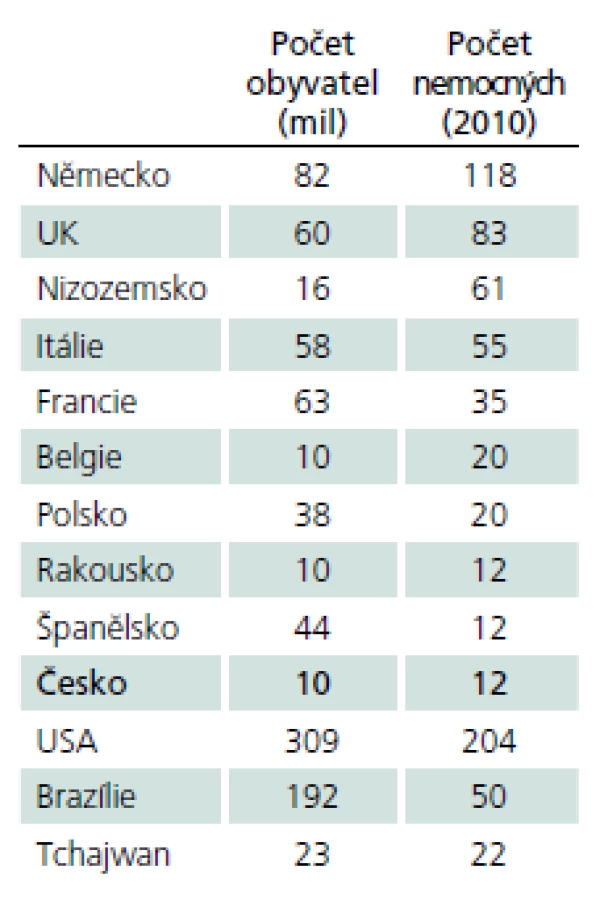
Pompeho choroba patří mezi vzácná, tzv. orphan onemocnění (definovány prevalencí 1 případ a méně na 2 000 osob), výskyt v různých zemích ukazuje tab. 2.
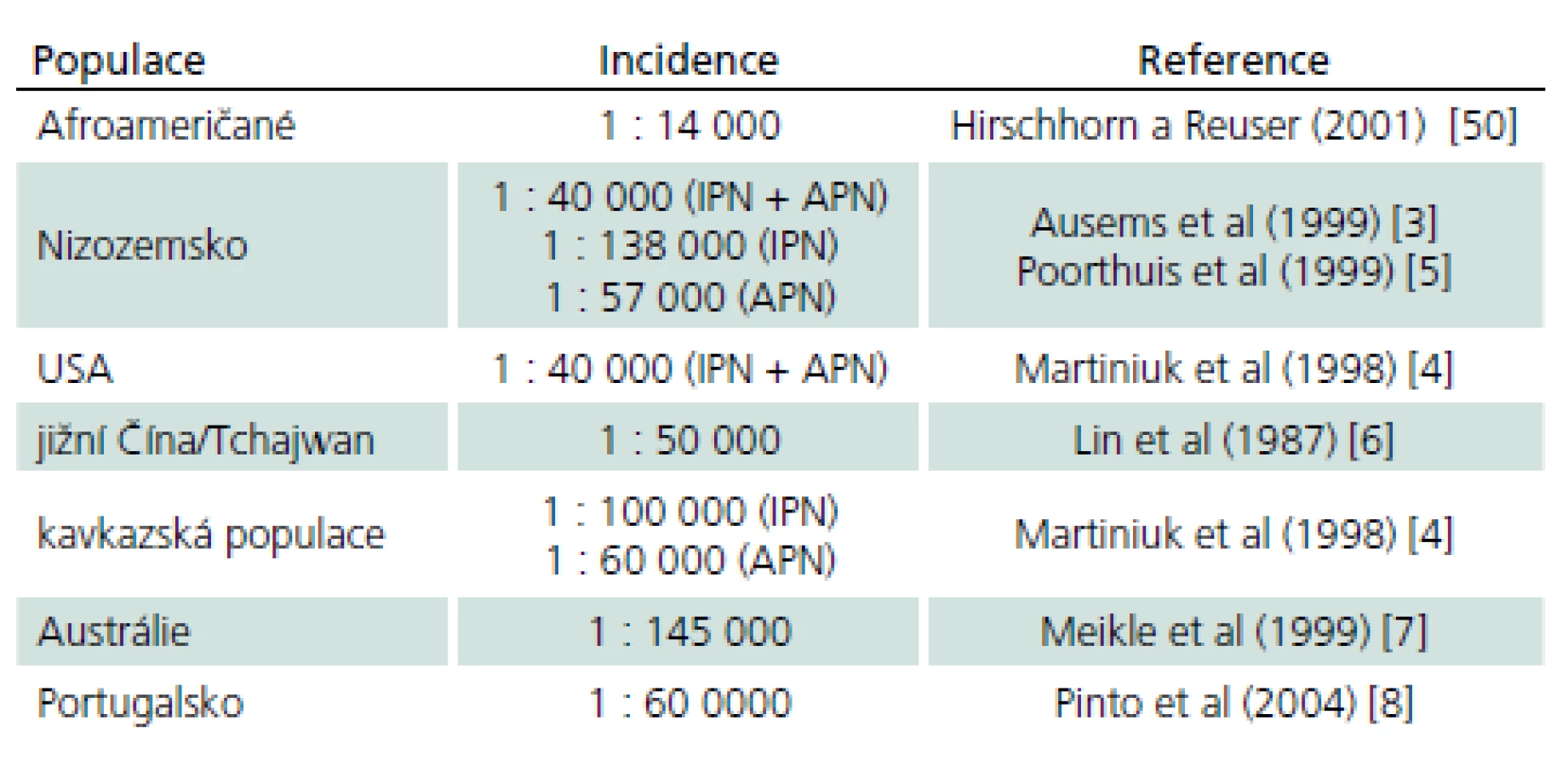
Choroba rovnoměrně postihuje obě pohlaví. Přesná prevalence choroby není vzhledem k vzácnosti a počtu nediagnostikovaných případů známa. Odhaduje se v širokém rozmezí 1 případ na 40 tis. až 1 případ na 300 tis. obyvatel [3]. Výskyt v různých populacích ukazuje tab. 3 [4– 8].
V posledních letech pozorujeme nárůst diagnostikovaných pacientů, což souvisí se zvýšeným zájmem o tuto chorobu, která je dána možnostmi účinné substituční terapie. Choroba je však stále zvláště v dospělém věku diagnostikována se značným zpožděním. Práce Hagemanse et al z roku 2005 [9] a Müller-Felbera et al z roku 2007 [10] prokazovaly interval mezi prvními příznaky a diagnózou sedm a 10 let, recentní data z globálního Pompe registru ukazují zpoždění osm let [11]. Situace v ČR je obdobná: u dospělých pacientů odpovídá zpoždění 5– 7 letům [12,13].
Klinický obraz
Klasická infantilní forma
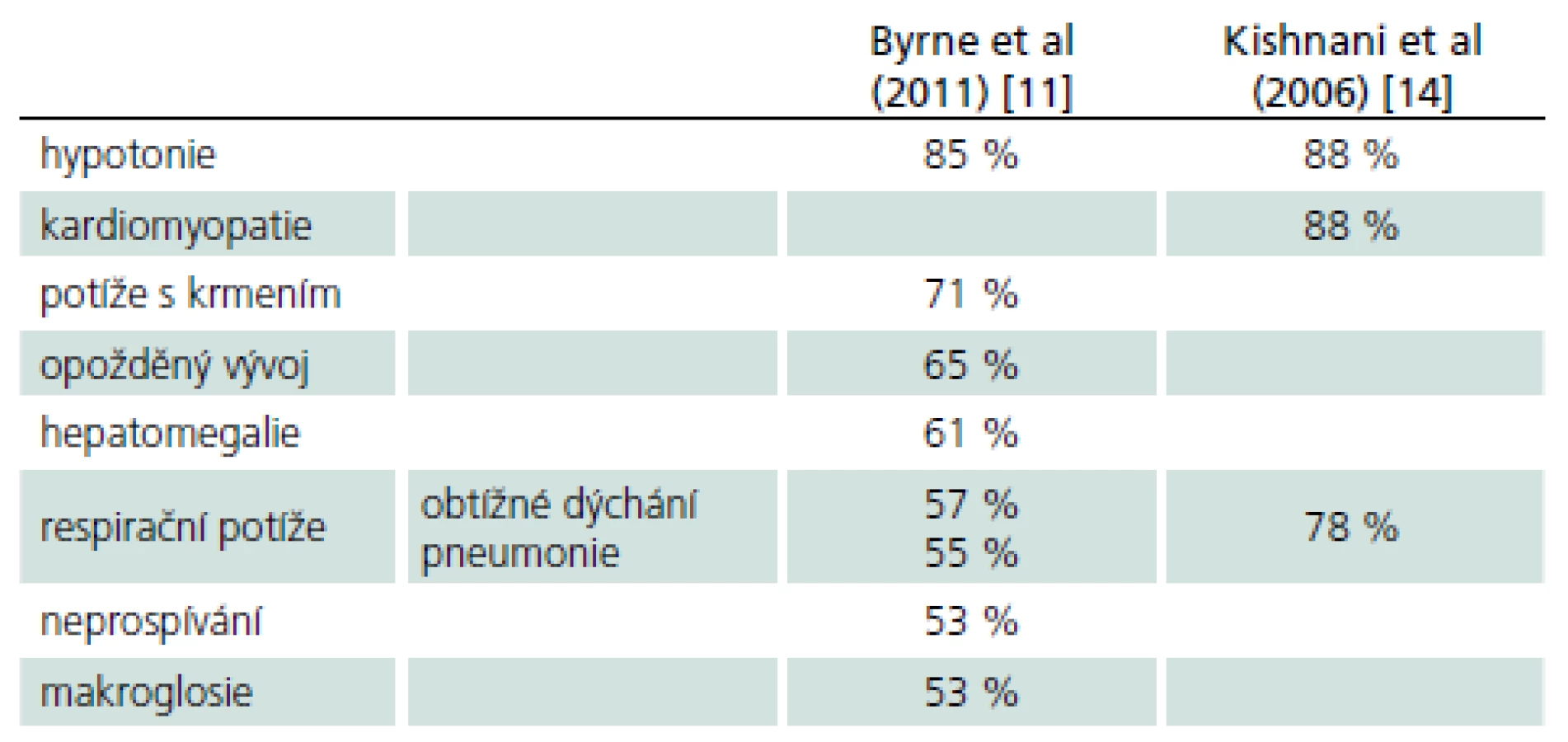
Rozvíjí se brzy po narození a rychle progreduje. Prakticky nulová aktivita GAA vede k výrazné kumulaci glykogenu ve tkáních, zvláště v kosterních svalech a myokardu. Před zavedením terapie většina dětí zemřela do jednoho roku od narození. Příčinou smrti je kardiorespirační insuficience. Typický je obraz hypotonického dítěte („floppy baby“), neprospívání, potíže s příjmem potravy a celkové motorické zaostávání [11,14]. Zřetelná je tachypnoe a ortopnoe pro slabost respiračního svalstva a bránice, časté jsou respirační infekty. Diagnostickým vodítkem je kardiomegalie – hypertrofická kardiomyopatie (tab. 4, 5), dále hepatomegalie a makroglosie. Laboratorně je výrazně zvýšená hladina kreatinkinázy (10krát).
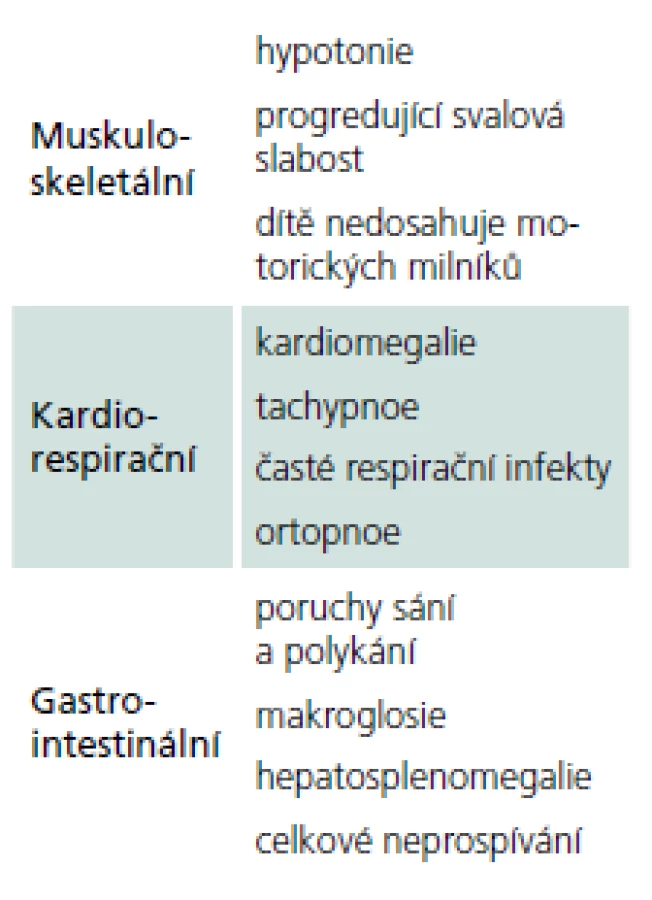
Pozdní varianta onemocnění
S manifestací v dětství, adolescenci a dospělosti je diagnosticky hůře uchopitelná, protože se projevuje jako postupně progredující, především kořenová a trupová svalová slabost [9,11]. Pacienti mají typické obtíže s chůzí do schodů, nastupováním do dopravních prostředků, zvedáním se z nízké židle apod. Postižení trupového svalstva vede k vzniku skoliózy, která se vyskytuje u 25 % nemocných [15], některé soubory ji ale udávají až v 77 % [16]. (Výskyt skoliózy koreluje s tíží postižení a časností manifestace choroby.) Postižení pletence ramenního se často projevuje odstávajícími lopatkami (obr. 1). Vzhledem k postižení respiračního svalstva, zvláště bránice, jsou pohybové aktivity provázeny námahovou dušností. Respirační insuficience se často nejprve manifestuje vleže (postižení bránice) a ve spánku, což vede k typickým příznakům po probuzení, jako jsou bolesti hlavy, tachykardie, a pocitu neosvěžujícího spánku. Trupová slabost se projevuje potížemi se vstáváním z lehu s typickými trikovými pohyby (tab. 6). Bez léčby vede choroba k těžké poruše hybnosti a relativně (na rozdíl od jiných svalových chorob) časné respirační insuficienci s nutností umělé plicní ventilace. Bývá zvýšená hladina svalových enzymů (CK), ale elevace je jen mírná a v některých případech (10 %) může být i normální [17]. Vývoj choroby je velmi pozvolný a nenápadný, s často nezachytitelným začátkem. Pacienti bývají mnohdy od dětství fyzicky nevýkonní a asteničtí. Svalové bolesti udává asi polovina pacientů, tento výskyt je ale přibližně stejný jako u jiných svalových chorob [9,10,18,19]. Od prvních příznaků k významné invalidizaci dělí pacienta většinou desítky roků [9,20].
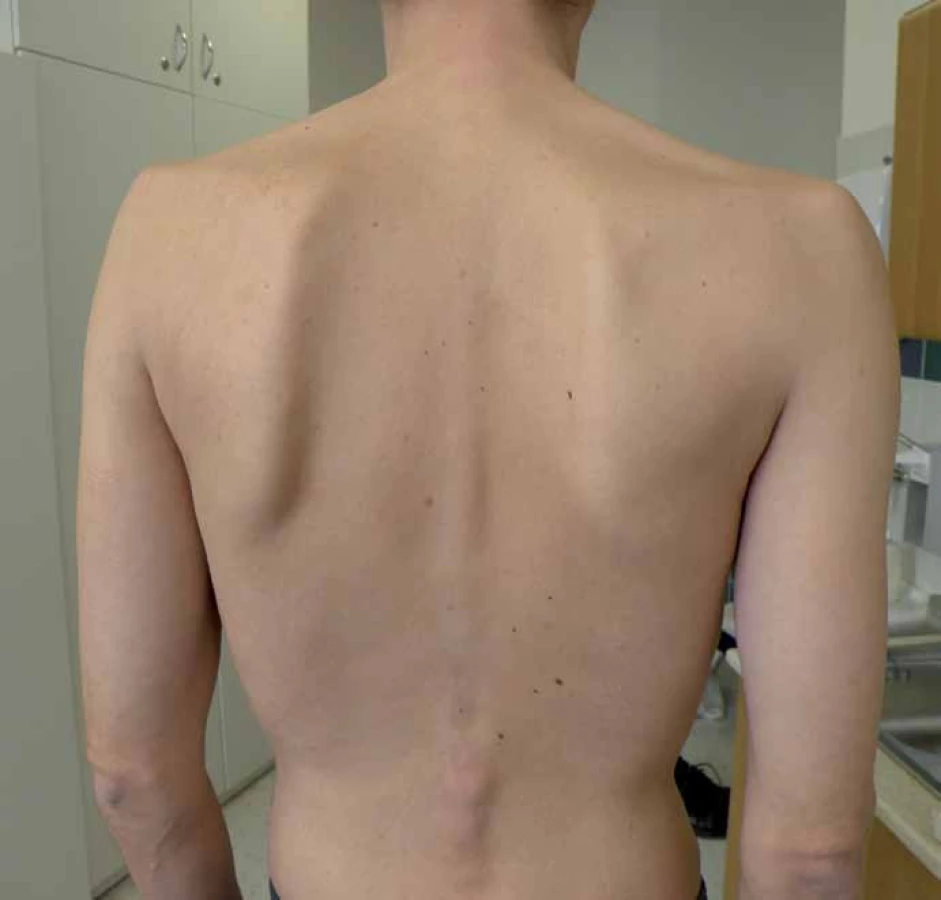

Adultní forma Pompeho nemoci se tak fenotypově podobá skupině pletencových svalových dystrofií či kongenitálních myopatií nebo jiných nemocí s kořenovou svalovou slabostí, jako jsou zánětlivé anebo endokrinní myopatie, popř. spinální svalové atrofie. Stanovení správné diagnózy dále komplikuje fakt, že k manifestaci může dojít prakticky v jakémkoliv věku. V souboru nizozemských autorů [21] byli i pacienti s manifestací choroby kolem 60. roku života. Spojení dědičná choroba = mladý věk je bohužel zakořeněný omyl a častá past při diagnostice kořenové svalové slabosti dospělého a vyššího věku [22].
Respirační funkce
Asi 60 % procent nemocných s PN má lehce redukovanou vitální kapacitu (< 80 % predikované vitální kapacity), 30– 40 % má pokles významnější (< 60 %). Stupeň respirační insuficience nekoreluje s hybným postižením. Někteří pacienti s výraznou poruchou hybnosti mají jen malou redukci vitální kapacity a naopak u některých pacientů jde o dominantní příznak bez zřetelné poruchy hybnosti [23,24]. U nemocných s PN nelze respirační problematiku redukovat jen na svalovou slabost respiračního svalstva s dominantním postižením bránice [25]. Významnou roli pravděpodobně hraje i postižení centrálních a periferních nervů. Depozita glykogenu byla nalezena i v autoptických vzorcích n. phrenicus a předních rozích míšních [1]. Také se spekuluje o centrální deregulaci dýchání na úrovni mozkového kmene: byla nalezena korelace mezi centrálními poruchami sluchu a respiračními potížemi u nemocných s PN [26]. To je také jedno z možných vysvětlení nestejnoměrného postižení hybnosti a dýchání. Respirační potíže každopádně patří mezi důležité příznaky PN. Nezřídka bývá dlouho omezena jen na námahovou dušnost nebo noční hypoventilaci s typickými ranními potížemi charakteru neosvěžujícího spánku, bolestí hlavy a denní spavosti.
Kardiální postižení
Zatímco u klasické infantilní formy je hypertrofická kardiomyopatie obligátní příznak, u pozdní formy není kardiální postižení přítomno, resp. je spíše zřídkavé a nevýznamné. Forsha et al [27] nalezli v kohortě 87 dospělých pacientů (průměrný věk 41 let) zkrácení PR intervalu v 10 %, v 7 % sníženou ejekční frakci levé komory a v 5 % zesílení levé komory při echokardiografii. Substituční léčba neměla na tyto abnormality vliv. Naopak v souboru Angeliniho et al [28,29], kteří prokázali v 14 % abnormality ve formě hypertrofie levé komory nebo septa, došlo po léčbě u jednoho pacienta (z devíti) k normalizaci morfologických parametrů. U ostatních zůstal nález nezměněn. V klasické práci Müllera- Felbera et al [10] byly v kohortě 38 dospělých pacientů zachyceny 5krát komorové extrasystoly při Holterově monitorování (Lown 1 až 3) a 3krát syndrom preexcitace Wolf- Parkinson- White. Kardiomyopatie nebyla echokardiograficky zachycena ani jednou.
Další systémové známky
Přibývá informací o postižení arteriálního řečiště. Dilatace ascendentní aorty a magistrálních mozkových tepen, disekce a zvýšený výskyt aneuryzmat vedou k vzniku cévního postižení mozku [30]. V mnichovské kohortě 44 nemocných [16] byly cerebrovaskulární příznaky nalezeny v 7 % případů. Prevalenční studie ukazují na signifikantně vyšší výskyt cerebrovaskulárních potíží u nemocných s PN [31].
Časté je také subklinické postižení sluchu, které bylo prokázáno asi u poloviny nemocných [26]. Vyskytují se oba typy postižení (kondukční i percepční) a jejich kombinace.
Průjmy a další gastrointestinální příznaky, které se u nemocných s PN vyskytují častěji než v běžné populaci [32], jsou důsledkem akumulace glykogenu v hladkých svalech trávicí trubice nebo gangliích Meissnerova či Auerbachova plexu [31]. Stejně tak se častěji vyskytuje inkontinence moči.
Z výše uvedeného vyplývá, že pozdní forma PN patří do obtížného diferenciálně diagnostického okruhu pletencových myopatií, resp. pletencové slabosti s řadou dalších nespecifických chronických potíží, z nichž žádná není pro chorobu určující. Řada nemocných se pravděpodobně skrývá pod jinými diagnózami a jsou neléčeni nebo léčeni nesprávně (tab. 7).
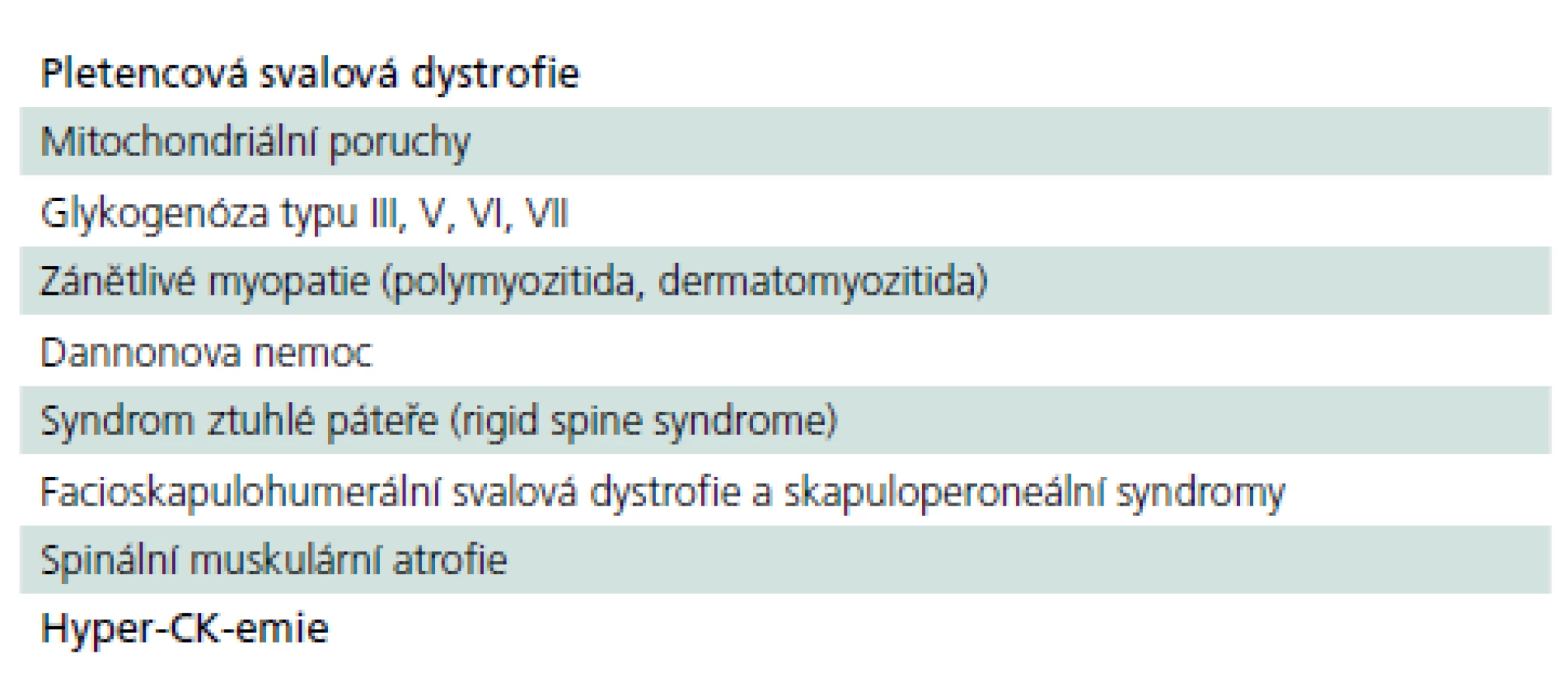
Diagnostika
Klinická diagnostika
Kořenovou a trupovou slabost vždy pečlivě kvantifikujeme pomocí MRC škály, dále vždy vyšetřujeme respirační funkce pomocí spirometrie. Základní vyšetření je usilovná vitální kapacita (FVC). Je důležité zdůraznit provedení nejen ve vertikální, ale i v horizontální poloze. Rozdíl o > 25 % [33] ukazuje na významnou slabost bránice, což je jeden z významných projevů PN. Pátráme po skolióze a zjišťujeme celkovou fyzickou výkonnost v dlouhodobém horizontu.
Svalové enzymy
Hyper-CK-emií se myslí vzestup nad 1,5násobek horního limitu normy, kdy je vyšetření opakováno alespoň 2krát a je vyloučen vliv cvičení (alespoň 72 hod). Evropské doporučení [34] říká, že asymptomatická nebo oligosymptomatická hyper-CK-emie by měla být vyšetřena svalovou biopsií, když je: 1) CK ≥ 3násobek normy nebo je: 2) přítomna intolerance zátěže či cvičením navozené bolesti nebo: 3) je přítomen „myopatický“ EMG nález nebo: 4) je nemocný mladší 25 let. Otázka je, zda a kdy bude toto doporučení modifikováno ve světle pokročilých DNA diagnostických metod. U mladých žen by každopádně biopsii mělo předcházet DNA vyšetření na přenašečství dystrofinopatie. Vzhledem k všeobecné dostupnosti spolehlivého skríningového vyšetření pomocí testu suché kapky (dried blood spot) je vhodné toto vyšetření provést před svalovou biopsií [35]. Pompeho nemoc byla shledána jako příčina hyper-CK-emie v 9 % dosud nediagnostikovaných případů [36].
CK je zvýšena u 3/ 4 nemocných [16], většinou nepřevyšuje 10 µkat/ l, i když jsou popsány i hodnoty přes 30 µkat/ l, které se blíží hodnotám obvyklým u svalových dystrofií s membránovým deficitem. Byly popsány i případy s elevací ALT a AST bez elevace CK [16].
EMG vyšetření
EMG vyšetření prokazuje nespecifické myogenní změny: zmenšení teritoria potenciálů motorických jednotek, předčasný nábor a spontánní aktivitu charakteru fibrilací, pozitivních ostrých vln a komplexních repetitivních výbojů. Vzhledem k chronickému průběhu lze u některých nemocných nalézt i zvětšené potenciály motorických jednotek, vzácně byly také zachyceny myotonické výboje [10].
Biopsie
Provedení svalové biopsie není při správně zaměřeném vyšetření nutné. Skríningové vyšetření pomocí suché kapky by mělo proběhnout před svalovou biopsií, pokud máme podezření na kořenovou slabost svalového původu a není z diferenciálně diagnostické rozvahy riziko z prodlení (např. zánětlivá myopatie). Nemocní s PN mají v bioptickém nálezu obraz vakuolární myopatie s akumulací glykogenu intralysozomálně i volně [10]. Množství vakuol odpovídá tíži choroby. Samozřejmě jsou přítomny i obecné známky svalového postižení: kolísání kalibru svalových vláken, centrální jádra, štěpení vláken, zmnožení intersticia atd. Nález je zpravidla výrazný, nicméně při vyšetření svalů málo postižených nebo naopak výrazně dystroficky změněných může být biopsie nevýtěžná. Nejčastěji vyšetřovaným a nejvýtěžnějším se jeví čtyřhlavý sval [1]. Někdy také nacházíme podobně jako u jiných svalových dystrofií zánětlivou celulizaci [1], která může vést k chybné diagnóze zánětlivé myopatie. Postiženy jsou svalové buňky kosterního svalstva, myokardu, ale i buňky hladké svaloviny cév či močového měchýře nebo jícnu [1], což vysvětluje další postižení jako cévní disekce, poruchy kontinence nebo polykání. Jako diagnostická se biopsie ukazuje asi v 70 % případů [17]. Při izolovaném spoléhání na svalovou biopsii tedy uniká asi 1/ 3 nemocných správné diagnóze a možnosti včasné terapie.
MR svalů
Jde o metodu, která je v současné době považována za vyšetření s nejvyšší senzitivitou a specificitou, obojí kolem 90 % [37]. Pomocí vyšetření lze dobře prokázat postižení respiračních svalů (bránice, břišní stěna, interkostální svaly) [38] i časné postižení paravertebrálních svalů. Müller- Felber et al [10] nalezli významné postižení svalů jak na přední, tak zadní straně stehna s výrazným ušetřenímm. rectus femoris. Za zmínku také stojí, že tam, kde byla podána kontrastní látka, došlo k významnému sycení. Není to tedy vhodná metoda k odlišení od zánětlivých myopatií. Pomocí celotělového MR vyšetření je možné dobře demonstrovat celkový rozsah svalového postižení včetně hluboko uložených svalů nebo např. svalstva jazyka [39,40]. Vlastní MR obraz se nijak neliší od jiných svalových dystrofií: zvýšení signálu (tuková degenerace) v T1 vážených obrazech bez známek objemové atrofie.
Stanovení hladiny klíčového enzymu: alfa glukosidázy
Jde o rozhodující diagnostický proces, který má dvě úrovně. První je skríningové vyšetření pomocí testu suché kapky (viz výše). K vyšetření stačí skutečně kapka kapilární nebo žilní krve, která se nechá zaschnout na speciálním filtračním papírku. Senzitivita, a díky testu s akarbózou, která inaktivuje ostatní neutrální glukosidázy, i specificita metody je vysoká. Oba parametry se blíží 100 % [41]. Potvrzení diagnózy se provádí vyšetřením aktivity GAA v leukocytech. Obě vyšetření se v ČR provádějí v Ústavu dědičných metabolických poruch v Praze (http:/ / udmp.lf1.cuni.cz/ ).
DNA diagnostika
DNA vyšetření je důležité pro detekci přenašečů v rodině, popřípadě v situaci hraničního enzymologického vyšetření. Shromažďují se data o korelaci genotyp- fenotyp a frekvenci jednotlivých mutovaných alel v populaci. U adultní formy je daleko nejčastější mutace charakteru nukleotidové substituce v intronu 1: c.– 32– 13T>G (IVS1– 13T>G), která se vyskytuje v 36– 90 % případů [21,42,43]. Jde o mutaci, jež způsobuje významně sníženou aktivitu alfa glukosidázy, ale úplně ji neblokuje. Většina pacientů v rodinách se stejným genotypem sdílí podobný fenotyp včetně doby začátku potíží. V některých případech se však ukazují významné rozdíly, které jdou patrně na vrub vnějším vlivům nebo dosud nepoznaným epigenetickým faktorům [44].
Přirozený vývoj
Před zavedením substituční terapie byla klasická infantilní forma PN onemocnění s katastrofickou prognózou. Patnáct měsíců života přežilo jen jedno dítě z 61 [4]. Medián délky života byl 8,7 měsíce.
U pozdní formy je průběh choroby podstatně pomalejší a velmi individuální. Z práce Van der Beekové et al [24] vyplývá, že průměrný pokles respiračních funkcí je jen 1,6 % ročně a svalové síly dokonce jen 1,2 % ročně (přičemž pokles není paralelní). Někteří pacienti ale ztratili 1/ 3 svalové síly za tři roky, zatímco u jiných nedošlo za 15 let k žádnému poklesu. Stejné je to u respiračních funkcí: někteří za 15 let neztratili téměř žádnou respirační kapacitu, kdežto jiní až 60 % predikované kapacity za pět let.
Terapie
První klinické studie byly zahájeny v roce 1999 a účinná látka byla registrována v Evropě i USA v roce 2006. Terapie spočívá v podávání i.v. infuze biotechnologicky vyrobené alfa glukosidázy (rhGAA) v týdenních nebo 14denních intervalech. Léčba infantilní formy redukuje riziko smrti nebo nutnosti plicní ventilace o 60– 90 % [4]. Přes tento významný pokrok, který změnil přirozený fatální vývoj choroby, nedochází k úplnému uzdravení a u mnoha dětí se vyvíjí nějaká forma „reziduálního“ pozdního onemocnění.
U pozdní formy choroby nejsou výsledky substituční terapie tak výrazné. V roce 2010 byla zveřejněna práce nizozemských autorů [45], která hodnotila skupinu 90 osob (30 randomizováno pro placebo, 60 pro účinnou látku – 20 mg rhGAA/ Kg t.hm. 1× za 14 dnů). Délka sledování byla šest let a hlavními hodnocenými parametry byl 6min test chůze a FVC. Ze studie vyplývá, že u nemocných léčených účinnou látkou došlo v prvních 12– 24 měsících k částečnému zlepšení sledovaných funkcí s následnou stabilizací stavu. Přes nesporný úspěch práce ukazuje na omezené regenerační schopnosti svalové tkáně a nastoluje otázku, v které fázi začít pacienty léčit. Každopádně je nutné nemocné velmi pečlivě kontrolovat a vyšetřovat pomocí kvantifikovaných testů, abychom zachytili a dokumentovali progresi choroby.
Recentní doporučení americké AANEM (The American Association of Neuromuscular & Electrodiagnostic Medicine) [46] lze shrnout do následujících bodů:
- neléčit presymptomatické pacienty,
- léčit symptomatické i při lehkých relevantních příznacích („subtle“),
- léčit i nemocné v závažném stavu (umělá plicní ventilace),
- rok po zahájení znovu zhodnotit,
- aplikovat multidisciplinární přístup.
Za relevantní příznaky je považován pokles FVC a proximální svalová slabost.
Pacienti by měli dostat kvalitní pravidelný rehabilitační program k zabránění dekondice a kontraktur, včetně dechové rehabilitace. Pokud to situace vyžaduje, tak i neinvazivní, event. invazivní umělou plicní ventilaci.
Respirační péče
Postižení dechového svalstva je jedním z prominentních příznaků PN. Pečlivé a pravidelné monitorování dechových funkcí je základní opatření [23] – na něj musí ale navazovat i cílená dechová rehabilitace, nácvik expektorace včetně asistenčních pomůcek. Vyžaduje-li to situace, tak zahájíme zavčas neinvazivní, popřípadě invazivní dechovou podporu. Cílem je dosažení normální saturace O2 a normalizace pCO2. Někdy je možné těchto cílů dosáhnout i neinvazivní podporou [47].
Rehabilitační péče
Zatím nemáme spolehlivá data založená na EBM ohledně efektu rehabilitační péče. Zdůrazňuje se používání submaximální aerobní zátěže k zabránění dekondice. Nemají se používat žádné metody k přetěžování fragilních svalů a mají být pečlivě monitorovány respirační funkce. Samozřejmě prevence kontraktur, dechové cvičení, nácvik správné postury a podle potřeby používání vhodných ortéz. Obecně je doporučována jízda na kole nebo plavání či jiné aktivity ve vodě, vždy s aktivní asistencí [2].
Na rozdíl od některých jiných metabolických chorob nejsou nutná žádná dietní opatření, obecně se u všech svalových chorob doporučuje vyšší příjem bílkovin asi o 30 %.
Dosavadní studie naznačují, že terapie vede u pozdní choroby nejen ke stabilizaci, ale i ke zlepšování stavu [48] (alespoň v krátkém, tříletém horizontu). Dlouhodobé výsledky nemáme a vzhledem k pomalému vývoji nemoci se stále hledají odpovídající klinické testy, které by ukázaly, zda je dlouhodobá léčba pro nemocné užitečná. Hovoří se o konceptu minimálních klinicky významných změn (Minimal Clinically Important Diference, MCID) [48]. Průkaz efektivity léčby infantilní formy je, vzhledem k rychlé infaustní prognóze, v tomto smyslu podstatně jednodušší a je známa i řada prognostických biomarkerů [49].
MUDr. Stanislav Voháňka, CSc., MBA
Neurologická klinika LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: svohanka@fnbrno.cz
Přijato k recenzi: 1. 9. 2014
Přijato do tisku: 21. 10. 2014
Recenzenti
doc. MUDr. Edvard Ehler, CSc.
doc. MUDr. Hana Ošlejšková, Ph.D.
doc. MUDr. Peter Špalek, Ph.D.
Autor deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prim. MUDr. Stanislav Voháňka, CSc., MBA

MUDr. Voháňka promoval na Masarykově univerzitě v roce 1982. Poté zahájil svoji lékařskou kariéru na Klinice dětské neurologie. Od roku 1989 pracuje na Neurologické klinice LF MU a FN Brno, kde se stal roku 1993 zástupcem přednosty pro LPP. Dlouhodobě se věnuje problematice nervosvalových onemocnění. Je zakládajícím členem Neuromuskulární sekce České neurologické společnosti ČLS JEP a od roku 2005 jejím předsedou. Je členem Treat-NMD a kurátorem dvou národních neuromuskulárních registrů. Prezentoval přes 450 přednášek a posterů a jako autor nebo spoluautor se podílel na více než 300 publikacích. Působí jako sekretář redakce časopisu Česká a slovenská neurologie a neurochirugie.
Zdroje
1. Hobson- Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late- onset Pompe disease: a case report and systematic review of the literature. Mol Genet Metab 2012; 106(4): 462– 469. doi: 10.1016/ j.ymgme.2012.05.007.
2. Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med 2006; 8(5): 318– 327.
3. Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999; 7(6): 713– 716.
4. Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998; 79(1): 69– 72.
5. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 1999; 105(1– 2): 151– 156.
6. Lin CY, Hwang B, Hsiao KJ, Jin YR. Pompe’s disease in Chinese and prenatal diagnosis by determination of alpha- glucosidase activity. J Inherit Metab Dis 1987; 10(1): 11– 17.
7. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. J Am Med Assoc 1999; 281(3): 249– 254.
8. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet 2004; 12(2): 87– 92.
9. Hagemans ML, Winkel LP, Van Doorn PA, Hop WJ,Loonen MC, Reuser AJ et al. Clinical manifestation and natural course of late- onset Pompe’s disease in 54 Dutch patients. Brain J Neurol 2005; 128(3): 671– 677.
10. Müller- Felber W, Horvath R, Gempel K, Podskarbi T, Shin Y, Pongratz D et al. Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord 2007; 17(9– 10): 698– 706.
11. Byrne BJ, Kishnani PS, Case LE, Merlini L, Müller- Felber W, Prasad S et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab 2011; 103(1): 1– 11. doi: 10.1016/ j.ymgme.2011.02.004.
12. Vohanka S, Oslejskova H, Hlavata J, Lukacs Z. Screening program for adult Pompe disease in Czech Republic. Neuromuscul Disord 2010; 20: 673.
13. Voháňka S, Ošlejšková H, Lukacs Z, Hlavatá J. Adultní forma Pompeho nemoci v ČR, rok druhý. Cesk slov Neurol N 2010; 73/ 106(4): S470.
14. Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile- onset Pompe disease. J Pediatr 2006; 148(5): 671– 676.
15. Roberts M, Kishnani PS, van der Ploeg AT, Müller- Felber W, Merlini L, Prasad S et al. The prevalence and impact of scoliosis in Pompe disease: lessons learned from the Pompe Registry. Mol Genet Metab 2011; 104(4): 574– 582. doi: 10.1016/ j.ymgme.2011.08.011.
16. Schüller A, Wenninger S, Strigl- Pill N, Schoser B. Toward deconstructing the phenotype of late- onset Pompe disease. Am J Med Genet C Semin Med Genet 2012; 160(1): 80– 88. doi: 10.1002/ ajmg.c.31322.
17. Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease: muscle biopsy vs blood-based assays. JAMA Neurol 2013; 70(7): 923– 927. doi: 10.1001/ 2013.jamaneurol.486.
18. Winkel LP, Hagemans ML, van Doorn PA, Loonen MC, Hop WJ, Reuser AJ et al. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 2005; 252(8): 875– 884.
19. Jensen MP, Hoffman AJ, Stoelb BL, Abresch RT,Carter GT, McDonald CM. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil 2008; 89(2): 320– 328. doi: 10.1016/ j.apmr.2007.08.153.
20. Hagemans ML, Hop WJ, Van Doorn PA, Reuser AJ,Van der Ploeg AT. Course of disability and respiratory function in untreated late- onset Pompe disease. Neurology 2006; 66(4): 581– 583.
21. Van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet 2008; 372(9646):1342– 1353. doi: 10.1016/ S0140- 6736(08)61555- X.
22. Echaniz- Laguna A, Mohr M, Lannes B, Tranchant C.Myopathies in the elderly: a hospital-based study. Neuromuscul Disord 2010; 20(7): 443– 447. doi: 10.1016/ j.nmd.2010.05.003.
23. Van der Ploeg AT. Monitoring of pulmonary function in Pompe disease: a muscle disease with new therapeutic perspectives. Eur Respir J 2005; 26(6): 984– 985.
24. Van der Beek NA, Hagemans ML, Reuser AJ, Hop WC,Van der Ploeg AT, Van Doorn PA et al. Rate of disease progression during long-term follow-up of patients with late- onset Pompe disease. Neuromuscul Disord 2009; 19(2): 113– 117. doi: 10.1016/ j.nmd.2008.11.007.
25. Fuller DD, ElMallah MK, Smith BK, Corti M, Lawson LA, Falk DJ et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol 2013; 189(2): 241– 249. doi: 10.1016/ j.resp.2013.06.007.
26. Musumeci O, Catalano N, Barca E, Ravaglia S, Fiumara A, Gangemi G et al. Auditory system involvement in late onset Pompe disease: A study of 20 Italian patients. Mol Genet Metab 2012; 107(3): 480– 484. doi: 10.1016/ j.ymgme.2012.07.024.
27. Forsha D, Li JS, Smith PB, van der Ploeg AT, Kishnani P, Pasquali SK. Cardiovascular abnormalities in late onset Pompe disease and response to enzyme replacement therapy. Genet Med 2011; 13(7): 625– 631. doi: 10.1097/ GIM.0b013e3182142966.
28. Angelini C, Nascimbeni AC, Semplicini C. Therapeutic advances in the management of Pompe disease and other metabolic myopathies. Ther Adv Neurol Disord 2013; 6(5): 311– 321. doi: 10.1177/ 1756285613487570.
29. Angelini C, Semplicini C, Ravaglia S, Bembi B, Servidei S, Pegoraro E et al. Observational clinical study in juvenile- adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 2012; 259(5): 952– 958. doi: 10.1007/ s00415- 011- 6293- 5.
30. El- Gharbawy AH, Bhat G, Murillo JE, Thurberg BL,Kampmann C, Mengel KE et al. Expanding the clinical spectrum of late- onset Pompe disease: dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol Genet Metab 2011; 103(4): 362– 366. doi: 10.1016/ j.ymgme.2011.04.009.
31. Filosto M, Todeschini A, Cotelli MS, Vielmi V, Rinaldi F, Rota S et al. Non- muscle involvement in late- onset glycogenosis II. Acta Myol 2013; 32(2): 91– 94.
32. Karabul N, Skudlarek A, Berndt J, Kornblum C, Kley RA, Wenninger S et al. Urge incontinence and gastrointestinal symptoms in adult patients with Pompe disease: a cross- sectional survey. JIMD Rep. In press 2014. doi: 10.1007/ 8904_2014_334.
33. Lechtzin N, Wiener CM, Shade DM, Clawson L, Diette GB. Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest 2002; 121(2): 436– 442.
34. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ et al. EFNS guidelines on the diag-nostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17(6): 767– 773. doi: 10.1111/ j.1468- 1331.2010.03012.x.
35. Lukacs Z, Nieves Cobos P, Mengel E, Hartung R, Beck M, Deschauer M et al. Diagnostic efficacy of the fluorometric determination of enzyme activity for Pompe disease from dried blood specimens compared with lymphocytes- possibility for newborn screening. J Inherit Metab Dis 2010; 33(1): 43– 50. doi: 10.1007/ s10545- 009- 9003- z.
36. Fernandez C, de Paula AM, Figarella- Branger D, Krahn M, Giorgi R, Chabrol B et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66(10): 1585– 1587.
37. Cardy CM, Potter T. The predictive value of creatine kinase, EMG and MRI in diagnosing muscle disease. Rheumatology (Oxford) 2007; 46(10): 1617– 1618.
38. Gaeta M, Barca E, Ruggeri P, Minutoli F, Rodolico C, Mazziotti S et al. Late- onset Pompe disease (LOPD): correlations between respiratory muscles CT and MRI features and pulmonary function. Mol Genet Metab 2013; 110(3): 290– 296. doi: 10.1016/ j.ymgme.2013.06.023.
39. Carlier RY, Laforet P, Wary C, Mompoint D, Laloui K, Pellegrini N et al. Whole- body muscle MRI in 20 patients suffering from late onset Pompe disease: involvement patterns. Neuromuscul Disord 2011; 21(11): 791– 799. doi: 10.1016/ j.nmd.2011.06.748
40. Horvath JJ, Austin SL, Case LE, Greene KB, Jones HN,Soher BJ et al. Correlation between quantitative whole- body muscle MRI and clinical muscle weakness in Pompe disease. Muscle Nerve. In press 2014. doi: 10.1002/ mus.24437.
41. Umapathysivam K, Hopwood JJ, Meikle PJ. Determination of acid a- glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin Chem 2001; 47(8): 1378– 1383.
42. Hermans MM, van Leenen D, Kroos MA, Beesley CE,Van Der Ploeg AT, Sakuraba H et al. Twenty- two novel mutations in the lysosomal alpha- glucosidase gene (GAA) underscore the genotype- phenotype correlation in glycogen storage disease type II. Hum Mutat 2004; 23(1): 47– 56.
43. Montalvo AL, Bembi B, Donnarumma M, Filocamo M, Parenti G, Rossi M et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum Mutat 2006; 27(10): 999– 1006.
44. Wens SC, van Gelder CM, Kruijshaar ME, de Vries JM, van der Beek NA, Reuser AJ et al. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis 2013; 8(1): 182. doi: 10.1186/ 1750- 1172- 8- 182.
45. van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ et al. A randomized study of alglucosidase alfa in late- onset Pompe’s disease. N Engl J Med 2010; 362(15): 1396– 1406. doi: 10.1056/ NEJMoa0909859.
46. Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ,Barohn RJ et al. Consensus treatment recommendations for late- onset Pompe disease. Muscle Nerve 2012; 45(3): 319– 333. doi: 10.1002/ mus.22329.
47. Mellies U, Stehling F, Dohna- Schwake C, Ragette R,Teschler H, Voit T. Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology 2005; 64(8): 1465– 1467.
48. Lachmann R, Schoser B. The clinical relevance of outcomes used in late- onset Pompe disease: can we do better? Orphanet J Rare Dis 2013; 8(1):160. doi: 10.1186/ 1750- 1172- 8- 160.
49. Patel TT, Banugaria SG, Case LE, Wenninger S, Schoser B, Kishnani PS. The impact of antibodies in late- onset Pompe disease: a case series and literature review. Mol Genet Metab 2012; 106(3): 301– 309. doi: 10.1016/j.ymgme.2012.04.027.
50. Hirschhorn R, Reuser AJ. Glycogen storage dinase type II; acid-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly W, Valle D (eds). The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill 2001, 3389–3420.
Štítky
Detská neurológia Neurochirurgia NeurológiaČlánok vyšiel v časopise
Česká a slovenská neurologie a neurochirurgie

2014 Číslo 6
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Neuromultivit v terapii neuropatií, neuritid a neuralgií u dospělých pacientů
- Antidepresivní efekt kombinovaného analgetika tramadolu s paracetamolem
- Geriatrická křehkost a léčba bolesti
Najčítanejšie v tomto čísle
- Diazepam i. m. – nejčastěji užívaný, ale nevhodný lék ke zvládání akutní úzkosti, agitovanosti a agresivity
- Střelné poranění hlavy replikou historické zbraně – patofyziologie a popis kazuistiky
- Adultní forma Pompeho nemoci
- Test neverbální fluence – Five Point Test: normativní data pro dospělé