
Nejnovější poznatky v mikroskopické diagnostice kardiomyopatií
Recent advances in microscopic diagnosis of cardiomyopathies
A substantial proportion of cardiomyopathies (CMP) harbour non-specific microscopic findings and the diagnosis is based on the clinical phenotype. Therefore a majority of dilated and hypertrophic CMP are encountered by a pathologist as explanted hearts or during autopsy. The indication for the endomyocardial biopsy usually follows clinical suspicion for infiltrative disease and plays an important role in paediatric patients, where the metabolic CMP are more frequent. Due to suggestive microscopic appearance of these diseases, a histopathological examination represents an important part of the diagnostic algorithm. The biopsy is relevant especially in case of restrictive CMP, because this disease is often caused by amyloid depositions. In case of hypertrophic CMP, the endomyocardial biopsy is considered usually in paediatric population since the majority of storage and mitochondrial disorders manifest hypertrophic phenotype. Diagnosis of dilated CMP is based on the clinical grounds and the main task for the pathologist is to rule out myocarditis.
Keywords:
immunohistochemistry – histopathology – endomyocardial biopsy – cardiomyopathy
Autori:
Ondřej Fabián 1; Cyril Štěchovský 2
Pôsobisko autorov:
Ústav patologie a molekulární medicíny 2. LF UK a FN Motol, Praha
1; Kardiologická klinika 2. LF UK a FN Motol, Praha
2
Vyšlo v časopise:
Čes.-slov. Patol., 55, 2019, No. 4, p. 224-230
Súhrn
Podstatná část kardiomyopatií (CMP) je s ohledem na jejich nespecifický mikroskopický nález diagnostikována na klinické úrovni a ve specifických případech doplněná o genetické vyšetření. Patolog se s nimi setká spíše v podobě explantovaných srdcí nebo v pitevní problematice. K odběru endomyokardiální biopsie se přistupuje zejména za účelem vyloučení infiltrativního onemocnění a důležitou roli hraje hlavně v pediatrické problematice. U dětí je vyšší zastoupení metabolických CMP, které mnohdy vykazují sugestivní mikroskopický obraz a histopatologické vyšetření je zde důležitou součástí diagnostického procesu. Biopsie má význam zejména v případě restrikční CMP, která je často způsobená depozicí amyloidu. V případě hypertrofické CMP se k bioptickému odběru přistupuje téměř výlučně v pediatrické populaci, jelikož podstatná část střádavých a mitochondriálních onemocnění se manifestuje právě hypertrofickým fenotypem. Diagnóza dilatační CMP stojí spíše na klinické bázi a úkolem patologa je zde vyloučit myokarditidu.
Klíčová slova:
kardiomyopatie – histopatologie – endomyokardiální biopsie – imunohistochemie
Kardiomyopatie (CMP) představují skupinu geneticky podmíněných nebo získaných onemocnění srdečního svalu a podstatná část z nich je s ohledem na jejich nespecifický mikroskopický nález diagnostikována na klinické úrovni. Ve specifických případech je diagnostika doplněna o genetické vyšetření. Patolog se s nimi setká spíše v podobě explantovaných srdcí nebo v pitevní problematice. Odběr endomyokardiální biopsie (EMB) je indikován zejména za účelem vyloučení infiltrativního onemocnění, například amyloidózy. Častěji se k biopsii přistupuje u dětí, kde do hry vstupuje široká paleta metabolických CMP. Patolog tak ve své bioptické praxi naráží na poněkud odlišné spektrum diagnóz než klinický kardiolog. Tento rozdíl respektuje i následující text, který volně navazuje na klinický článek „Štechovský, Adla, Bonaventura: Myokarditidy a kardiomyopatie z pohledu kardiologa“ a referuje o histopatologické diagnostice CMP spolu se zaměřením na vzácnější formy metabolických a geneticky podmíněných CMP (tab. 1). Jejich morfologie je totiž mnohdy sugestivní a mikroskopické vyšetření zde hraje zásadní roli.
ZPRACOVÁNÍ MATERIÁLU
U tak nesourodé skupiny onemocnění jakými CMP bezesporu jsou nelze podat žádné obecné doporučení stran zpracování materiálu. Indikace jednotlivých specializovaných vyšetření vychází z klinicky zvažované diagnózy a nálezu v základním barvení. V případě jakéhokoliv podezření na CMP, zejména u dětí, je vždy vhodné jeden vzorek ponechat v nativním stavu na případný průkaz tukových vakuol nebo imunofluorescenční typizaci amyloidu. I barvení PAS a PAS s diastázou je lepší provést na nativní materiál. Při podezření na metabolickou CMP je pak vhodné jeden vzorek fixovat v glutaraldehydu pro potřeby elektronové mikroskopie.
DILATAČNÍ KARDIOMYOPATIE (DCMP)
Ačkoliv nejčastější fenotypickou variantu představuje hypertrofická CMP, s dilatační CMP se v klinické i patologické praxi setkáváme mnohem častěji, protože většina pacientů dospěje do chronického srdečního selhání. Role patologa je zde limitovaná vzhledem k obvykle necharakteristickému mikroskopickému nálezu. S výjimkou malé skupiny metabolických CMP, které se někdy mohou fenotypem DCMP prezentovat (1), zůstává definitivní diagnóza na klinické úrovni a úkolem patologa, pakliže je přistoupeno k odběru EMB, je tak spíše vyloučit myokarditidu, respektive zánětlivou CMP. Svou roli hraje i špatná genotypicko-fenotypická korelace, jelikož řadu mutací dříve považovaných za specifické pro DCMP nacházíme i u jiných forem CMP. DCMP tak nejspíše představuje „end stage“ fázi řady různých, geneticky podmíněných i získaných chorob (2). V neposlední řadě situaci komplikuje fakt, že kauzální mutaci se podaří prokázat jen v malém procentu případů. Nicméně, v posledních letech se objevují první slibné práce využívající imunohistochemii (IHC) v diagnostice jednotlivých geneticky podmíněných typů DCMP (viz níže).
Z morfologického pohledu DCMP charakterizuje dilatace všech srdečních oddílů s maximem v levé komoře. Dilatace síní bývá mírnější než u restrikční CMP. Koronární arterie jsou obvykle volné a díky extrémní dilataci komor se jeví napnuté. Ačkoliv je zde téměř vždy určitá míra hypertrofie, komory mívají kvůli dilataci často normální tloušťku nebo jsou i tenčí. Celkově srdce působí ochablým až hadrovitým dojmem a na řezu kolabuje. Mnohdy jsou přítomny okrsky makroskopicky viditelné fibrotizace až větších jizev (2). Lineární jizva na spodní stěně levé komory je charakteristická pro DCMP s mutací genu pro dystrofin a v diferenciální diagnóze v tomto případě přichází (při vyloučení koronárního postižení) zejména levostranně predominantní arytmogenní CMP (3). Endokard bývá fibrotizovaný, zejména v oblasti apexu, často i s nástěnnými tromby. Semilunární chlopně mají většinou normální morfologii, atrioventirkulární pak vykazují sekundární změny při relativní insuficienci způsobené dilataci fibrózních anulů (stočení a zesílení volných okrajů cípů chlopně) (2).
Mikroskopie bývá obvykle nespecifická a odpovídá obrazu excentrické hypertrofie např. u ischemické choroby srdeční. Kardiomyocyty jsou hypertrofické, s hyperchromními kvadratickými jádry a objemnou sarkoplasmou, často s patrnou ztrátou myofibril, vakuolární dystrofií a někdy až rozvojem myocytolýzy. Někdy bývají kardiomyocyty zvlněné následkem dilatace komor, na příčném průřezu tak nemusí nutně působit hypertroficky. V intersticiu je fibrotizace, někdy i splývající jizvy a v jejich okolí může být velmi mírný disperzní chronický zánět (2). Jak již bylo zmíněno výše, histopatologie neodliší geneticky podmíněné a získané formy DCMP. Nicméně, v poslední době se objevují první práce, využívající IHC průkaz různých sarkolemálních a cytoskeletálních proteinů. Například ztráta nebo diskontinuální exprese laminu A/C na jaderné membráně může být známkou laminopatií (4). Jako DCMP se často projevují i dystrofinopatie (5). Fenotyp může být omezený pouze na srdce (tzv. X-vázaná DCMP) nebo se může rozvinout spolu s kosterní myopatií v rámci Duchennovy či Beckerovy muskulární dystrofie. V přehledném barvení je morfologie opět nespecifická, v IHC lze však prokázat výpadky exprese dystrofinu, které bývají v případě Beckerovy muskulární dystrofie ložiskové a v případě Duchennovy dystrofie či X-vázané CMP extenzivnější až kompletní (4).
V posledních letech byla popsána tzv. mitogenní CMP. Jedná se o raritní formu DCMP manifestující se v kojeneckém věku, obvykle s rychlým a fatálním průběhem. Název choroby vyplývá z nálezu velmi početných mitotických figur v kardiomyocytech, které dosahují počtu až 4-5/1 HPF a obvykle lze zastihnout i atypické figury. Proliferační index stanovený IHC expresí antigenu Ki67 dosahuje až 20 % (norma pro kojenecký věk je cca 1 %) (6). U těchto pacientů byla popsána mutace genu Alström, jehož funkce doposud nebyla objasněna (7).
Myokarditida vs. zánětlivá kardiomyopatie
Určitý zmatek v klasifikaci CMP přinesl termín zánětlivá CMP. Světová zdravotnická organizace ji v roce 1995 definovala jako zánětlivé onemocnění myokardu spojené se srdeční dysfunkcí (8). Jakákoliv myokarditida vedoucí k alteraci myokardu a následnému rozvoji srdeční dysfunkce tak může být nazývána kardiomyopatií, což je svým způsobem logické vzhledem k velmi vágní a široké definici kardiomyopatií obecně (9,10). Toto doporučení následovala i European Society of Cardiology, která v roce 2008 tuto jednotku zařadila do skupiny DCMP (12). Ani v jednom případě však pod zánětlivou CMP nespadají případy myokarditidy s fulminantním průběhem nebo ty, které nevykazují známky srdeční dysfunkce. Čistě z histopatologického pohledu je zánětlivá CMP de facto chronickou myokarditidou, kde se kombinuje obraz zánětlivého postižení myokardu a znaků rozvíjející se CMP v podobě hypertrofie a fibrotizace (13). Nicméně nomenklatura není zcela ustálená, jelikož například American Heart Association od roku 2006 řadí jakoukoliv myokarditidu mezi primární získané DCMP (do skupiny infekčních, toxických a autoimunitních DCMP) (14).
Pro patologa je zánětlivá CMP kontroverzní jednotkou. Jde totiž o klinický termín, který mikroskopie nezná, a může do něj spadat obraz aktivní i borderline myokarditidy. Nález je tak vhodné interpretovat například jako „Obraz aktivní myokarditidy, konzistentní s klinickou diagnózou zánětlivé CMP“. Někdy je patolog postaven před otázku, jestli lze mikroskopickým vyšetřením diagnostikovat pozánětlivou DCMP. Odpověď zní nikoliv, jakmile vymizí známky zánětu (jinými slovy zánětlivá CMP přejde do pozánětlivé DCMP), nelze proběhlou myokarditidu mikroskopicky rozpoznat.
HYPERTROFICKÁ KARDIOMYOPATIE (HCMP)
Dle starších údajů drtivou většinu HCMP tvořily geneticky podmíněné formy, většinou na podkladě sarkomerických proteinů. V dnešní době je část těchto dříve kauzálních mutací považována za varianty nejasného významu a skutečně pozitivní genotyp má méně než 50 % HCMP. Morfologii takovýchto srdcí určuje kombinace charakteristické asymetrické formy hypertrofie a postižení chlopní a koronárních arterií. Hypertrofie, která může v některých případech přesahovat 30 mm tloušťky levé komory, postihuje všechny srdeční oddíly, maximum však nacházíme v oblasti bazální a ventrální části komorového septa a anterolaterální části volné stěny levé komory. Cca v 10% případů jde o predominantně apikální formu (2). Nicméně až pětina pacientů s geneticky potvrzenými sarkomerickými mutacemi má normální tloušťku komor (14). Z chlopní je nejčastěji postižená mitrální, s hypertrofií a atypickou lokalizací papilárních svalů, zkrácením šlašinek, inzercí šlašinek do volné stěny komory (což je fyziologické pouze pro trikuspidální chlopeň) a prodloužením cípů chlopně. Někdy je patrná ploška fibrózně zesíleného endokardu v místě styku předního cípu mitrální chlopně s vyklenujícím se komorovým septem (15). Poměrně častou doprovodnou anomálií jsou intramyokardiální můstky, neboli segmenty koronární arterie (většinou ramus interventricularis anterior), probíhající v myokardu namísto epikardiálně (16). Z mikroskopického pohledu je pak zcela charakteristickým nálezem tzn. disarray, neboli ložiska neuspořádanosti myokardu. S využitím „měkotkáňové“ terminologie lze mluvit o dvou základních histopatologických vzorcích: 1) storiformní uspořádání, které je častější a odpovídá tradičně známému obrazu nepravidelně se větvících svazků hypertrofických kardiomyocytů s fibrotizací v okolí; 2) stromečkovité (v angličtině „herringbone“) uspořádání s pravoúhlou orientací svazků kardiomyocytů. Doprovodným nálezem pak bývají dysplastické intramyokardiální větve koronárních arterií s hypertrofickou a fibrotizovanou svalovinou, fibroplázií intimy, nepravidelným pilovitým lumen a perivaskulární fibrózou (17,18).
Jak již bylo zmíněno, diagnostika HCMP je v dnešní době převážně klinická a patolog se tak s HCMP setkává spíše v podobě srdečních explantátů nebo při pitvě. Pro nález disarray a potvrzení klinické diagnózy HCMP je zapotřebí pečlivý sampling, ideálně s provedením příčných řezů přes komorové septum, kolmo na dlouhou osu srdce. Důležitá je i klinická korelace, drobné okrsky neuspořádanosti lze totiž nalézt např. i u vrozených srdečních malformací (typicky hypoplastické levé srdce, které disarray doprovází až v 80 % případů). V oblasti apexu a přechodu komorového septa do volných stěn komor je tento nález dokonce fyziologický (19). Absolutní množstevní kritéria pro disarray nejsou pevně daná, obecně je přijímána hranice minimálně 5 % objemu komorového septa (20). Patolog se dále může setkat s materiálem z myektomie. Cílem mikroskopického vyšetření je v tomto případě vyloučení jiné konkrétní jednotky, zejména střádavé choroby. Disarray zde zachytíme jen asi v 60 % případů, zbylé případy pak čistě z morfologického hlediska nelze odlišit od kongenitální subvalvární stenózy a zapotřebí je pečlivá klinická korelace (21,22).
V pediatrické populaci hraje histopatologické vyšetření mnohem důležitější roli. Hypertrofický fenotyp totiž velmi často vykazují metabolické CMP, například střádavá onemocnění typu glykogenóz nebo primární mitochondriální poruchy (23,24). HCMP spolu s převodními poruchami přichází v rámci glykogenózy III. typu (Corriho choroba) nebo infantilní formy Pompeho choroby (v dospělosti je postižení myokardu u Pompeho choroby méně časté) (25). Mikroskopicky bývá přítomná výrazná vakuolizace cytoplazmy kardiomyocytů, někdy až do obrazu pletiva, s pozitivním průkazem depozit glykogenu elektronmikroskopicky nebo v barvení PAS. Obdobný obraz mají i HCMP s mutacemi v genech LAMP2 (Danonova choroba) a PRKAG2, které typicky doprovází Wolf-Parkinson-Whiteův (WPW) syndrom a často chybí extrakardiální příznaky nebo jsou subklinické (26-28). Mikroskopický obraz glykogenózy myokardu u dítěte s novorozeneckou nebo juvenilní formou HCMP a WPW syndromem by tak měl vést k cílenému vyšetření těchto genů. Při nálezu PAS+ vakuol je vždy nutné vyloučit možnost i lipidózy, v případě srdce pak zejména Fabryho choroby. Napomůže průkaz koncentrických lamelárních tělísek v ultrastruktuře kardiomyocytů a nález vakuol v endotelu větví koronárních arterií (endoteliální depozita jsou pro Fabryho chorobu typická obecně) (29,30).
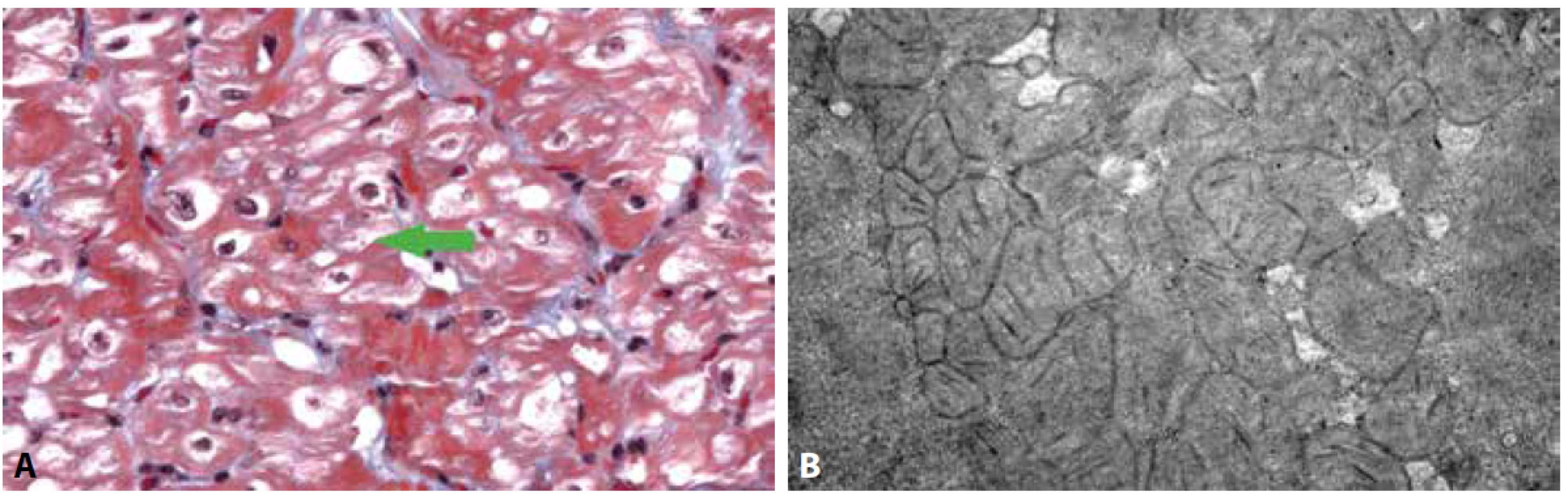
Mitochondriální CMP jsou obvykle doprovázeny neurologickými symptomy a dalšími znaky, budícími podezření na mitochondriální poruchu (např. laktátová acidóza). Mikroskopický nález je obdobný jako v jiných orgánech (typicky například v játrech) a lze pozorovat vakuolizaci kardiomyocytů s přítomností hojných zmnožených mitochondrií včetně tzv. obřích forem, které z definice přesahují průměr 2 um (v praxi se udává velikost 1/4 erytrocytu). Zásadní roli zde hraje elektronová mikroskopie, která ozřejmí početné atypické mitochondrie abnormálních tvarů i rozměrů, často s krystaloidními inkluzemi a denzními tělísky v matrix (31-34). Lze si pomoci i histochemickou analýzou aktivity enzymů sukcinátdehydrogenázy (SDH) nebo cyklooxygenázy (COX). Nejde o běžnou praxi, diagnostika většinou spočívá na genetickém vyšetření, případně svalové biopsii, lze jej však využít v případě izolovaného srdečního postižení. Nutné je vzít v úvahu, že ložiskové výpadky COX doprovází terminálně selhávající srdce i z jiných příčin (35).
Obrazem HCMP se může prezentovat i Noonanův syndrom, typicky v kombinaci s pulmonální stenózou nebo polyvalvární dysplázií, nebo může přicházet v rámci Friedrichovy ataxie (36,37). Z klinického hlediska je důležité mít na paměti, že přechodná hypertrofie srdce, která má tendenci ke spontánní úpravě, bývá přítomná i u dětí diabetických matek nebo u nedonošených novorozenců jako následek kortikoterapie při indukci plicní zralosti (38,39).
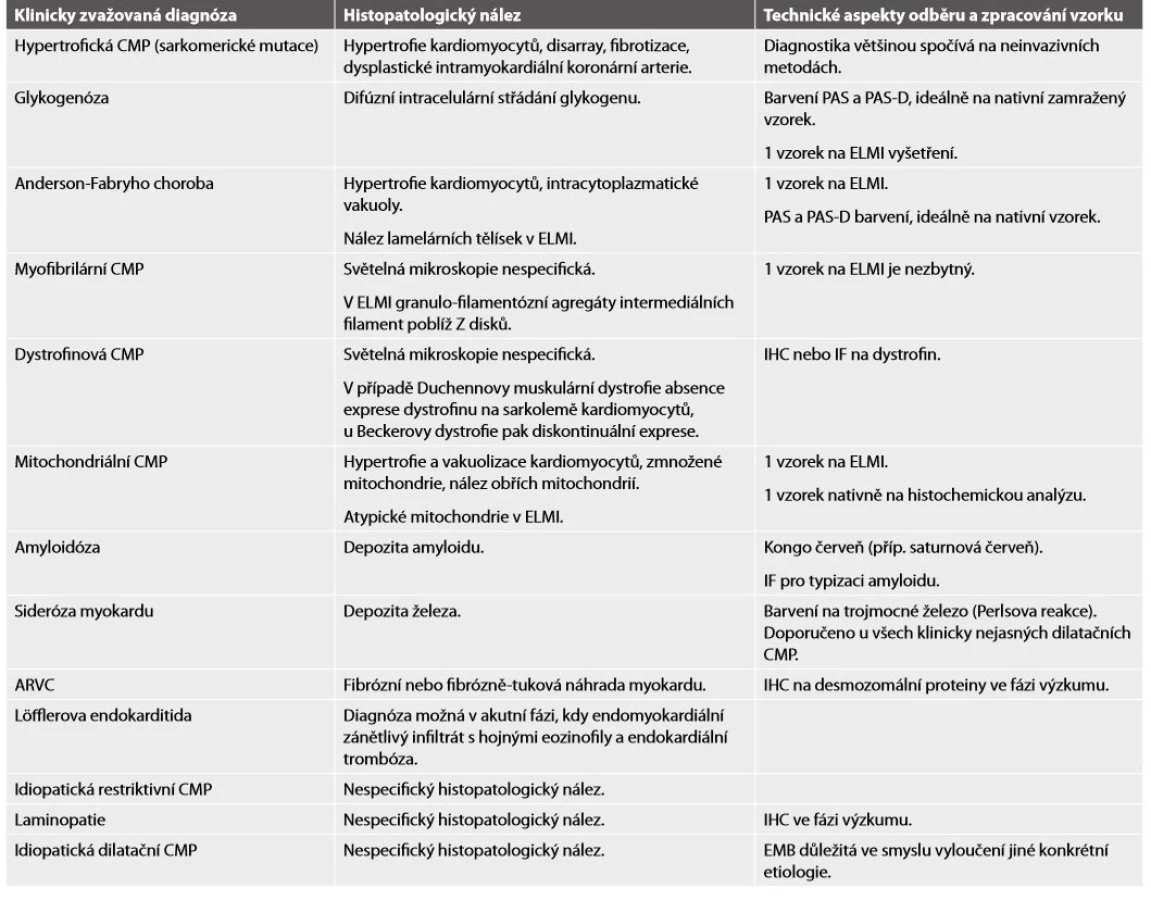
A: V makroskopickém nálezu dominuje výrazná hypertrofie všech srdečních oddílů s maximem v levém komoře a mezikomorovém septu.
B, C: Charakteristický obraz disarray s nepravidelně se křížícími hypertrofickými kardiomyocyty a intersticiální fibrózou (hematoxylin & eosin, Massonův
trichrom, 200x).
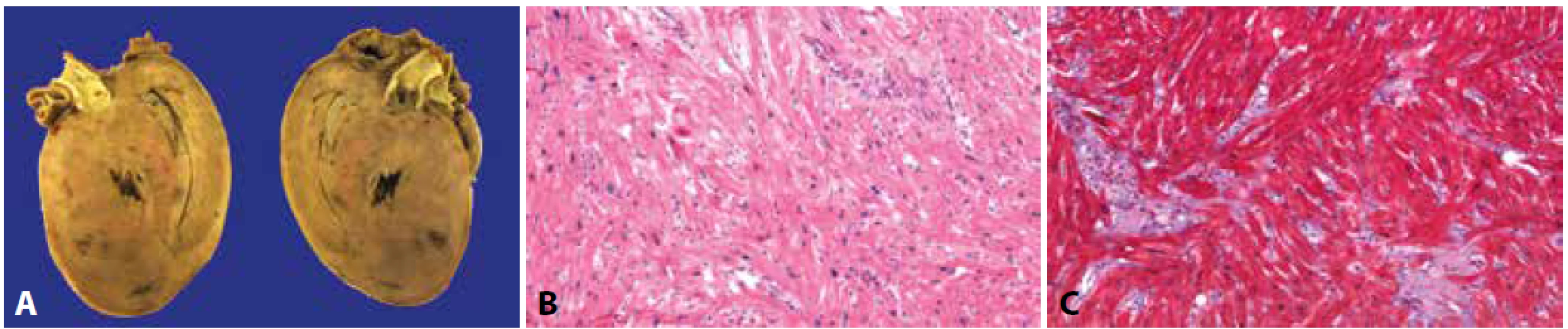

A: Depozita amyloidu intersticiálně i difúzně se zánikem kardiomyocytů (hematoxylin & eosin, 200x).
B: Průkaz amyloidu kongo červení (200x).
C: Pozitivita amyloidu v polarizovaném světle (200x).

RESTRIKČNÍ KARDIOMYOPATIE (RCMP)
Typický fenotyp RCMP odpovídá abnormální dilataci srdečních síní při normálním nebo lehce sníženém objemu komor a často i normální tloušťce stěn (11,13). Definice RCMP je však funkční, nikoliv morfologická, a stojí na průkazu restrikčního plnění levé komory (viz článek „Štěchovský, Adla, Bonaventura: Myokarditidy a kardiomyopatie z pohledu kardiologa“). Nejčastější příčinou RCMP je amyloidóza. Detailní popis problematiky amyloidóz přesahuje hranice tohoto článku. Ve stručnosti lze zmínit, že srdce bývá nejčastěji postiženo při AL amyloidóze (udává se, že až 20 % pacientů tímto typem amyloidózy má srdeční postižení) a poměrně častá je i transthyretinová RCMP, buď v rámci familiární amyloidové polyneuropatie nebo jako systémová senilní amyloidóza, případně i izolovaná síňová amyloidóza (40-43). Srdce je makroskopicky tuhé, matné, voskového vzhledu. V terminální fázi onemocnění bývá pravidlem i určitá míra hypertrofie komor. Mikroskopicky pozorujeme depozita amyloidu zejména intersticiálně, obkružující jednotlivé kardiomyocyty nebo tvořící drobné noduly, lze však nalézt i depozita subendokardiálně, na chlopních nebo ve stěně větví koronárních arterií (2,42).
Primární geneticky podmíněné RCMP jsou mnohem vzácnější. Část z nich je obdobně jako HCMP podmíněna mutacemi sarkomerických proteinů. Restriktivní fenotyp v těchto případech může být primární manifestací nebo může progredovat z předchozího obrazu HCMP. Mikroskopicky zde dominuje fibrotizace a zánik kardiomyocytů, někdy lze zastihnout reziduální úseky disarray (2).
Fenotypem RCMP se obvykle manifestují i myofibrilární CMP, ačkoliv jejich morfologie může být i hypertrofická či ARVC-like (44). Jde o skupinu vzácných onemocnění způsobených mutacemi genů pro proteiny Z-disků, nejčastěji desminu, vzácněji např. alfa-B krystalinu nebo BAG3. Onemocnění mohou být familiární i sporadická, manifestovat se v dětském i dospělém věku a většinou je doprovází i různá míra postižení kosterního svalstva (45-48). Základní mikroskopie je nespecifická, lze však využít IHC průkaz desminu, který ozřejmí jeho abnormální distribuci s tvorbou agregátů desminových filament v blízkosti Z-disků (což lze ověřit i elektronmikroskopicky). Klinicky toto onemocnění často doprovází AV blok, je proto vhodné provést IHC průkaz desminu u všech pacientů s touto formou arytmie, restrikčním fenotypem CMP a negativním průkazem amyloidu (49).
Pod RCMP spadají i některá onemocnění, postihující primárně endokard a případně přilehlou vrstvu myokardu. Pod diagnózu tzv. endomyokardiální fibrózy spadají dvě jednotky – Löfflerova (fibroplastická) endokarditida a tropická endomyokardiální fibróza (Daviesova choroba). První jmenovaná přichází v rámci hypereosinofilního syndromu a obvykle koexistuje s eosinofilní nekrotizující myokarditidou jako Löfflerova endomyokarditida. Zánět obvykle začíná postižením myokardu a postupně přechází na endokard. Charakteristický je objemný murální trombus, který je eosinofilním zánětem také prostoupen a následná endokardiální fibroprodukce pak do sebe trombus zaujímá a vytváří až 1 cm tlustý plak (50-52). Tropická endomyokardiální fibróza je endemitní v oblasti severního a jižního tropického pásu a způsobuje ji nejspíše hypersenzitivní reakce na malárii nebo proběhlá virová infekce. Charakterizuje ji zánět a fibroprodukce endokardu a přilehlého myokardu s maximem v apexu, vtokovém úseku obou komor a často i cípatých chlopní. Eosinofily v zánětu nejsou podmínkou (51,53).
Klinicky důležitou formou RCMP je i sideróza myokardu. Čistě mikroskopicky však nelze rozlišit hemochromatózu a sekundární siderózu (4,54).
A: Vakuolizace kardiomyocytů s patrnými zmnoženými mitochondriemi a záchytem obřích mitochondrií (šipka) (Massonův trichrom, 400x).
B: Ultrastruktura cytoplazmy kardiomyocytů s přítomností hojných atypických mitochondrií, které vykazují tvarové abnormality, paralelně uspořádané
kristy a denzní tělíska v mitochondriální matrix.
ARYTMOGENNÍ KARDIOMYOPATIE (ARVC)
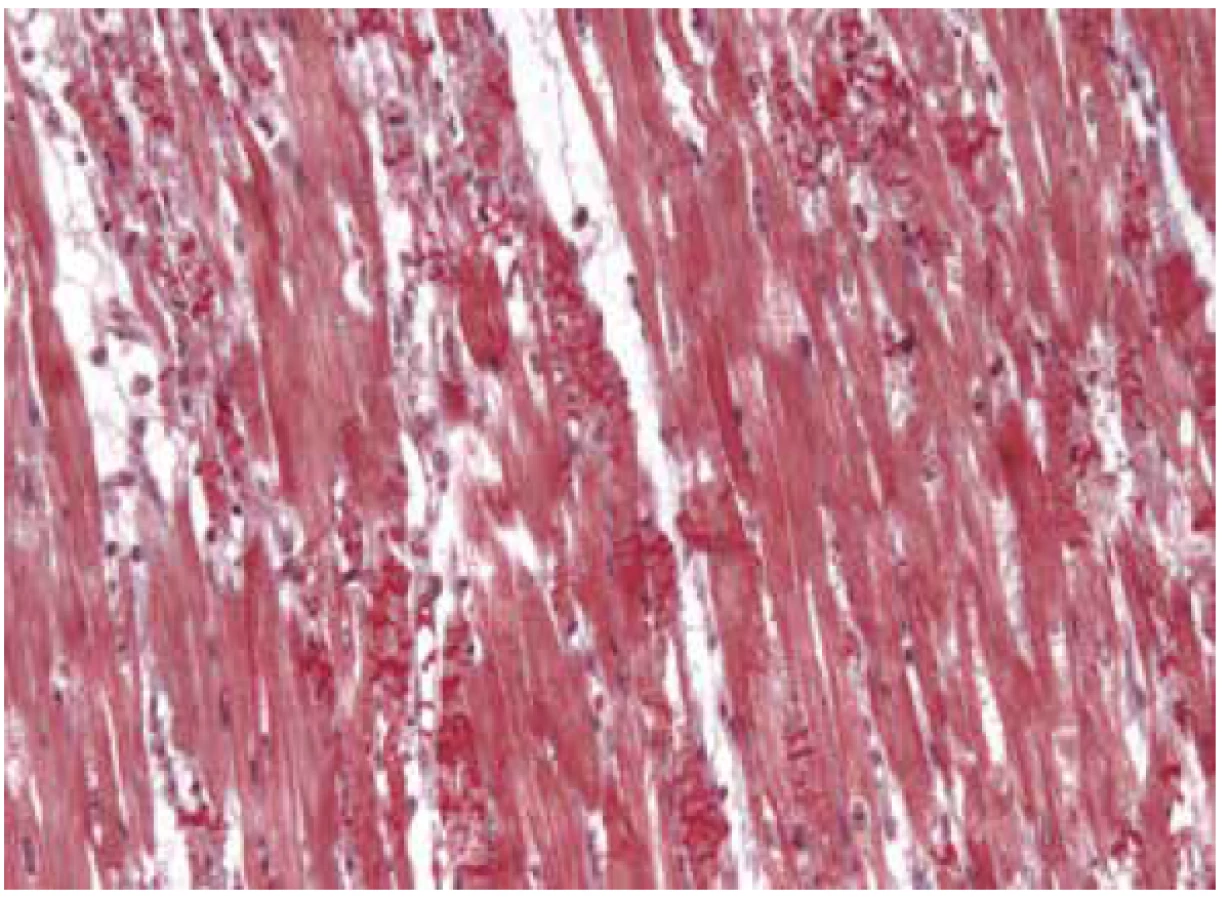
Arytmogenní kardiomyopatie pravé komory (dříve arytmogenní dysplázie) zdaleka není izolované postižení pravého srdce a téměř vždy je v určité míře postižena i levá komora. Existuje i levostranně predominantní typ (55). Proto byl název choroby zkrácen na arytmogenní kardiomyopatii (56). Podstatou choroby jsou mutace v desmosomálních proteinech kardiomyocytů, které vedou k jejich zániku a následné náhradě myokardu vazivovou a tukovou tkání (57,58). Oproti dřívějším doporučením je dnes z histopatologického pohledu určující právě fibrotizace, tuková náhrada je nekonstantní nález a není pro diagnózu nezbytná. Naopak čistá lipomatóza myokardu bez doprovodné fibrózy diagnózu ARVC vylučuje.
Makroskopie se liší v závislosti na fázi choroby. V pokročilém stadiu je patrná tukově-vazivová náhrada myokardu s maximem v oblasti ventrální nebo ventrokaudální stěny pravé komory a plicního infundibula. Stěna komory je ztenčená a srdce bývá dilatované. Fatální arytmie však může předcházet morfologicky viditelné strukturální abnormality a takové srdce pak může vypadat zcela normálně. Někdy bývá přítomná mírná dilatace plicního infundibula, která může v případě nejasných úmrtí vést k suspekci na tuto chorobu (2).
Diagnóza ARVC je multidisciplinární a opírá se o tzv. Task force kritéria z roku 2010 (59), podle kterých je histopatologický nález považovaný za velké kritérium pakliže fibrózní nebo fibrózně-tuková náhrada postihuje alespoň 60 % myokardu v alespoň 1 ze všech vzorků EMB. Postižení nad 25 % myokardu je pak malé kritérium. Jelikož se EMB obvykle provádí z pravé strany komorového septa, které téměř nikdy nebývá postiženo, využívá se biopsie navigovaná elektroanatomickým mapováním (60). Nicméně, procento falešně negativních výsledků je i tak vysoké a někdy může odběr vést i k mylné diagnóze myokarditidy, jelikož v případě ARVC okolní myokard zcela běžně vykazuje známky zánětu, někdy i s destrukcí kardiomyocytů. Díky těmto diagnostickým obtížím a vysokému riziku perforace ztenčené stěny komory je od EMB v dnešní době upouštěno a na většině pracovišť se již prakticky neprovádí. Ve fázi výzkumu je použití IHC a imunofluorescence na desmosomální proteiny (zejména plakoglobin) (61).
NEKLASIFIKOVANÉ KARDIOMYOPATIE
Za zmínku stojí nonkompaktní CMP (left ventricular non-compaction, spongiformní myokard), která může být izolovaná nebo doprovázet některé vrozené srdeční malformace (zejména konotrunkální vady) či některá systémová onemocnění (typicky Barthův nebo DiGeorgův syndrom) (62). Určující je zde makroskopie s nálezem spongiformní přeměny komor. Stěny komor jsou ztluštělé, s extrémně hlubokými a parožnatě větvenými recesy, anastomozujícími trabekulami a absencí normálně vyvinutých papilárních svalů (2). Nález je nejvýraznější v oblasti apexu (63). Nonkompakce by měla z definice postihovat alespoň 50 % tloušťky myokardu.
Histiocytoidní CMP (dříve hamartom z Purkyňových buněk) je forma infantilní metabolické CMP na podkladě mitochondriální poruchy. Mikroskopicky jsou přítomny okrsky a noduly kardiomyocytů s eosinofilní granulární cytoplazmou. Jedná se v zásadě o onkocytárně změněné kardiomyocyty, které většinou sledují průběh převodního systému (64).
Stresová (tako-tsubo) CMP je čistě klinickou diagnózou. Mikroskopický nález odpovídá obrazu katecholaminové toxicity s myocytolýzou a prominentními kontrakčními pruhy (65).
ZÁVĚR
Histopatologická diagnostika CMP je v konkrétních indikovaných případech nezbytnou součástí diagnostického procesu a zejména v pediatrické problematice přináší důležité informace stran dalšího terapeutického postupu. Obvykle vyžaduje využití specializovaných metod v podobě elektronové mikroskopie, IHC či vyšetření nativní tkáně a nezbytná je i pečlivá korelace s celkovým klinickým stavem pacienta. Výsledná interpretace je tak odrazem úzké spolupráce patologa a klinického kardiologa.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Práce byla podpořena MZ ČR – RVO, FN v Motole 00064203.
MUDr. Ondřej Fabián
Ústav patologie a molekulární medicíny 2. LF UK a FN Motol
V Úvalu 84
150 06, Praha 5
tel.: +420 224 435 645
e-mail: Ondrej.Fabian2@fnmotol.cz
Zdroje
1. Schwartz ML, Cox GF, Lin AE et al. Clinical approach to genetic cardiomyopathy in children. Circulation 1996; 94(8): 2021–2038.
2. Buja LM, Butany J. Cardiovascular Pathology (4th ed). United States: Academic Press; 2015: 437-445, 448, 458, 461-463, 469, 470, 477.
3. Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology 2003; 99(1): 1-19.
4. Leone O, Veinot JP, Angelini A et al. 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc Pathol 2012; 21(4): 245-274.
5. McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med 2007; 58: 75-88.
6. Chang KTE, Taylor GP, Meschino WS et al. Mitogenic cardiomyopathy: a lethal neonatal familial cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol 2010; 41(7): 1002–1008.
7. Shenje LK, Andersen P, Halushka MK et al. Mutations in Alström protein impair terminal differentiation in cardiomyocytes. Nat Commun 2014; 5: 3416.
8. Richardson P, McKenna W, Bristow M et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93(5): 841-842.
9. Caforio AL, Pankuweit S, Arbustini E et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34(33): 2636-2648.
10. Chimenti C, Frustaci A. Histopathology of myocarditis. Diagnostic Histopathology 2008; 14(8): 401-407.
11. Elliott P, Andersson B, Arbustini E et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29(2): 270-276.
12. Maisch B, Portig I, Ristic A, Hufnagel G, Pankuweit S. Definition of inflammatory cardiomyopathy (myocarditis): on the way to consensus. A status report. Herz 2000; 25(3): 200-209.
13. Maron BJ, Towbin JA, Thiene G et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113(14): 1807-1816.
14. Olivotto I, Girolami F, Ackerman MJ et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 2008; 83(6): 630-638.
15. Klues HG, Roberts WC, Maron BJ. Anomalous insertion of papillary muscle directly into anterior mitral leaflet in hypertrophic cardiomyopathy. Significance in producing left ventricular outflow obstruction. Circulation 1991; 84(3): 1188-1197.
16. Basso C, Thiene G, Mackey-Bojack S, Frigo AC, Corrado D, Maron BJ. Myocardial bridging, a frequent component of the hypertrophic cardiomyopathy phenotype, lacks systematic association with sudden cardiac death. Eur Heart J 2009; 30(13): 1627-1634.
17. Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Tajik AJ, Holmes DR. Myocardial bridging in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2003; 42(5): 889-894.
18. Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1): 36-44.
19. Khong TY, Malcombson RDG. Keelings Fetal and Neonatal Pathology (5th edn). Switzerland: Springer; 2015: 511.
20. Burke A, Tavola F. Practical Cardiovascular Pathology (Har/Psc ed). United States: Lippincott Williams & Wilkins; 2011: 163.
21. Lamke GT, Allen RD, Edwards WD, Tazelaar HD, Danielson GK. Surgical pathology of subaortic septal myectomy associated with hypertrophic cardiomyopathy. A study of 204 cases (1996-2000). Cardiovasc Pathol 2003; 12(3): 149-158.
22. Tazelaar HD, Billingham ME. The surgical pathology of hypertrophic cardiomyopathy. Arch Pathol Lab Med 1987; 111(3): 257-260.
23. Bates MG, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J 2012; 33(24): 3023-3033.
24. Giordano C, Perli E, Orlandi M et al. Cardiomyopathies due to homoplasmic mitochondrial tRNA mutations: morphologic and molecular features. Hum Pathol 2013; 44(7): 1262-1270.
25. Servidei S, Bertini E, DiMauro S. Hereditary metabolic cardiomyopathies. Adv Pediatr 1994; 41: 1–32.
26. Arad M, Moskowitz IP, Patel VV et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002; 109(3): 357–362.
27. Arad M, Maron BJ, Gorham JM et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 2005; 352(4): 362–372.
28. Maron BJ, Roberts WC, Arad M. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009; 301(12); 1253-1259.
29. O’Mahony C, Elliott P. Anderson-Fabry disease and the heart. Prog Cardiovasc Dis 2010; 52(4): 326-335.
30. Thurberg BL, Fallon JT, Mitchell R, Aretz T, Gordon RE, O’Callaghan MW. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation 2009; 119(19): 2561-2567.
31. Marin-Garcia J, Ananthakrishnan R, Goldenthal MJ et al. Cardiac mitochondrial dysfunction and DNA depletion in children with hypertrophic cardiomyopathy. J Inherit Metab Dis 1997; 20(5): 674–679.
32. Marin-Garcia J, Goldenthal MJ. Mitochondrial cardiomyopathy: molecular and biochemical analysis. Pediatr Cardiol 1997; 18(4): 251–260.
33. Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging and pathology. Cardiovasc Res 2005; 68(3): 355–365.
34. Taylor GP. Neonatal mitochondrial cardiomyopathy. Pediatr Dev Pathol 2004; 7(6): 620–624.
35. Kim UK, Kim HS, Oh BH et al. Analysis of mitochondrial DNA deletions in four chambers of failing human heart: hemodynamic stress, age, and disease are important factors. Basic Res Cardiol 2000; 95(2): 163-171.
36. Friedrich FW, Wilding BR, Reischmann S et al. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet 2012; 21(14): 3237–3254.
37. Sreeram N, Kitchener D, Smith A. Spectrum of valvular abnormalities in Noonan’s syndrome – a pathologic study. Cardiol Young 1994; 4(1): 62–66.
38. McMahon JN, Berry PJ, Joffe HS. Fatal hypertrophic cardiomyopathy in an infant of a diabetic mother. Pediatr Cardiol 1990; 11(4): 211–212.
39. Israel BA, Sherman FS, Guthrie RD. Hypertrophic cardiomyopathy associated with dexamethasone therapy for chronic lung disease in preterm infants. Am J Perinatol 1993; 10(4): 307–310.
40. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32(1): 45-59.
41. Rapezzi C, Quarta CC, Obici L et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 2013; 34(7): 520-528.
42. Leone O, Longhi S, Quarta CC et al. New pathological insights into cardiac amyloidosis: implications for non-invasive diagnosis. Amyloid 2012; 19(2): 99-105.
43. Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med 2011; 124(11): 1006-1015.
44. Otten E, Asimaki A, Maass A et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010; 7(8): 1058–1064.
45. Olivé M, Kley RA, Goldfarb LG. Myofibrillar myopathies: new developments. Curr Opin Neurol 2013; 26(5): 527-535.
46. Kostera-Pruszczyk A, Pruszczyk P, Kaminska A et al. Diversity of cardiomyopathy phenotypes caused by mutations in desmin. Int J Cardiol 2008; 131: 146-147.
47. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 2004; 127(Pt 2): 439–451.
48. Schröder R, Schoser B. Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol 2009; 19(3): 483-492.
49. Arbustini E, Pasotti M, Pilotto A et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail 2006; 8(5): 477-483.
50. Ogbogu PU, Rosing DR, Horne 3rd MK. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin N Am 2007; 27: 457-475.
51. Arnold M, McGuire L, Lee JC. Loeffler’s fibroplastic endocarditis. Pathology 1988; 20(1): 79-82.
52. Fauci AS, Harley JB, Roberts WC et al. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 1982; 97(1): 78–92.
53. Iglezias SD, Benvenuti LA, Calabrese F et al. Endomyocardial fibrosis: pathological and molecular findings of surgically resected ventricular endomyocardium. Virchows Arch 2008; 453(3): 233-241.
54. Wood JC. Cardiac iron across different transfusion-dependent diseases. Blood Rev 2008; 22 (Suppl 2): 14-21.
55. Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 2012; 9(4): 223-233.
56. Basso C, Corrado D, Marcus FI et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009; 373(9671): 1289-1300.
57. Pilichou K, Remme CA, Basso C et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med 2009; 206(8): 1787-1802.
58. D’Amati G, Leone O, diGioia CR et al. Arrhythmogenic right ventricular cardiomyopathy: clinicopathologic correlation based on a revised definition of pathologic patterns. Hum Pathol 2001; 32(10): 1078–1086.
59. Marcus FI, McKenna WJ, Sherrill D et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31(7): 806-814.
60. Tandri H, Saranathan M, Rodriguez ER et al. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol 2005; 45(1): 98-103.
61. Asimaki A, Tandri H, Huang H et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009; 360(11): 1075-1084.
62. Burke A, Mont E, Kutys R, Virmani R. Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol 2005; 36(4): 403-411.
63. Sarma RJ, Chana A, Elkayam U. Left ventricular noncompaction. Prog Cardiovasc Dis 2010; 52(4): 264-273.
64. Malhotra V, Ferrans VJ, Virmani R. Infantile histiocytoid cardiomyopathy: three cases and literature review. Am Heart J 1994; 128(5): 1009–1021.
65. Wybraniec M, Mizia-Stec K, Krzych L. Stress cardiomyopathy: yet another type of neurocardiogenic injury: ‘stress cardiomyopathy’. Cardiovasc Pathol 2014; 23(3): 113-120.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2019 Číslo 4
Najčítanejšie v tomto čísle
- Myokarditida a kardiomyopatie z pohledu kardiologa
- Fumarát hydratáza deficientní karcinom z renálních buněk a jemu podobný karcinom z renálních buněk: Komparativní studie 23 geneticky testovaných případů
- Inflamatorní myofibroblastický tumor dělohy – kazuistika
- Nová učebnice PATOLOGIE je tady