
Význam mikroprostředí kostní dřeně v patogenezi mnohočetného myelomu
Implication of Bone Marrow Microenvironment in Pathogenesis of Multiple Myeloma
Multiple myeloma is a hematooncological disease characterized by malignant proliferation of plasma cells. These cells accumulate in the bone marrow where they suppress physiological hematopoiesis; at the same time, these cells interact with a wide variety of cytokines, growth factors and adhesion molecules. It is obvious that the bone marrow microenvironment plays an important role in disease pathogenesis as well as treatment resistance.
Key words:
multiple myeloma – bone marrow – IL‑6
This study was supported by scientific program of the Czech Ministry of Education, Youth and Sports No. MSM0021622434, by grant of Czech Science Foundation No. GAP304/10/1395 and by grant of Internal Grant Agency of the Czech Ministry of Health No. NT11154 and NT12130.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
26. 4. 2012
Accepted:
10. 5. 2012
Autori:
B. Fišerová; L. Kubiczková; S. Ševčíková; R. Hájek
Pôsobisko autorov:
Babákova myelomová skupina, Ústav patologické fyziologie, LF MU Brno
Vyšlo v časopise:
Klin Onkol 2012; 25(4): 234-240
Kategória:
Přehledy
Súhrn
Mnohočetný myelom je hematoonkologické onemocnění charakterizované maligní proliferací plazmatických buněk. Tyto buňky se hromadí v kostní dřeni, kde potlačují fyziologickou krvetvorbu a zároveň interagují s celou škálou cytokinů, růstových faktorů a adhezivních molekul. Je zřejmé, že právě mikroprostředí kostní dřeně hraje velkou roli v patogenezi onemocnění, ale i v rezistenci k léčbě.
Klíčová slova:
mnohočetný myelom – kostní dřeň – IL‑6
Úvod
Mnohočetný myelom (MM) je maligní hematologické onemocnění charakterizované monoklonální expanzí plazmatických (myelomových) buněk. Toto onemocnění se týká hlavně starších pacientů s mediánem věku stanovení diagnózy 69 let, incidence v ČR je 4/ 100 000. Myelomové buňky jsou lokalizovány v kostní dřeni, kde narušují fyziologický proces hematopoezy a zároveň narušují strukturu kosti, což vede ke vzniku osteolytických lézí, které jsou pro pacienty s MM hlavním zdrojem obtíží [1,2].
MM je vhodným modelem pro studium interakcí tumoru a mikroprostředí ze tří důvodů: na rozdíl od normálních buněk se maligní plazmatické buňky hromadí zejména v kostní dřeni, což znamená, že stromální buňky poskytují jedinečné mikroprostředí pro růst maligních buněk. Dalším důvodem je přítomnost mnoha adhezivních molekul na povrchu myelomových buněk [3], normální buňky povrchové markery téměř neprodukují. Třetím důvodem je bezesporu možnost kultivace heterogenních populací adherentních buněk odebraných z kostní dřeně pacientů s MM v podmínkách in vitro [4].
Interakce adhezivních molekul
Adhezivní molekuly umožňují přímé propojení mezibuněčné hmoty s myelomovými buňkami a buňkami navzájem. Po navázání dalších molekul se spouští řada dějů, které mohou ovlivňovat vývoj buněčných složek nebo aktivovat signalizační kaskády. Je také podporována aktivace osteoklastů a růst maligních buněk, které jsou ještě více zadržovány v kostní dřeni [5] (obr. 1).
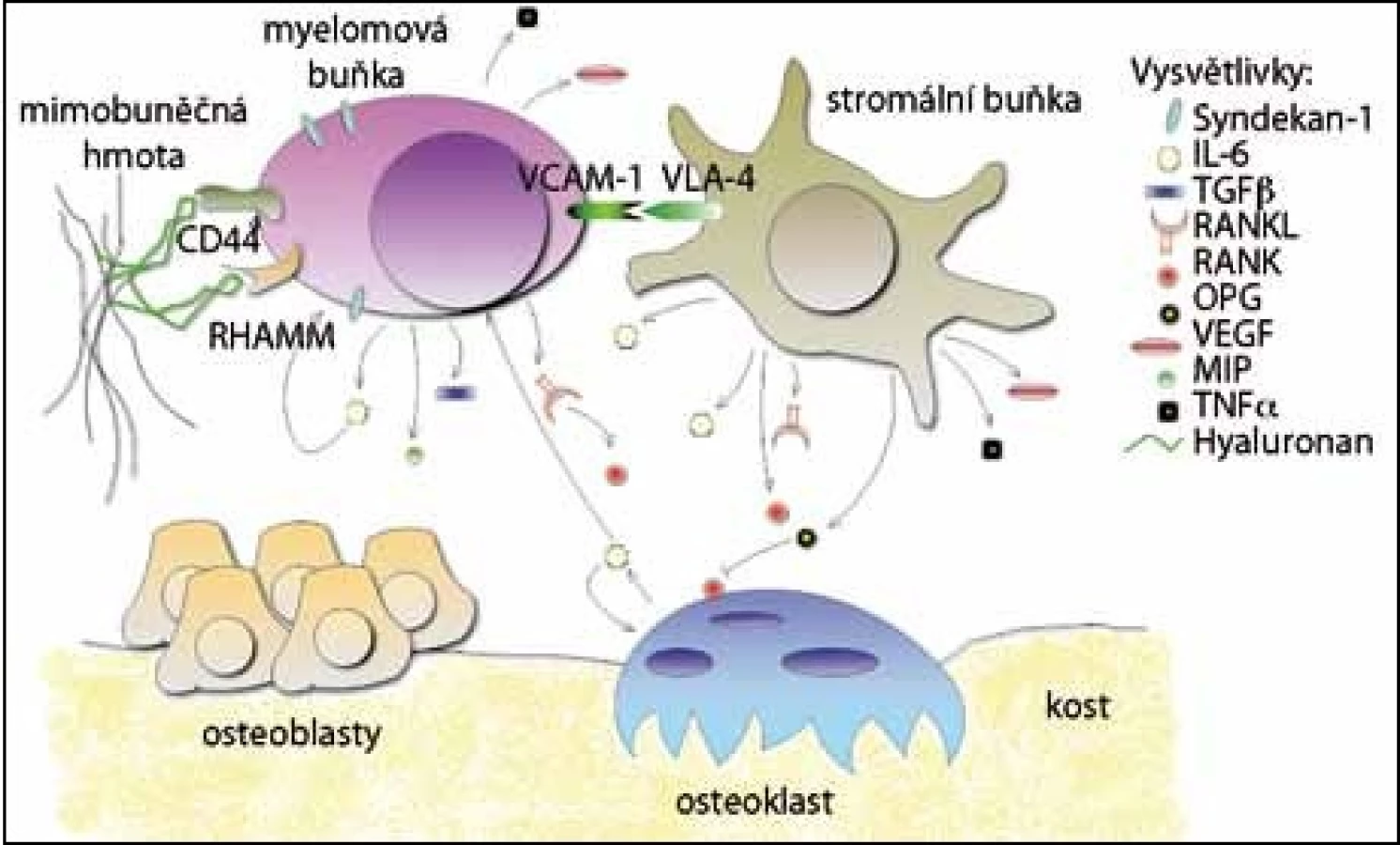
Jedním ze specifických povrchových markerů myelomových buněk je syndekan– 1 (CD138). Je to transmembránový proteoglykan, který se může přímo vázat na proteiny mezibuněčné hmoty, u MM se váže na kolagen typu I [6]. V kostní dřeni je syndekan– 1 detekován pouze na buňkách z B lymfoidní linie a jeho exprese se mění se stupněm diferenciace. U myší byl nalezen na povrchu pre‑B buněk, u zralých B buněk se nevyskytoval a opět byl produkován u plazmatických buněk [7]. U pacientů s MM se vyskytuje pouze na povrchu myelomových buněk [8], inhibuje osteoklastogenezi a pozitivně ovlivňuje diferenciaci osteoblastů [9]. U myelomových buněk, které procházejí apoptózou, se však rychle ztrácí [10]. Jelikož je syndekan– 1 produkován na povrchu životaschopných myelomových buněk, byly vyvinuty specifické protilátky, které dnes umožňují identifikaci a purifikaci myelomových buněk ze vzorků pacientů [8].
Za interakci myelomových buněk s hyaluronanem jsou zodpovědné dva receptory, a to CD44 [11] a RHAMM [12]. Standardní forma receptoru CD44 se na povrchu myelomových buněk vyskytuje zřídka, ovšem některé nestandardní receptorové varianty jsou zde velmi časté. Byly detekovány například varianty 3v, 4v, 6v a 10v, které nejsou přítomné u zdravých jedinců [13]. Receptor RHAMM napomáhá pohybu myelomových buněk po hyaluronovém substrátu. U MM jsou vylučovány tři formy: RHAMMFL, což je běžný typ, a dále dva deleční mutanti, RHAMM– 48, (delece 48 bp) a RHAMM– 147 (delece 147 bp). Oba deleční mutanti jsou přítomni jen u B buněk a plazmatických buněk MM, ale ne u zdravých jedinců. Výskyt delečních mutantů mění intracelulární signalizaci v buňkách MM [12].
Cytokiny, růstové faktory
Myelomové i stromální buňky produkují látky, které ovlivňují vývoj MM. Mezi tyto látky se řadí cytokiny a chemokiny, které mohou působit jako promotory nádorového vývoje, růstové faktory nebo chemoatraktanty. Váží se na receptory, a tím aktivují různé signální kaskády. Pacienti s MM vykazují typické rozpustné faktory v mikroprostředí kostní dřeně: IL‑16, IL‑2R, MCP– 1, HGF, IL‑1RA, MIG, IP– 10, EGF [14].
Stěžejním cytokinem MM je interleukin‑6 (IL‑6), který je produkován mnoha buňkami včetně osteoblastů, monocytů, makrofágů a stromálních buněk kostní dřeně. Za fyziologických podmínek je jeho hladina nízká nebo nedetekovatelná, ale bylo prokázáno, že u MM pacientů s osteolytickými lézemi je hladina IL‑6 zvýšená [15].
U MM je IL‑6 hlavním cytokinem, který zprostředkovává růst, přežívání a lékovou rezistenci myelomových buněk (obr. 2). I když některé myelomové buňky produkují IL‑6 autokrinně [16], primárně je produkován stromálními buňkami kostní dřeně a působí parakrinně na růst a diferenciaci myelomových buněk [17]. Produkce IL‑6 ve stromálních buňkách je tedy indukována buď adhezí myelomových buněk [18], nebo prostřednictvím jiných cytokinů, jako tumor nekrotizující faktor α (TNFα) [19] a vaskulární endoteliální růstový faktor (VEGF) [20]. Zmíněné faktory následně aktivují např. signální dráhu jaderného faktoru kB (NF– kB), která má vliv na přežívání a růst myelomových buněk [21].
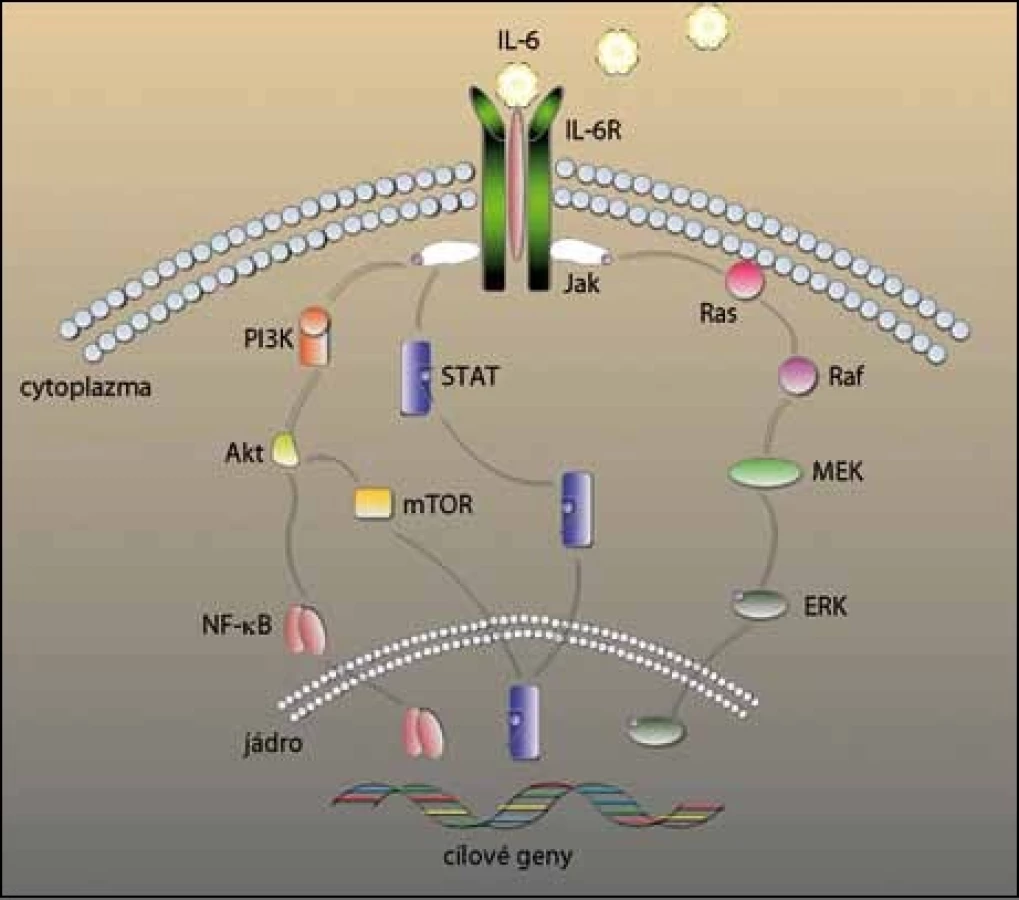
Osteolýza, další patologický proces podporovaný přítomností IL‑6, je navozena hned několika mechanizmy. Za prvé, IL‑6 indukuje produkci RANKL v mezenchymálních buňkách kostní dřeně a osteoblastech. Vazbou RANKL na RANK je navozeno dozrávání osteoklastů a aktivace signálních drah [22]. Za druhé, IL‑6 indukuje zvýšení hladin proteinů zapojených do procesu kostní resorpce, např. peptidů vázajících paratyroidní hormon PTHrP [23]. Za třetí, IL‑6 inhibuje osteogenezi zprostředkovanou Wnt, ještě více ruší homeostázu v kosti a posouvá rovnováhu směrem k degradaci kosti [24].
Hladina IL‑6 odráží stupeň rozvoje monoklonálních gamapatií, jak bylo prokázáno ve studii, do které bylo začleněno 131 pacientů. Ze skupiny 22 nemocných s MGUS, což je prekancerózní stadium předcházející MM, byl IL‑6 detekován pouze u jednoho jedince, u MM to bylo už 35 % pacientů a u nejagresivnějšího stadia zvaného plazmocytární leukemie se IL‑6 vyskytoval ve vysoké koncentraci u všech pacientů. Navíc se hodnoty IL‑6 lišily i mezi MM pacienty, podíl pacientů s vyšší hladinou IL‑6 byl jiný při diagnóze (37 %), během intenzivního vývoje (60 %) a během stabilní fáze (13 %) [25]. Z druhé strany bylo prokázáno, že agresivní mimokostní stadia MM mohou být nezávislá na hladině IL‑6 [26]. V klinických studiích byl testován účinek anti‑IL‑6 mAB (např. CNTO 328), který ale neprokázal zásadní vliv na léčbu MM.
RANKL, ligand z rodiny TNF, je produkován nezralými osteoblasty, stromálními buňkami a T lymfocyty [27,28]. Osteoklasty a jejich prekurzory produkují jeho receptor RANK. Vazbou RANKL na RANK se aktivují signální dráhy, které jsou důležité při diferenciaci osteoklastů z jejich prekurzorových buněk, RANKL také reguluje diferenciaci, funkci a přežívání osteoklastů [22,29]. Osteoprotegerin (OPG) je antagonistou RANKL, inhibuje vazbu RANK– RANKL, inhibuje diferenciaci a aktivaci osteoklastů, a tím brání degradaci kosti [30].
U MM je RANKL hlavním osteoklastogenním faktorem podílejícím se na lytické kostní nemoci. Jeho vysoká produkce ve stromálních buňkách pacientů s MM má důležitou úlohu v patogenezi MM [31]. Některé studie ukázaly, že RANKL je produkován i myelomovými buňkami [32]. V mikroprostředí kostní dřeně MM se interakcí stromálních a myelomových buněk produkce RANKL zvyšuje a OPG snižuje, což podporuje kostní resorpci a osteolýzu [33]. Hladiny těchto molekul korelují s klinickou aktivitou MM a závažností kostní nemoci [34]. Denosumab, monoklonální protilátka proti RANKL, byla schválena FDA k ochraně kostí před dalším poškozením u pacientů s nádory prostaty a prsu s kostními metastázami. V současné době probíhají klinické studie fáze III u pacientů s MM.
Myelomové buňky také vylučují transformující růstový faktor β (TGFβ), pleiotropní cytokin, který za normálních podmínek mimo jiné inhibuje imunitní odpověď tím, že brání proliferaci a diferenciaci B lymfocytů a sekreci Ig [35]. Na rozdíl od účinku na B lymfocyty TGFβ nesnižuje proliferaci myelomových buněk, a při vysokých koncentracích dokonce podporuje vylučování IL‑6 těmito buňkami. Tímto zčásti zprostředkovává růst myelomu a podporuje patogenezi [36]. Navíc je TGFβ důležitý v nerovnovážné kostní remodelaci u MM, je aktivní při zvýšené kostní resorpci a inhibuje osteoblasty. Inhibice TGFβ podporuje diferenciaci osteoblastů, které pak inhibují růst a přežívání myelomových buněk. Naopak potlačení diferenciace osteoblastů urychluje ztrátu kostní tkáně [37].
Signální dráhy důležité v mikroprostředí kostní dřeně MM
V patogenezi MM je mnoho regulátorů, proteinových kináz a růstových faktorů, pomocí kterých buňky komunikují. Tyto procesy nejsou jednoduché, právě přímým kontaktem strukturních a buněčných složek nebo podporou autokrinní a parakrinní produkce cytokinů se v mikroprostředí kostní dřeně aktivuje široké spektrum signálních drah [18,21,38,39].
Wnt/β– katenin
Důležitou roli při růstu, vývoji a fungování osteoblastů hraje signální dráha Wnt/ β– katenin (obr. 3). Glykoproteiny Wnt se váží na koreceptory LRP– 5 nebo LRP– 6 a Frizzled receptor a aktivují Wnt dráhu. Přenos signálu stabilizuje b– katenin, který je translokován do jádra a zde stimuluje expresi genů zodpovědných za diferenciaci osteoblastů. Bez přítomnosti signálu je b– katenin fosforylován a degradován v proteazomu.
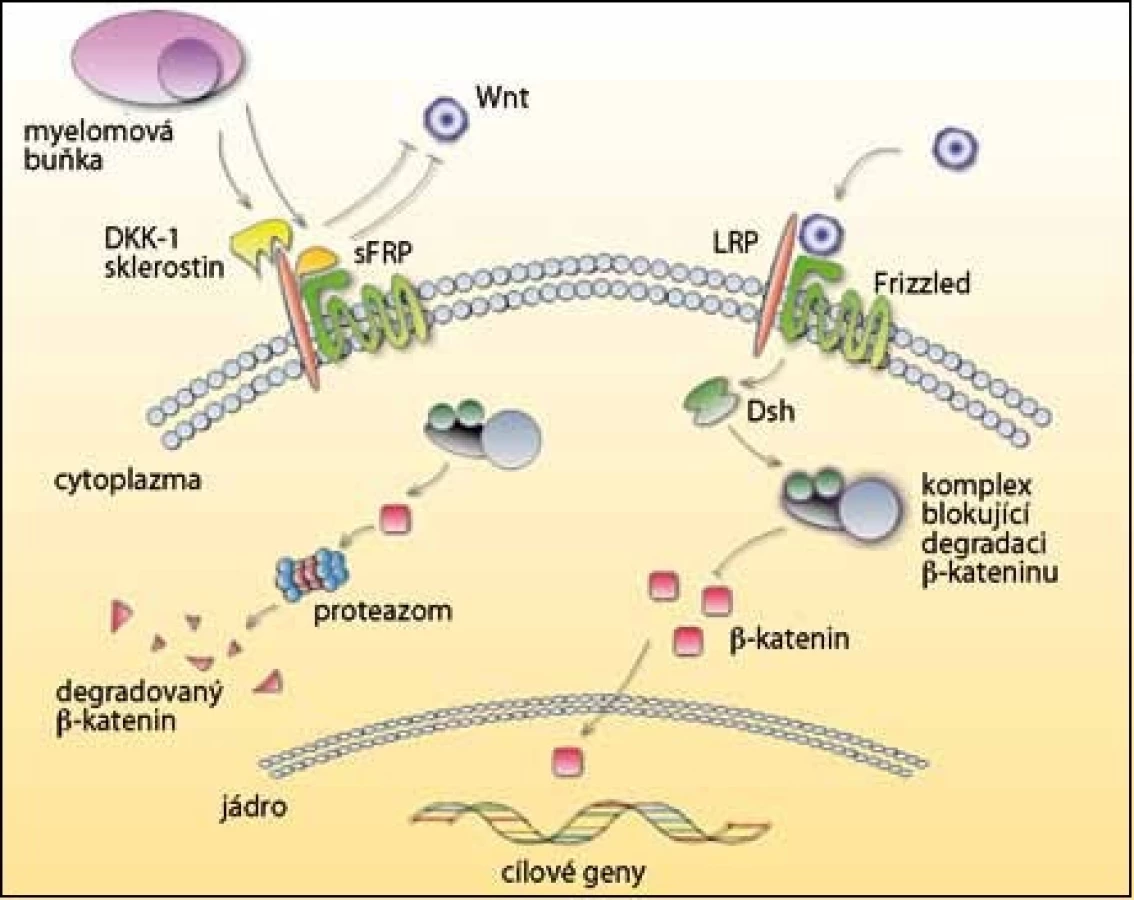
Existují dvě funkční skupiny antagonistů Wnt signalizace, a to sFRP a Dickkopf (DKK), po jejichž navázání se naruší funkce osteoblastů [40,41]. Významným inhibitorem u MM je DKK– 1, který se váže na LRP– 5. Je vylučován myelomovými buňkami a v jejich přítomnosti také stromálními buňkami a osteoblasty. DKK– 1 se vyskytuje především u pacientů s osteolytickými lézemi a jeho hladina koreluje s rozšířením osteolytických ložisek [24,42,43]. Kromě potlačení diferenciace osteoblastů také podporuje osteoklastogenezi zvýšenou expresí RANKL a sníženou expresí OPG [44]. V současnosti probíhají klinické studie fáze I/ II, které testují anti‑DKK– 1 monoklonální protilátku (BHQ880) u MM.
Další skupina inhibitorů sFRP blokuje vazbu k receptoru Frizzled. Myelomové buňky produkují sFRP– 2 a sFRP– 3 a ty významně potlačují diferenciaci osteoblastů a tvorbu kostí [45,46]. Existuje však i studie, která ukazuje, že hladiny DKK– 1 a sFRP u pacientů s MM nepotlačují diferenciaci lidských osteoblastů. To znamená, že nemusí být jedinými faktory zodpovědnými za inhibici osteoblastů [47].
Sklerostin je produkován osteocyty a působí na dráhu Wnt podobně jako DKK– 1, tedy navázáním na LRP– 5 [48]. Inhibuje aktivitu osteoblastů a indukuje jejich apoptózu, je negativním regulátorem tvorby kostní tkáně [49]. Teprve nedávno bylo potvrzeno, že sklerostin je vylučován i myelomovými buňkami, a tak přispívá k patogenezi MM [50].
Dráha NF– κB
Obecně je tato dráha důležitá pro proliferaci, přežívání a vývoj nádorových buněk. U MM působí aktivace signalizace NF– κB pozitivně na růst, rezistenci k lékům a přežívání myelomových buněk v mikroprostředí kostní dřeně [51]. Aktivace může probíhat jak klasickým způsobem, tak i alternativně. Bylo prokázáno, že mikroprostředí kostní dřeně u MM spouští tuto signalizaci prostřednictvím adheze i vylučováním cytokinů a chemokinů. Například růstový a anti‑-apoptotický faktor IL‑6, jehož produkce je vyšší při adhezi myelomových a stromálních buněk, může indukovat dráhu NF– κB [21]. Také TNFα aktivuje NF– κB: za prvé u stromálních buněk, čímž podporuje vylučování IL‑6 těmito buňkami, a za druhé u myelomových buněk, kde podporuje adhezi buněk a zvyšuje produkci intracelulární adhezivní molekuly 1 (ICAM‑1) a vaskulární adhezivní molekuly 1 (VCAM‑1) [19]. Aktivace je u MM možná oběma způsoby. Již dříve bylo zjištěno, že klasická dráha může být blokována inhibicí IKKβ proteinu [51], ovšem to neplatí u alternativního způsobu aktivace. Proto byla také zjišťována inhibice jiné molekuly, a to IKKa, která se vyskytuje u obou způsobů aktivace. Růst buněk byl sice zpomalen, ale aktivita signalizace NF–κB byla vyšší než u kontroly, což naznačuje, že inhibiční efekt IKKα je nezávislý na aktivitě NF– κB [52].
Dráha PI3K/ Akt
Fosfatidylinositol– 3– kináza (PI3K)/ Akt je jedna z nejčastěji aktivních drah u lidských nádorů. Mnoho proteinových kináz a transkripčních faktorů, které se účastní této signalizace, ovlivňuje rezistenci myelomových buněk k léčbě. Dráha PI3K/ Akt reguluje průběh buněčného cyklu a apoptózu, indukuje syntézu DNA a působí na přežívání a migraci myelomových buněk. Je propojena s dráhou NF– κB přes kinázu Akt, která, podobně jako IKK u dráhy NF– κB, fosforyluje a degraduje IkBa. To vede k přesunutí NF– κB do jádra, kde může indukovat transkripci anti‑apoptotických genů. Tedy Akt může inhibovat apoptózu aktivací NF– κB [53].
Aktivátory této dráhy jsou IL‑6 a růstový faktor podobný inzulinu 1 (IGF‑1); aktivace PI3K je důležitá při proliferaci a anti‑apoptotické odpovědi myelomových buněk na tyto cytokiny [54]. IL‑6 nejen spouští dráhu PI3K/ Akt, ale tato interakce má navíc regulační účinky na buněčný cyklus, chrání buňky před apoptózou způsobenou léky a ovlivňuje růst MM [55].
Důležitou součástí této dráhy je kináza mTOR, která se u savců vyskytuje ve dvou rozdílných komplexech, mTORC1 a mTORC2, které ovšem mají rozdílné funkce. Akt aktivuje mTORC1, který fosforyluje další molekuly regulující syntézu proteinů, a tak kontroluje buněčný růst. Naopak mTORC2 v odpovědi na růstové faktory aktivuje Akt a reguluje přežívání buněk [56]. V souvislosti s MM byl identifikován DEPTOR, který za fyziologických podmínek inhibuje mTORC1 a mTORC2. I když jeho vysoká produkce u MM inhibuje mTORC1, překvapivě také vede k aktivaci dráhy PI3K/ mTORC2/ Akt. Tento nepřímý způsob aktivace je důležitý ku příkladu u myelomových buněk, kterým chybí mutace aktivující PI3K [57]. V současnosti probíhají klinické studie, ve kterých jsou testovány kombinace lenalidomidu a everolimu (RAD001), inhibitoru mTOR jak v solidních nádorech, tak u refraktorního MM.
Dráhy Ras/ Raf/ MEK/ MAPK a Jak2/ STAT3
Dalšími důležitými drahami jsou Ras/ Raf/ MEK/ MAPK a Jak2/ STAT3, které mohou být aktivovány IL‑6. Inhibice IL‑6R sice blokuje fosforylaci STAT3, ale neovlivňuje aktivaci dráhy MAPK, z čehož vyplývá, že v mikroprostředí kostní dřeně se dráha STAT3 aktivuje prostřednictvím IL‑6 a dráha MAPK mechanizmy nezávislými na IL‑6 [58].
K aktivaci dráhy Ras/ Raf/ MEK/ MAPK, která stimuluje angiogenezi, proliferaci buněk a jejich apoptózu, jsou potřebné jak adhezivní interakce, tak vylučování růstových faktorů. Hlavními aktivačními faktory této dráhy jsou IL‑6 a IGF‑1, které aktivují Ras [59,60]. Následně dojde k aktivaci i ostatních složek – Raf, MEK a ERK. Tuto dráhu spouštějí také další faktory, jako VEGF [61]. Bylo dokázáno, že inhibicí aktivity ERK se sníží produkce VEGF, která vede ke snížené tvorbě nových cév v kostní dřeni indukované myelomovými buňkami [62].
Dráha Jak2/ STAT3 má vliv na přežívání myelomových buněk, aktivace této dráhy indukuje proliferaci a inhibici apoptózy. STAT3 přímo přispívá k malignímu rozvoji MM tím, že chrání myelomové buňky před apoptózou a podporuje přežívání [63]. Stimulace buněk IL‑6 vede k signalizaci přes IL‑6R a spouští fosforylaci STAT3 přes Jak, STAT3 je přenesen do jádra, kde aktivuje transkripci daných anti‑apoptotických genů. Aktivace pomocí IL‑6 tedy reguluje přežívání myelomových buněk vylučováním anti‑-apoptotických proteinů z rodiny Bcl– 2, například Bcl– XL, Mcl– 1 [63,64]. Bylo zjištěno, že v myelomových buňkách je STAT3 neustále aktivován a inhibice této dráhy indukuje apoptózu in vitro [58,65].
Rezistence k lékům způsobená mikroprostředím MM
Rezistentní fenotyp způsobený mikroprostředím MM může být dvojího typu: léková rezistence zprostředkovaná interakcemi cytokinů (cytokine mediated drug resistance – CM– DR), nebo léková rezistence zprostředkovaná adhezivním kontaktem buněk (cell adhesion– mediated drug resistance – CAM– DR). Oba mechanizmy mají zásadní význam v patogenezi MM. Výraz CAM– DR byl poprvé použit ve studii MM, kde byla pozorována zvýšená produkce α4, β1 a β7 integrinů, které jsou vylučovány myelomovými buňkami. Adheze maligních buněk k fibronektinu může přispět ke vzniku rezistence de novo, chrání buňky před apoptózou způsobenou léky [38].
S adhezí myelomových buněk k fibronektinu je také asociována zvýšená hladina p27kip1. Tento protein je důležitý pro udržení rezistentního fenotypu, má význam v buněčném cyklu, kde zadržuje buňky ve fázi G1. Bylo prokázáno, že přerušením adheze se hladina p27kip1 sníží, buňky pokračují v S fázi buněčného cyklu a opět se stávají citlivými k lékům. Pokusy s inhibicí produkce p27kip1 neměly vliv na adhezi, ale zvrátily lékovou rezistenci. To dokazuje, že vyšší produkce tohoto proteinu přispívá k CAM– DR [66].
U MM je také nadměrně vylučována adhezivní molekula P– selektin a její ligand PSGL– 1. Kromě jiných funkcí, jako jsou adheze a osídlování myelomových buněk v mikroprostředí kostní dřeně, je PSGL– 1 důležitý v rozvoji lékové rezistence myelomových buněk in vivo a in vitro. Inhibice interakcí tohoto ligandu k selektinu podporuje citlivost myelomových buněk k bortezomibu, inhibitoru proteazomu [67].
Hladina HSP70 je také zmíněnými interakcemi zvýšena, navíc podporuje vyšší produkci IL‑6, který pak napomáhá přežívání myelomových buněk pomocí aktivace signálních drah. Inhibice HSP70 potlačuje adhezi myelomových buněk k fibronektinu a způsobuje apoptózu rezistentních buněk. Fenotyp lékové rezistence je tak změněn a buňky se stávají citlivými k lékům. Tato studie ukázala, že inhibice HSP70 může způsobit apoptózu buněk, které vykazují rezistenci de novo i rezistenci získanou během léčby [68].
S rezistencí je také asociována dráha NF–κB, jejíž aktivace je stimulována adhezí k fibronektinu [39]. Neustálá aktivita této dráhy vzdoruje inhibujícím účinkům bortezomibu, i v jeho přítomnosti přispívá jinými mechanizmy k rezistenci. V této studii bylo navíc zjištěno, že pokud se myelomové buňky kultivují se stromálními buňkami pacienta s MM, aktivita dráhy NF– κB, a tím i rezistence k bortezomibu, je ještě více podporována [69]. Pozdější výsledky potvrdily, že aktivace dráhy NF– κB je významně vyšší ve stromálních buňkách a tyto buňky se významně liší od fyziologicky normálních buněk [70].
Závěr
V posledních letech bylo provedeno mnoho studií týkajících se mikroprostředí kostní dřeně u pacientů s MM, které dostávají toto téma do popředí zájmu a ukazují na jeho význam v patogenezi nemoci. Nicméně mechanizmy probíhající v tomto mikroprostředí nebyly doposud zcela objasněny, proto je nutné pokračovat v dalším výzkumu. Na základě nových poznatků o aktivitě buněk kostní dřeně a procesech podílejících se na vzniku myelomových buněk bude možno identifikovat nové potenciální cíle a markery pro nové léky a stanovit efektivnější léčbu pro pacienty.
Práce byla podpořena výzkumným záměrem MŠMT ČR č. MSM0021622434, grantem GA ČR
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Sabina Ševčíková, Ph.D.
Babákova myelomová skupina
Ústav patologické fyziologie
LF MU Brno
Kamenice 5, A3
625 00 Brno
e-mail: sevcik@med.muni.cz
Obdrženo: 26. 4. 2012
Přijato: 10. 5. 2012
Zdroje
1. Hájek R, Mužík J, Maisnar V et al. Mnohočetný myelom, MNK klasifikace a Národní onkologický registr České republiky. Klin Onkol 2007; 20 (Suppl 1): 147– 151.
2. Hájek R, Krejčí M, Pour L et al. Mnohočetný myelom. Klin Onkol 2011; 24 (Suppl 1): S10– S13.
3. Uchiyama H, Barut BA, Chauhan D et al. Characterization of adhesion molecules on human myeloma cell lines. Blood 1992; 80(9): 2306– 2314.
4. Caligaris– Cappio F, Bergui L, Gregoretti MG et al. Role of marrow stromal cells in the growth of human multiple myeloma. Blood 1991; 77(12): 2688– 2693.
5. Gregoretti MG, Gottardi D, Ghia P et al. Characterization of bone marrow stromal cells from multiple myeloma. Leuk Res 1994; 18(9): 675– 662.
6. Ridley RC, Xiao H, Hata H et al. Expression of syndecan regulates human myeloma plasma cell adhesion to type I collagen. Blood 1993; 81(3): 767– 774.
7. Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul 1989; 1(1): 27– 35.
8. Wijdenes J, Vooijs WC, Clément C et al. A plasmocyte selective monoclonal antibody (B– B4) recognizes syndecan– 1. Br J Haematol 1996; 94(2): 318– 323.
9. Dhodapkar MV, Abe E, Theus A et al. Syndecan– 1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood 1998; 91(8): 2679– 2688.
10. Jourdan M, Ferlin M, Legouffe E et al. The myeloma cell antigen syndecan– 1 is lost by apoptotic myeloma cells. Br J Haematol 1998; 100(4): 637– 646.
11. van Driel M, Günthert U, Stauder R et al. CD44 isoforms distinguish between bone marrow plasma cells from normal individuals and patients with multiple myeloma at different stages of disease. Leukemia 1998; 12(11): 1821– 1828.
12. Crainie M, Belch AR, Mant MJ et al. Overexpression of the receptor for hyaluronan– mediated motility (RHAMM) characterizes the malignant clone in multiple myeloma: identification of three distinct RHAMM variants. Blood 1999; 93(5): 1684– 1696.
13. Eisterer W, Betcher O, Hilbe W et al. CD44 isoforms are differentially regulated in plasma cell dyscrasias and CD44v9 represents a new independent prognostic parametr in multiple myeloma. Leuk Res 2001; 25(12): 1051– 1057.
14. Cao Y, Luetkens T, Kobold S et al. The cytokine/ chemokine pattern in the bone marrow environment of multiple myeloma patients. Exp Hematol 2010; 38(10): 860– 867.
15. Sati H, Apperley JF, Greaves M et al. Interleukin‑6 is expressed by plasma cells from patients with multiple myeloma and monoclonal gammopathy of undetermined significance. Br J Haematol 1998; 101(2): 287– 295.
16. Villunger A, Egle A, Kos M et al. Contituents of autocrine IL‑6 loops in myeloma cell lines and their twargeting for suppression of neoplastic growth by antibody strategies. Int J Cancer 1996; 65(4): 498– 505.
17. Klein B, Zhang XG, Jourdan M et al. Paracrine rather then autocrine regulation of myeloma– cell growth and differentiation by interleukin‑6. Blood 1989; 73(2): 517– 526.
18. Uchiyama H, Barut BA, Mohrbacher AF et al. Adhesion of human myeloma– derived cell lines to bone marrow stromal cells stimulates interleukin‑6 secretion. Blood 1993; 82(12): 3712– 3720.
19. Hideshima T, Chauhan D, Schlossman R et al. The role of tumor necrosis factor a in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 2001; 20(33): 4519– 4527.
20. Dankbar B, Padró T, Leo R et al. Vascular endothelial factor and interleukin‑6 in paracrine tumor– stromal cell interactions in multiple myeloma. Blood 2000; 95(8): 2630– 2636.
21. Chauhan D, Uchiyama H, Akbarali Y et al. Multiple myeloma adhesion‑induced interleukin‑6 expression in bone marrow stromal cells involves activation of NF– kB. Blood 1996; 87(3): 1104– 1112.
22. Kim K, Lee J, Kim JH et al. Protein inhibitor of activated STAT3 modulates osteoclastogenesis by down– regulation of NFATc1 and osteoclast‑associated receptor. J Immunol 2007; 1178(9): 5588– 5594.
23. de la Mata J, Uy HL, Guise TA et al. Interleukin‑6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone‑related protein in vivo. J Clin Invest 1995; 95(6): 2846– 2852.
24. Gunn WG, Conley A, Deininger L et al. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK– 1 and interleukin‑6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 2006; 24(4): 986– 991.
25. Bataille R, Jourdan M, Zhang XG et al. Serum levels of interleukin‑6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J Clin Invest 1989; 84(6): 2008– 2011.
26. Asaoku H, Kawano M, Iwato K et al. Decrease in BSF– 2/ IL‑6 response in advanced cases of multiple myeloma. Blood 1988; 72(2): 429– 432.
27. Yasuda H, Shima N, Nakagawa N et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/ osteoclastogenesis‑inhibitory factor and is identical to TRANCE/ RANKL. Proc Natl Acad Sci USA 1998; 95(7): 3597– 3602.
28. Atkins GJ, Kostakis P, Pan B et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res 2003; 18(6): 1088– 1098.
29. Lacey DL, Timms E, Tan HL et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93(2): 165– 176.
30. Simonet WS, Lacey DL, Dunstan CR et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997; 89(2): 309– 319.
31. Roux S, Meignin V, Quillard J et al. RANK (receptor activator of nuclear factor– kB) and RANKL expression in multiple myeloma. Br J Haematol 2002; 117(1): 86– 92.
32. Croucher PI, Shipman CM, Lippitt J et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood 2001; 98(13): 3534– 3540.
33. Giuliani N, Bataille R, Mancini C et al. Myeloma cells induce imbalance in the osteoprotegerin/ osteoprotegerin ligand systém in the human bone marrow microenvironment. Blood 2001; 98(13): 3527– 3533.
34. Terpos E, Szydlo R, Apperley JF et al. Soluble receptor activator of nuclear factor kB ligand– osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood 2003; 102(3): 1064– 1069.
35. Kehrl JH, Roberts AB, Wakefield LM et al. Transforming growth factor b is an important immunomodulatory protein for human B lymphocytes. J Immunol 1986; 137(12): 3855– 3860.
36. Urashima H, Ogata A, Chauhan D et al. Transforming growth factor– b1: differential effects on multiple myeloma versus normal B cells. Blood 1996; 87(5): 1928– 1938.
37. Takeuchi A, Abe M, Hiasa M et al. TGF‑β inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLos ONE 2010; 5(3): e9870.
38. Damiano JS, Cress AE, Hazlehurst LA et al. Cell adhesion mediated drug resistance (CAM– DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999; 93(5): 1658– 1667.
39. Landowski TH, Olashaw NE, Agrawal D et al. Cell adhesion– mediated drug resistance (CAM– DR) is associated with activation of NF– kB (RelB/ p50) in myeloma cells. Oncogene 2003; 22(16): 2417– 2421.
40. Gong Y, Slee RB, Fukai N et al. Osteoporosis– Pseudoglioma Syndrome Collaborative Group. LDL receptor‑related protein 5 (LRP5) affects bone accural and eye development. Cell 2001; 107(4): 513– 523.
41. Bain G, Müller T, Wang X et al. Activated b– catenin induces osteoblast differentiation of C3H10T1/ 2 cells and participates in BMP2 mediated signal transduction. Biochem Biophys Res Commun 2003; 301(1): 84– 91.
42. Tian E, Zhan E, Walker R et al. The role of the WNT– signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 2003; 349(26): 2483– 2494.
43. Politou M, Heath DJ, Rahemtulla A et al. Serum concentrations of Dickkopf– 1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int J Cancer 2006; 119(7): 1728– 1731.
44. Qiang YW, Barlogie B, Rudikoff S et al. Dkk1‑induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 2008; 42(4): 669– 680.
45. Oshima T, Abe M, Asano J et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP– 2. Blood 2005; 106(9): 3160– 3165.
46. Giuliani N, Morandi F, Tagliaferri S et al. Production of Wnt inhibitors by myeloma cells: potantial effects on canonical Wnt pathway in the bone microenvironment. Cancer Res 2007; 67(16): 7665– 7674.
47. Giuliani N, Colla S, Morandi F et al. Myeloma cells block RUNX2/ CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005; 106(7): 2472– 2483.
48. Semënov M, Tamai K, He X. SOST is a ligand for LRP5/ LRP6 and a Wnt signaling inhibitor. J Biol Chem 2005; 280(29): 26770– 26775.
49. Sutherland MK, Geoghegan JC, Yu C et al. Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone 2004; 35(4): 828– 835.
50. Colucci S, Brunetti G, Oranger A et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J 2011; 1(6): e27.
51. Hideshima T, Chauhan D, Richardson P et al. NF– kB as a therapeutic target in multiple myeloma. J Biol Chem 2002; 277(19): 16639– 16647.
52. Hideshima T, Chauhan D, Kiziltepe T et al. Biologic sequelae of IkB kinase (IKK) inhibition in multiple myeloma: therapeutic implications. Blood 2009; 113(21): 5228– 5236.
53. Madrid LV, Wang CY, Guttridge DC et al. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/ p65 sibunit of NF– kB. Mol Cell Biol 2000; 20(5): 1626– 1638.
54. Tu Y, Gardner A, Lichtenstein A. The phosphatidylinositol 3– kinase/ AKT kinase pathway in multiple myeloma plasma cells: roles in cytokine– dependent survival and proliferative responses. Cancer Res 2000; 60(23): 6763– 6770.
55. Hideshima T, Nakamura N, Chauhan D et al. Biologic sequelae of interleukin‑6 induced PI3– K/ Akt signaling in multiple myeloma. Oncogene 2001; 20(42): 5991– 6000.
56. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12(1): 9– 22.
57. Peterson TR, Laplante M, Thoreen CC et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137(5): 873– 886.
58. Chatterjee M, Stühmer T, Herrmann P et al. Combined disruption of both the MEK/ ERK and the IL‑6R/ STAT3 pathways is required to induce apoptosis of multiple myeloma cells in the presence of bone marrow stromal cells. Blood 2004; 104(12): 3712– 3721.
59. Nakafuku M, Satoh T, Kkaziro Y. Differentiation fectors, including nerve growth factor, fibroblast growth factor, and interleukin‑6, induce an accumulation of an active Ras•GTP complex in rat pheochromocytoma PC12 cells. J Biol Chem 1992; 267(27): 19448– 19454.
60. Ge NL, Rudikoff S. Insulin‑like growth factor I is a dual effector of multiple myeloma cell growth. Blood 2000; 96(8): 2856– 2861.
61. Podar K, Tai Y, Davies FE et al. Vascular endothelial factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001; 98 (2): 428– 435.
62. Giuliani N, Lunghi P, Morandi F et al. Downmodulation of ERK protein kinase activity inhibits VEGF secretion by human myeloma cells and myeloma‑induced angiogenesis. Leukemia 2004; 18(3): 628– 635.
63. Catlett– Falcone R, Landowski TH, Oshiro MM et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999; 10(1): 105– 115.
64. Puthier D, Bataille R, Amiot M. IL‑6 up– regulates mcl– 1 human myeloma cells through JAK/ STAT rather then ras/ MAP kinase pathway. Eur J Immunol 1999; 29(12): 3945– 3950.
65. Bharti AC, Shishodia S, Reuben JM et al. Nuclear factor– kB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004; 103(8): 3175– 3184.
66. Hazlehurst LA, Damiano JS, Buyuksal I et al. Adhesion to fibronectin via b1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM– DR). Oncogene 2000; 19(38): 4319– 4327.
67. Azab AK, Quang P, Azab F et al. P– selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood 2012; 119(6): 1468– 1478.
68. Nimmanapalli R, Gerbino E, Dalton WS et al. HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br J Haematol 2008; 142(4): 551– 561.
69. Markovina S, Callander NS, O’Connor SL et al. Bortezomib‑resistant nuclear factor– kB activity in multiple myeloma cells. Mol Cancer Res 2008; 6(8): 1356– 1364.
70. Markovina S, Callender NS, O’Connor SL et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF– kB activity in myeloma cells. Mol Cancer 2010; 9: 176.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2012 Číslo 4
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Nejasný stín na plicích – kazuistika
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Antidepresivní efekt kombinovaného analgetika tramadolu s paracetamolem
- Kombinace metamizol/paracetamol v léčbě pooperační bolesti u zákroků v rámci jednodenní chirurgie
Najčítanejšie v tomto čísle
- Hepatocelulární karcinom – dlouhodobě léčitelné onemocnění
- Mutace genu EGFR u pacientů s pokročilým NSCLC
- Léčba nízkostupňového gliomu supratentoriální oblasti mozku po neurochirurgické subtotální resekci s projevy objemového up-gradingu
- CT kolonografie – přehled vývoje metodiky a indikací